
Optimization of HIV Sequencing Method Using Vela Sentosa Library on Miseq Ilumina Platform

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Test Validation Samples
2.2. RNA Extraction
2.3. PCR Amplification
2.4. Library Preparation for Ion Torrent PGM Platform
2.5. Library Preparation for Illumina Miseq Platform
2.6. Sequence Data Analysis
- (1)
- The paired-end detection of read files, if uploaded in the same batch;
- (2)
- Technology-specific quality filtering to trim or remove low-quality reads;
- (3)
- The establishment of a work list of the batch.
- (1)
- Quality filtering: R1 and R2 files are merged for Illumina data, then a sliding window (size of 25 nt) is applied to reads, enhancing the trimming of poor-quality sections having a low Phred score (<23 for the present study) and filtering short reads as well (<20 nt in this study);
- (2)
- Read mapping: reads passing the quality filters are mapped against HIV-1 profiles for PR, RT, and INT, generating a frame-aware nucleic acid alignment, using a proprietary alignment method of combined global and local alignments developed by SmartGene, followed by the translation of the corrected alignments into amino acid sequences;
- (3)
- Mapped reads are used for the creation of a frequency matrix;
- (4)
- Mutations are determined and validated with respect to the reference (here, HxB2, accession no. K03455) and their frequencies are calculated.
2.7. Statistical Analysis
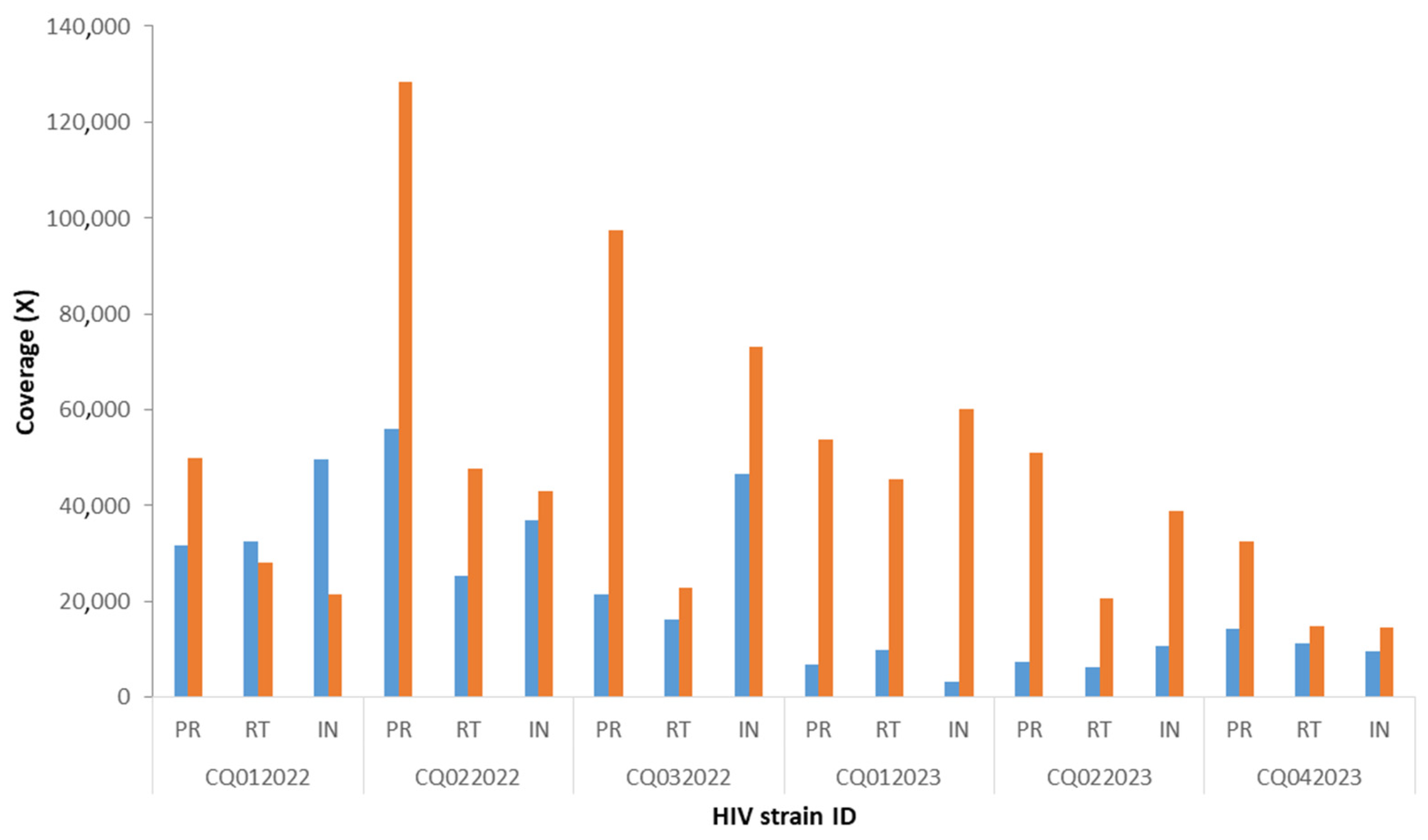
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Joint United Nations Programme on HIV/AIDS (UNAIDS). The Path that Ends AIDS: UNAIDS Global AIDS Update 2023; Joint United Nations Programme on HIV/AIDS: Geneva, Switzerland, 2023; Available online: https://thepath.unaids.org/wp-content/themes/unaids2023/assets/files/2023_report.pdf (accessed on 5 January 2024).
