
A Novel 3-Gene Signature for Identifying COVID-19 Patients Based on Bioinformatics and Machine Learning

Abstract
:1. Introduction
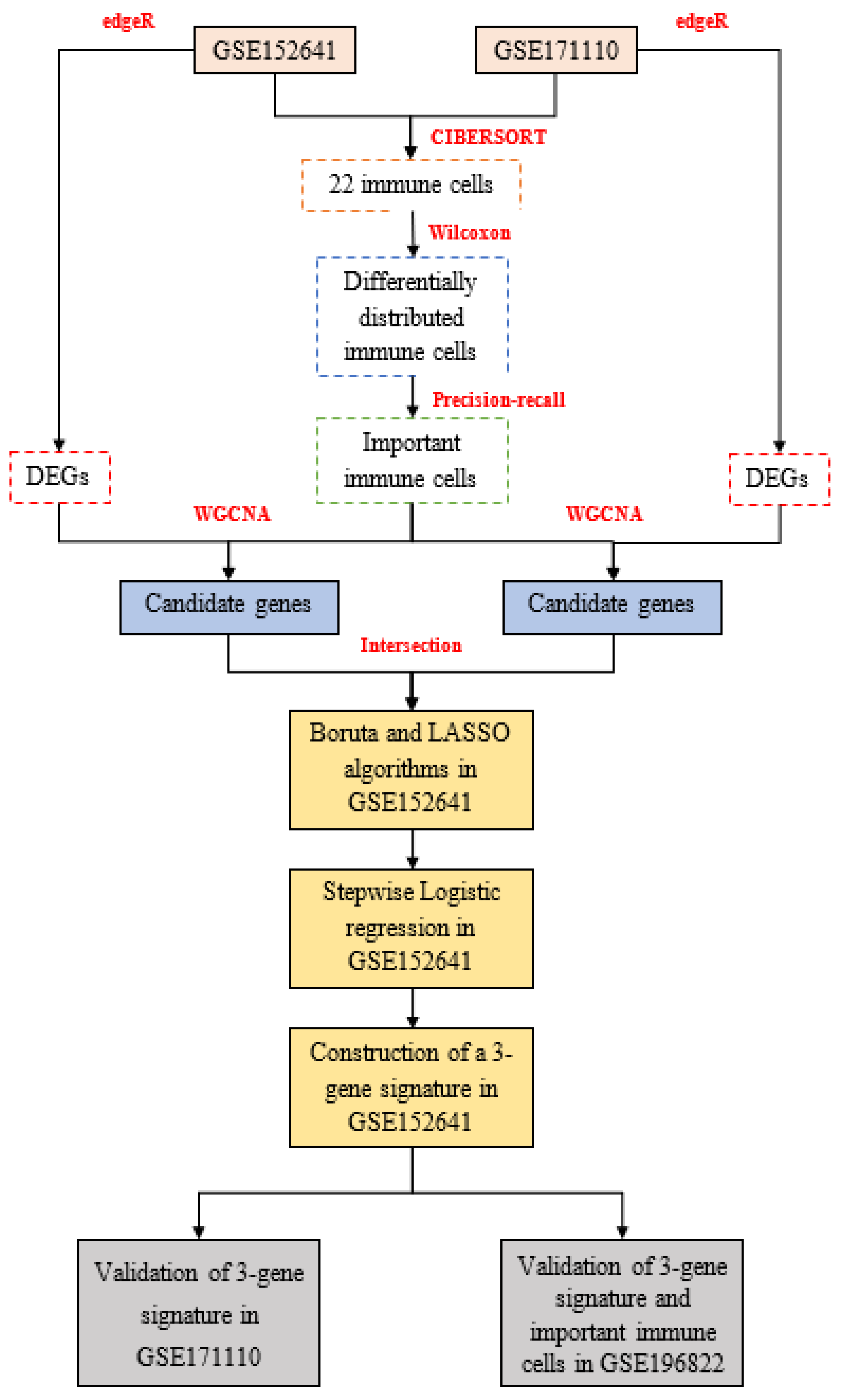
2. Materials and Methods