- Santé Publique France. Infection par le VIH. Bulletin de Santé Publique VIH-IST. November 2023. Available online: https://www.santepubliquefrance.fr/ (accessed on 5 January 2024).
- UNAIDS. HIV Treatment. Geneva, Switzerland: Joint United Nations Programme on HIV/AIDS. 2024. Available online: https://www.unaids.org/en/topic/treatment (accessed on 5 January 2024).
- Furman, P.A.; Fyfe, J.A.; St. Clair, M.H.; Weinhold, K.; Rideout, J.L.; Freeman, G.A.; Lehrman, S.N.; Bolognesi, D.P.; Broder, S.; Mitsuya, H. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interactions of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 1986, 83, 8333–8337. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.J.; Orr, D.C.; Penn, C.R.; Figueiredo, H.T.; Gray, N.M.; Boehme, R.E.; Cameron, J.M. Effects of (-)-2′-deoxy-3′-thiacytidine (3TC) 5′-triphosphate on human immunodeficiency virus reverse transcriptase and mammalian DNA polymerases α, β, and γ. Antimicrob. Agents Chemother. 1992, 36, 1688–1694. [Google Scholar] [CrossRef] [PubMed]
- Spence, R.A.; Kati, W.M.; Anderson, K.S.; Johnson, K.A. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science 1995, 267, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Quan, Y.; Li, Z.; Arts, E.J.; Wainberg, M.A. Effects of non-nucleoside inhibitors of human immunodeficiency virus type 1 in cell-free recombinant reverse transcriptase assays. J. Biol. Chem. 1995, 270, 31046–31051. [Google Scholar] [CrossRef]
- Boden, D.; Markowitz, M. Resistance to human immunodeficiency virus type 1 protease inhibitors. Antimicrob. Agents Chemother. 1998, 42, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Assoumou, L.; Bocket, L.; Pallier, C.; Grude, M.; Ait-Namane, R.; Izopet, J.; Raymond, S.; Charpentier, C.; Visseaux, B.; Wirden, M.; et al. Anrs Ac- Resistance Study Group. Stable prevalence of transmitted drug resistance mutations and increased circulation of non-B subtypes in antiretroviral-naive chronically HIV-infected patients in 2015/2016 in France. J. Antimicrob. Chemother. 2019, 74, 1417–1424. [Google Scholar] [CrossRef]
- Aulicino, P.C.; Zapiola, I.; Kademian, S.; Valle, M.M.; Fernandez Giuliano, S.; Toro, R.; Barbas, G.; Canizal, A.M.; Mayon, P.; Golemba, M.D.; et al. Pre-treatment drug resistance and HIV-1 subtypes in infants from Argentina with and without exposure to antiretroviral drugs for prevention of mother-to-child transmission. J. Antimicrob. Chemother. 2019, 74, 722–730. [Google Scholar] [CrossRef]
- Hertogs, K.; de Béthune, M.P.; Miller, V.; Ivens, T.; Schel, P.; Van Cauwenberge, A.; Van Den Eynde, C.; Van Gerwen, V.; Azijn, H.; Van Houtte, M.; et al. A rapid method for simultaneous detection of phenotypic resistance to inhibitors of protease and reverse transcriptase in recombinant human immunodeficiency virus type 1 isolates from patients treated with antiretroviral drugs. Antimicrob. Agents Chemother. 1998, 42, 269–276. [Google Scholar] [CrossRef]
- Johnston, E.; Dupnik, K.M.; Gonzales, M.J.; Winters, M.A.; Rhee, S.Y.; Imamichi, T.; Shafer, R.W. Panel of prototypical infectious molecular HIV-1 clones containing multiple nucleoside reverse transcriptase inhibitor resistance mutations. AIDS 2005, 19, 731–733. [Google Scholar] [CrossRef]
- Weber, J.; Vazquez, A.C.; Winner, D.; Rose, J.D.; Wylie, D.; Rhea, A.M.; Henry, K.; Pappas, J.; Wright, A.; Mohamed, N.; et al. Novel method for simultaneous quantification of phenotypic resistance to maturation, protease, reverse transcriptase, and integrase HIV inhibitors based on 3′Gag(p2/p7/p1/p6)/PR/RT/INT-recombinant viruses: A useful tool in the multitarget era of antiretroviral therapy. Antimicrob. Agents Chemother. 2011, 55, 3729–3742. [Google Scholar] [CrossRef]
- Saladini, F.; Giannini, A.; Boccuto, A.; Vicenti, I.; Zazzi, M. Agreement between an in-house replication competent and a reference replication defective recombinant virus assay for measuring phenotypic resistance to HIV-1 protease, reverse transcriptase, and integrase inhibitors. J. Clin. Lab. Anal. 2018, 32, e22206. [Google Scholar] [CrossRef] [PubMed]
- Yeni, P.G.; Hammer, S.M.; Carpenter, C.C.; Cooper, D.A.; Fischl, M.A.; Gatell, J.M.; Gazzard, B.G.; Hirsch, M.S.; Jacobsen, D.M.; Katzenstein, D.A.; et al. Antiretroviral treatment for adult HIV infection in 2002: Updated recommendations of the International AIDS Society-USA Panel. JAMA 2002, 288, 222–235, Erratum in JAMA 2003, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Quiros-Roldan, E.; Signorini, S.; Castelli, F.; Torti, C.; Patroni, A.; Airoldi, M.; Carosi, G. Analysis of HIV-1 mutation patterns in patients failing antiretroviral therapy. J. Clin. Lab. Anal. 2001, 15, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, P.; Opravil, M.; Hirschel, B.; Chave, J.P.; Furrer, H.J.; Sax, H.; Perneger, T.V.; Perrin, L.; Kaiser, L.; Yerly, S. Impact of drug resistance mutations on virologic response to salvage therapy. Swiss HIV Cohort Study. AIDS 1999, 13, F17–F21. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, J.G.; Condra, J.H. Genotypic analysis methods for detection of drug resistance mutations in the HIV-1 proteinase and reverse transcriptase genes. Antiviral Ther. 1999, 4, 135–142. [Google Scholar] [CrossRef]
- Stella-Ascariz, N.; Arribas, J.R.; Paredes, R.; Li, J.Z. The role of HIV-1 drug-resistant minority variants in treatment failure. J. Infect. Dis. 2017, 216 (Suppl. S9), S847–S850. [Google Scholar] [CrossRef]
- May, S.; Adamska, E.; Tang, J. Evaluation of Vela Diagnostics HIV-1 genotyping assay on an automated next generation sequencing platform. J. Clin. Virol. 2020, 127, 104376. [Google Scholar] [CrossRef]
- Bonifacio, M.A.; Genchi, C.; Lagioia, A.; Talamo, V.; Volpe, A.; Mariggiò, M.A. Analytical assessment of the Vela Diagnostics NGS assay for HIV genotyping and resistance testing: The Apulian experience. Int. J. Mol. Sci. 2022, 23, 2727. [Google Scholar] [CrossRef]
- Weber, J.; Volkova, I.; Sahoo, M.K.; Tzou, P.L.; Shafer, R.W.; Pinsky, B.A. Prospective evaluation of the Vela Diagnostics Next-Generation Sequencing platform for HIV-1 genotypic resistance testing. J. Mol. Diagn. 2019, 21, 961–970. [Google Scholar] [CrossRef]
- Tzou, P.L.; Ariyaratne, P.; Varghese, V.; Lee, C.; Rakhmanaliev, E.; Villy, C.; Yee, M.; Tan, K.; Michel, G.; Pinsky, B.A.; et al. Comparison of an in vitro diagnostic Next-Generation Sequencing assay with Sanger sequencing for HIV-1 genotypic resistance testing. J. Clin. Microbiol. 2018, 56, e00105–e00118. [Google Scholar] [CrossRef]
- Raymond, S.; Nicot, F.; Abravanel, F.; Minier, L.; Carcenac, R.; Lefebvre, C.; Harter, A.; Martin-Blondel, G.; Delobel, P.; Izopet, J. Performance evaluation of the Vela Dx Sentosa next-generation sequencing system for HIV-1 DNA genotypic resistance. J. Clin. Virol. 2020, 122, 104229. [Google Scholar] [CrossRef]
- Raymond, S.; Nicot, F.; Carcenac, R.; Lefebvre, C.; Jeanne, N.; Saune, K.; Delobel, P.; Izopet, J. HIV-1 genotypic resistance testing using the Vela automated next-generation sequencing platform. J. Antimicrob. Chemother. 2018, 73, 1152–1157. [Google Scholar] [CrossRef]
- Descamps, D.; Calvez, V.; Chaix, M.L.; Charpentier, C.; Delaugerre, C.; Flandre, P.; Izopet, J.; Marcellin, A.G.; Morand-Joubert, L.; Peytavin, G.; et al. HIV-1 Genotypic Drug Resistance Interpretation’s Algorithms. Paris, France: HIV French Resistance and French ANRS-MIE (National Agency for AIDS Research—Emergent Infectious Diseases) AC43 Resistance Group. 2023. Available online: https://pnmvh.org/outil-clinique/hiv-french-resistance/ (accessed on 17 January 2024).