2.1. Data Acquisition
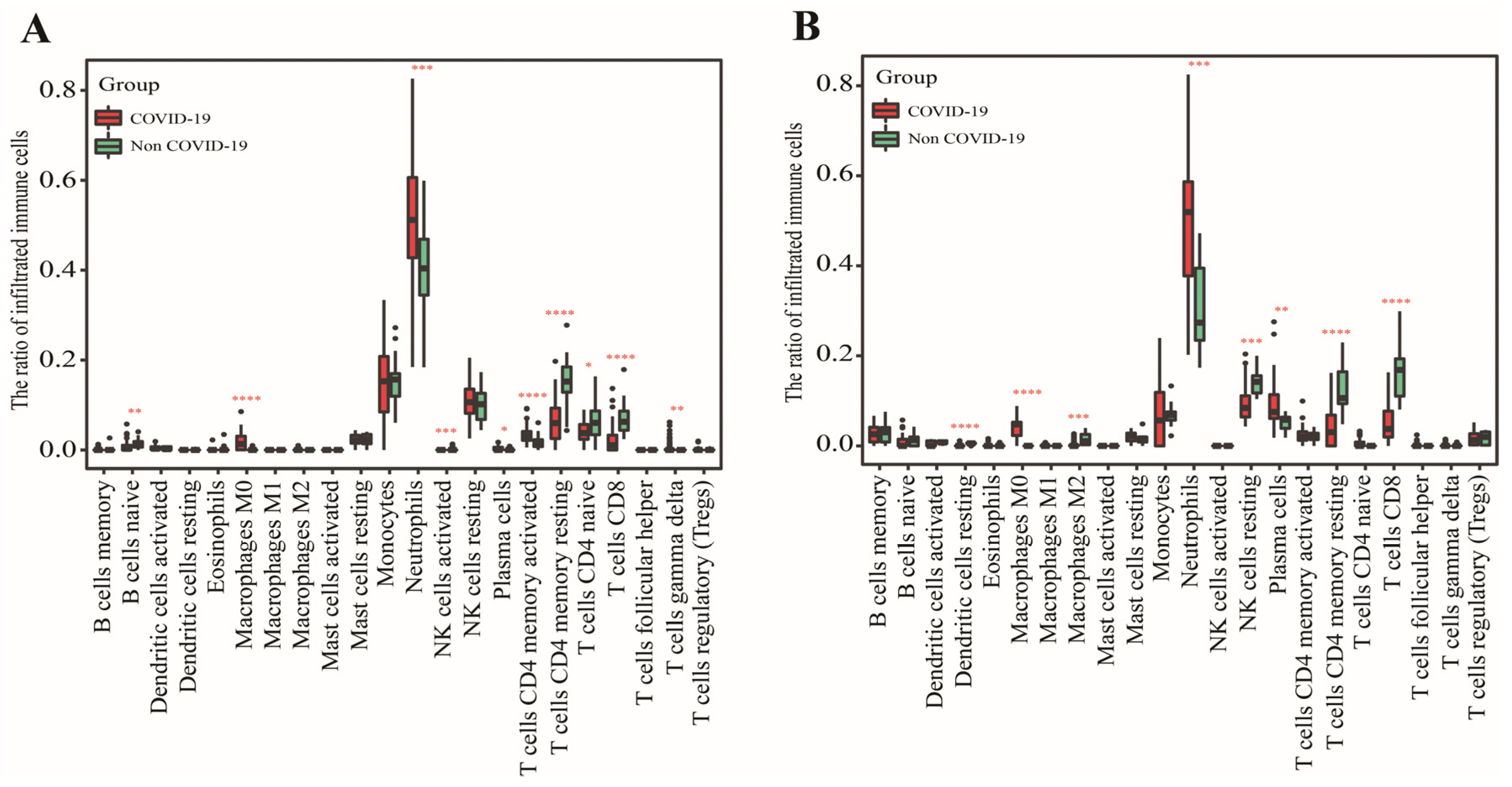
2.2. Calculation of Immune Cell