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef]
- Clutter, D.S.; Jordan, M.R.; Bertagnolio, S.; Shafer, R.W. HIV-1 drug resistance and resistance testing. Infect. Genet. Evol. 2016, 46, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Rapport d’expert VIH, 2016. Résistance du VIH-1 aux Antéroviraux. Available online: https://cns.sante.fr›experts-vih_resistance (accessed on 13 January 2024).
- Chaix, M.L.; Seng, R.; Frange, P.; Tran, L.; Avettand-Fenoël, V.; Ghosn, J.; Reynes, J.; Yazdanpanah, Y.; Raffi, F.; Goujard, C.; et al. Increasing HIV-1 non-B subtype primary infections in patients in France and effect of HIV subtypes on virological and immunological responses to combined antiretroviral therapy. Clin. Infect. Dis. 2013, 56, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Thermo Fisher Scientific. Ion Torrent. Available online: https://www.thermofisher.com/fr/fr/home/brands/ion-torrent.html (accessed on 5 January 2024).
- Dessilly, G.; Goeminne, L.; Vandenbroucke, A.T.; Dufrasne, F.E.; Martin, A.; Kabamba-Mukadi, B. First evaluation of the Next-Generation Sequencing platform for the detection of HIV-1 drug resistance mutations in Belgium. PLoS ONE 2018, 13, e0209561, Erratum in PLoS ONE 2019, 14, e0211587. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.T.; Simmon, K.E.; Mallory, M.A.; Hymas, W.C.; Stevenson, J.; Barker, A.P.; Hillyard, D.R. HIV-1 drug resistance assay using Ion Torrent Next Generation Sequencing and on-instrument end-to-end analysis software. J. Clin. Microbiol. 2022, 60, e0025322. [Google Scholar] [CrossRef] [PubMed]
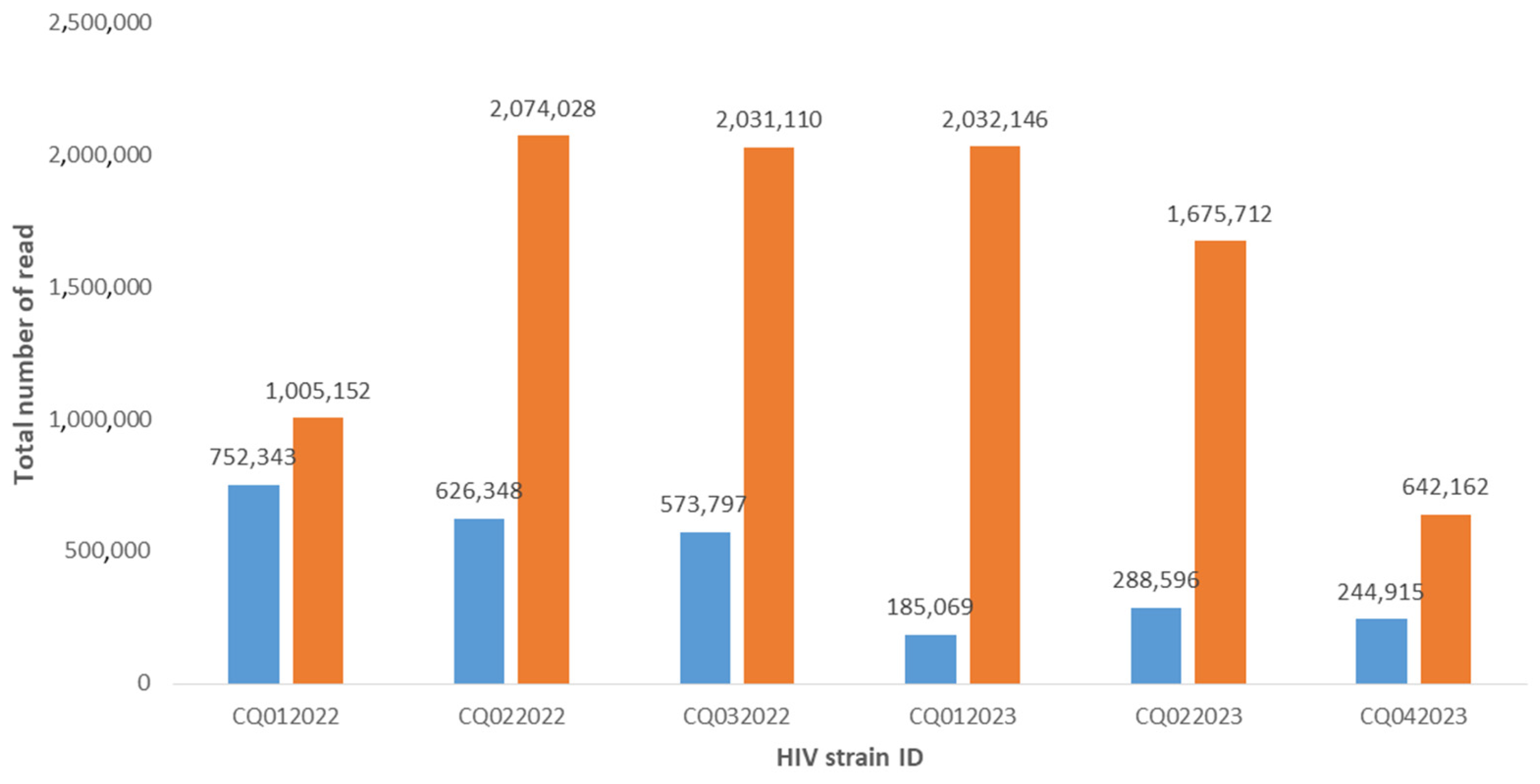
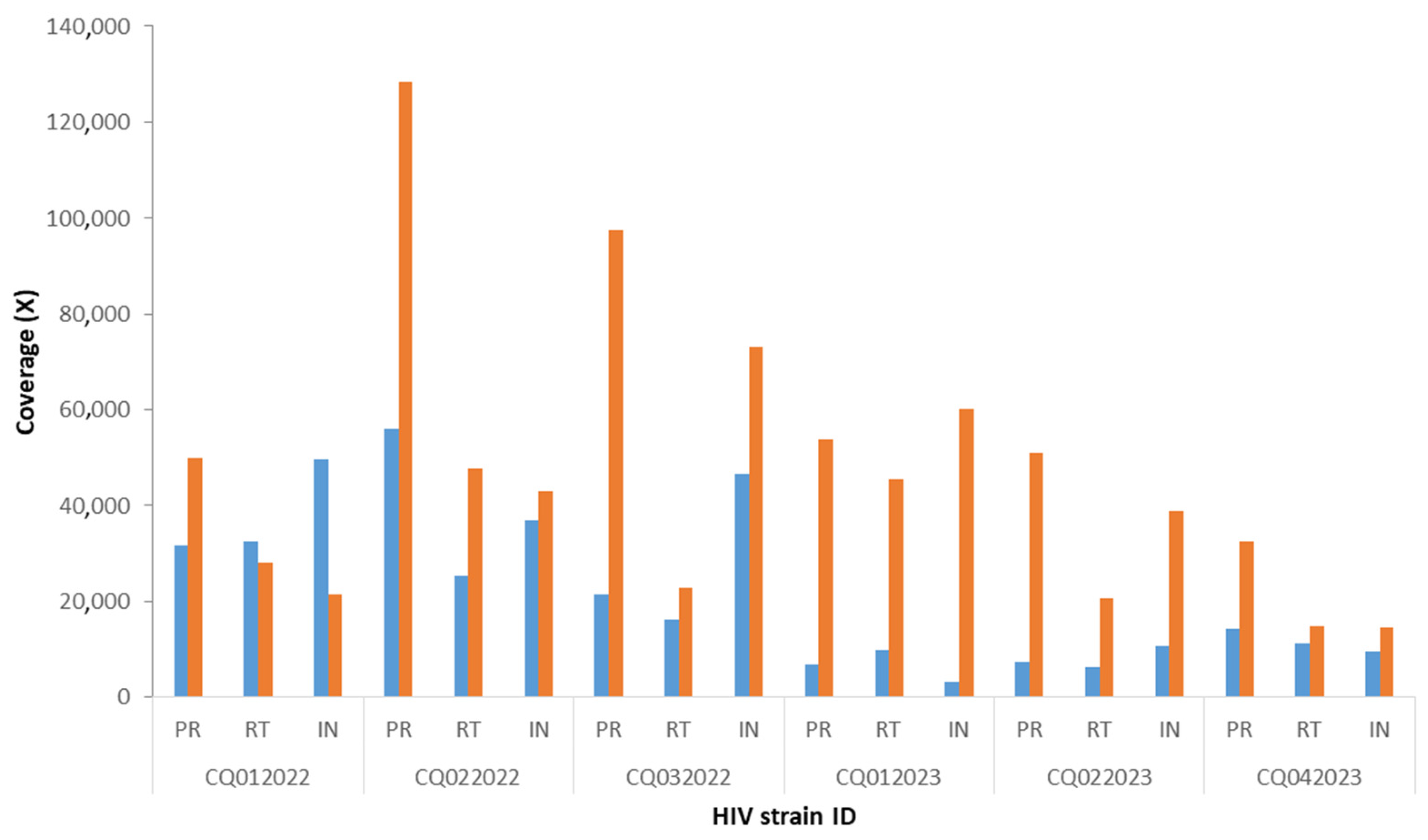
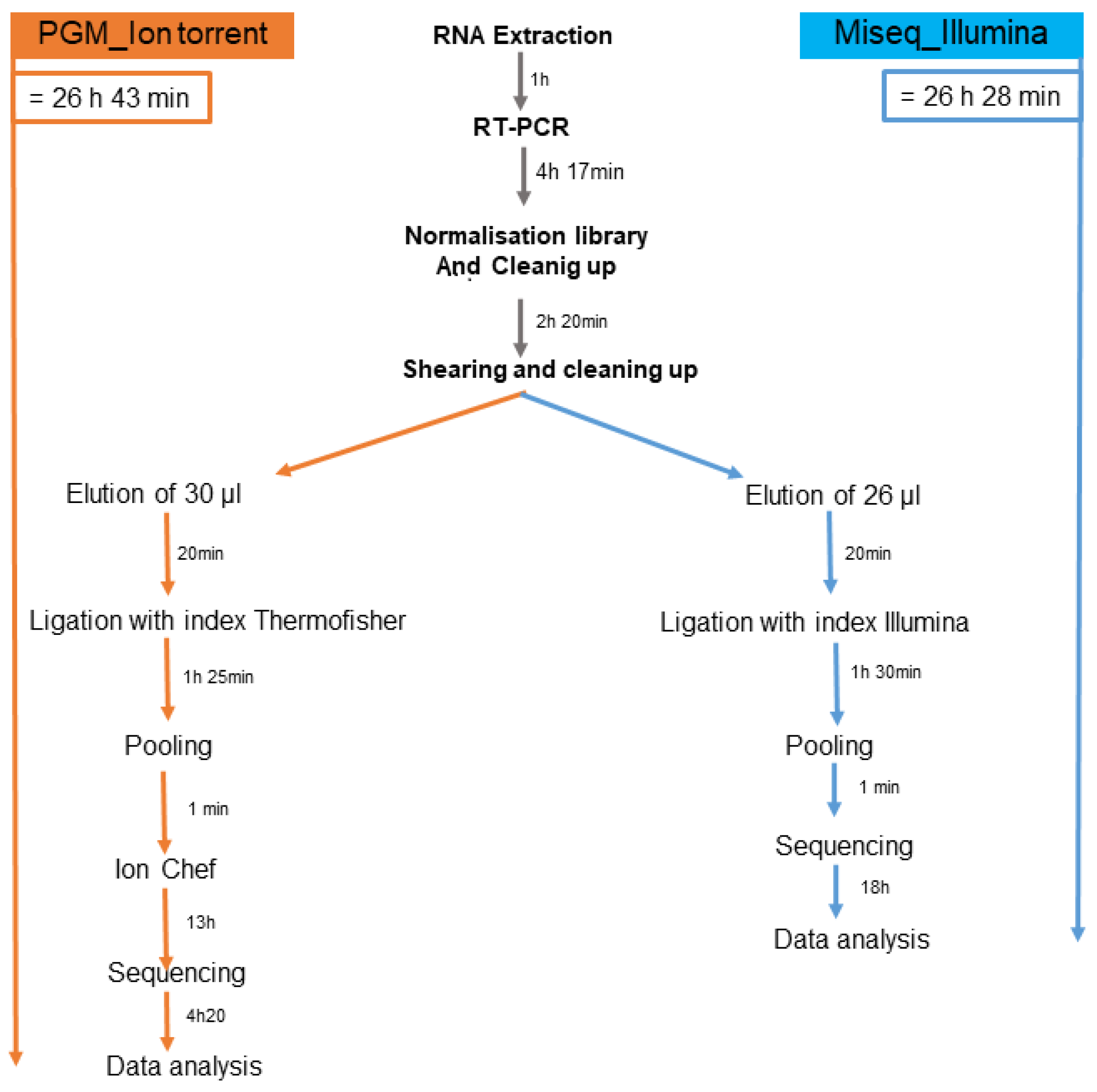

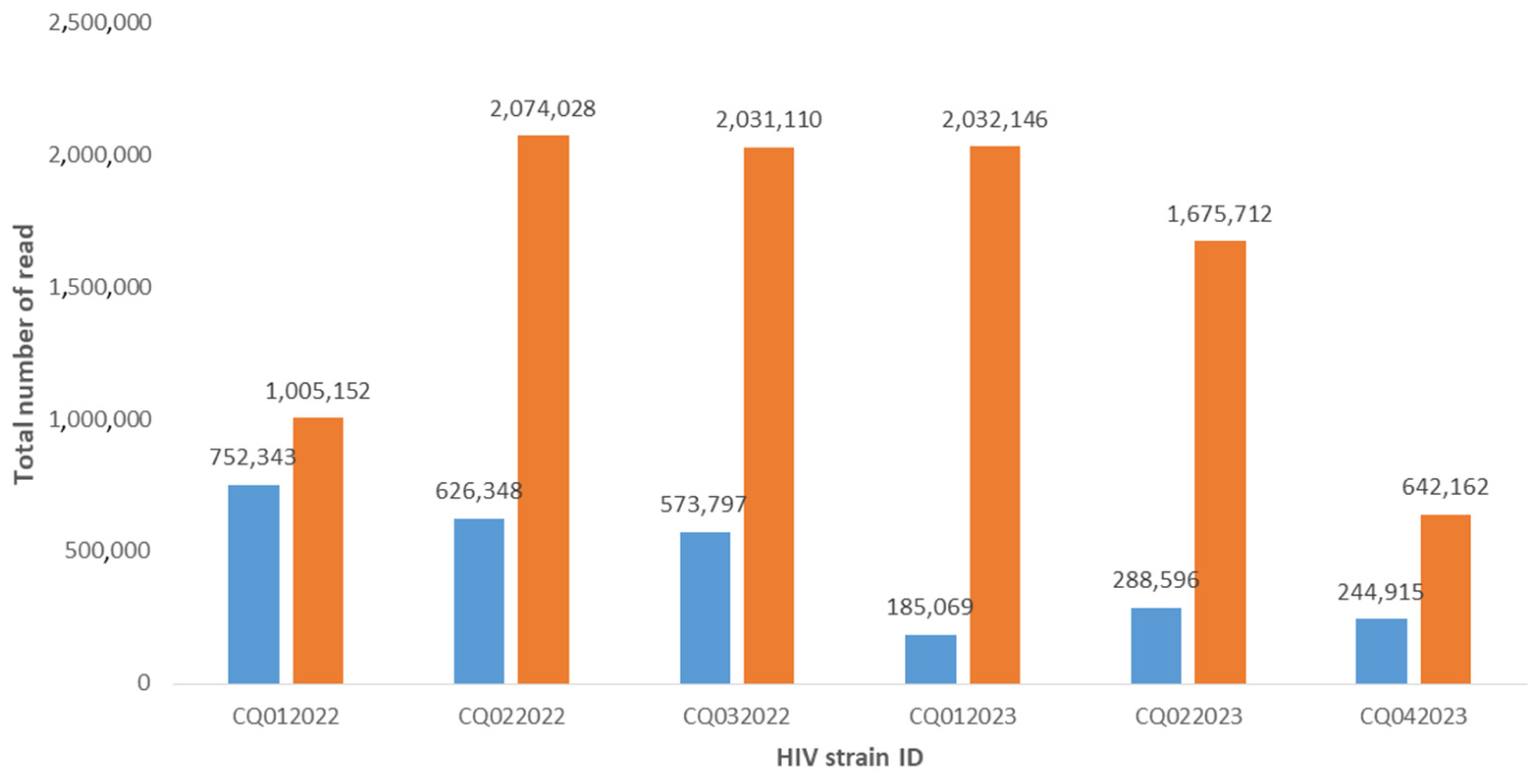{kind=link}
{kind=link}
{kind=link}
| Gene | ID HIV Strain Number | Mutation Number PGM > 20% | Mutation Number Miseq > 20% | Number of Discordance | Resistance Yes or No | Concordance (%) |
|---|---|---|---|---|---|---|
| Protease | CQ012022 | 24 | 23 | 1 | no | 99 |
| CQ022022 | 18 | 18 | 0 | yes | 100 | |
| CQ032022 | 19 | 20 | 1 | no | 99 | |
| CQ012023 | 24 | 24 | 0 | yes | 100 | |
| CQ022023 | 11 | 11 | 0 | yes | 100 | |
| CQ042023 | 14 | 14 | 0 | yes | 100 | |
| Reverse Transcriptase | CQ012022 | 43 | 43 | 0 | yes | 100 |
| CQ022022 | 38 | 37 | 1 | no | 99.5 | |
| CQ032022 | 22 | 23 | 1 | yes | 99.5 | |
| CQ012023 | 31 | 31 | 0 | yes | 100 | |
| CQ022023 | 34 | 34 | 0 | yes | 100 | |
| CQ042023 | 26 | 25 | 1 | no | 99.5 | |
| Integrase | CQ012022 | 20 | 20 | 0 | yes | 100 |
| CQ022022 | 10 | 10 | 0 | yes | 100 | |
| CQ032022 | 18 | 18 | 0 | yes | 100 | |
| CQ012023 | 22 | 22 | 0 | yes | 100 | |
| CQ022023 | 21 | 21 | 0 | yes | 100 | |
| CQ042023 | 15 | 15 | 0 | yes | 100 |
| Library Reagent | Manufacturer | Price (EUR) | Price, 1 run (EUR) | Total Price | |
|---|---|---|---|---|---|
| PGM_Ion Torrent | Miseq_Illumina | ||||
| Sentosa SQ HIV DRM Library Kit (96 samples) | Vela | 2644 | 220.33 | 1685.05 | 1640.24 |
| Sentosa SQ Virus Solution Prep Kit (96 samples) | Vela | 974.05 | 81.17 | ||
| MiseqR Reagent kit v2 (300 cycles) | Illumina | 1273 | 1273 | ||
| IDT for Illumina—TruSeq DNA UD Indexes v2 (96 samples) | Illumina | 788.88 | 65.74 | ||
| Ion Xpress Barcode Adapters Kit (96 samples) | ThermoFisher | 1757.97 | 146.49 | ||
| Ion PGMTM Hi-QTM View Chef Reagent (four reactions) | ThermoFisher | 3171.92 | 792.98 | ||
| Ion PGMTM Hi-QTM View Chef Supplies (four reactions) | ThermoFisher | ||||
| Ion PGMTM Hi-QTM View Chef Solutions (four reactions) | ThermoFisher | ||||
| Ion PGMTM Hi-QTM View Sequencing Supplies (four reactions) | ThermoFisher | ||||
| Ion PGM Hi-Q View Sequencing Solutions (four reactions) | ThermoFisher | ||||
| Ion PGM Hi-Q View Sequencing dNTPs (four reactions) | ThermoFisher | ||||
| Sentosa SQ 318 Chip Kit (one reaction) | Vela | 3552.6 | 444.07 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papa Mze, N.; Fernand-Laurent, C.; Daugabel, S.; Zanzouri, O.; Marque Juillet, S. Optimization of HIV Sequencing Method Using Vela Sentosa Library on Miseq Ilumina Platform. Genes 2024, 15, 259. https://doi.org/10.3390/genes15020259
Papa Mze N, Fernand-Laurent C, Daugabel S, Zanzouri O, Marque Juillet S. Optimization of HIV Sequencing Method Using Vela Sentosa Library on Miseq Ilumina Platform. Genes. 2024; 15(2):259. https://doi.org/10.3390/genes15020259
Chicago/Turabian StylePapa Mze, Nasserdine, Cécile Fernand-Laurent, Solen Daugabel, Olfa Zanzouri, and Stéphanie Marque Juillet. 2024. "Optimization of HIV Sequencing Method Using Vela Sentosa Library on Miseq Ilumina Platform" Genes 15, no. 2: 259. https://doi.org/10.3390/genes15020259
APA StylePapa Mze, N., Fernand-Laurent, C., Daugabel, S., Zanzouri, O., & Marque Juillet, S. (2024). Optimization of HIV Sequencing Method Using Vela Sentosa Library on Miseq Ilumina Platform. Genes, 15(2), 259. https://doi.org/10.3390/genes15020259