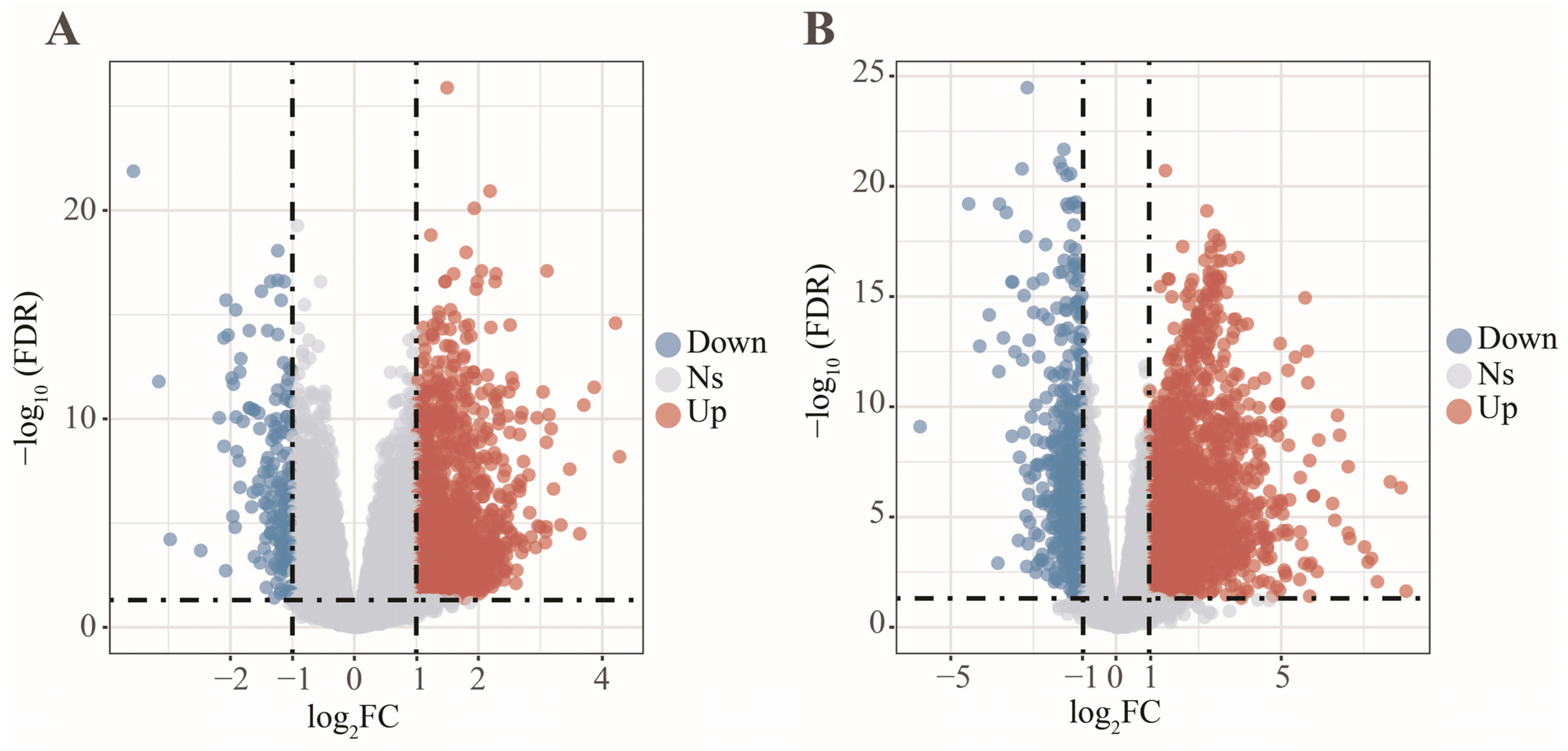
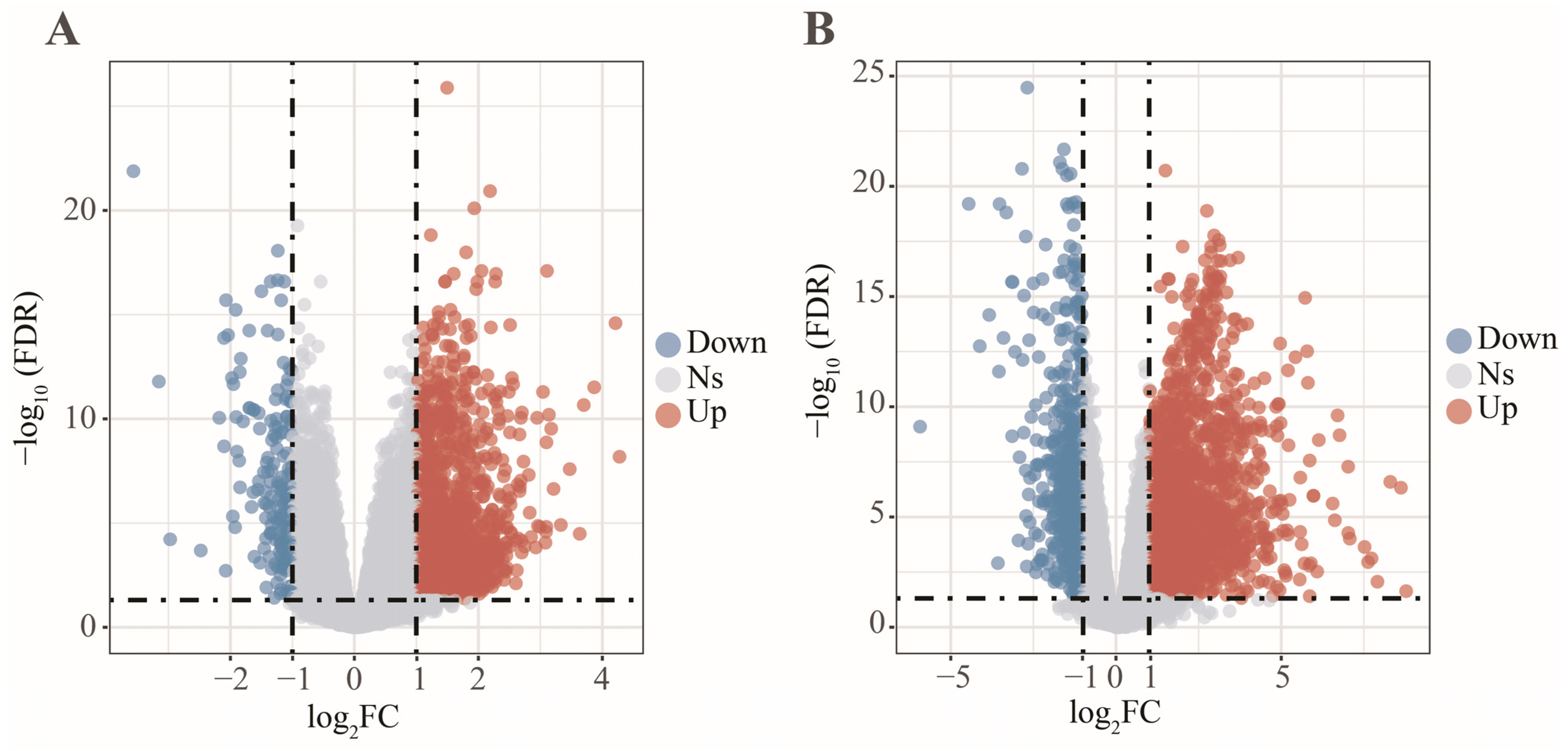
2.3. Differentially Expressed Analysis
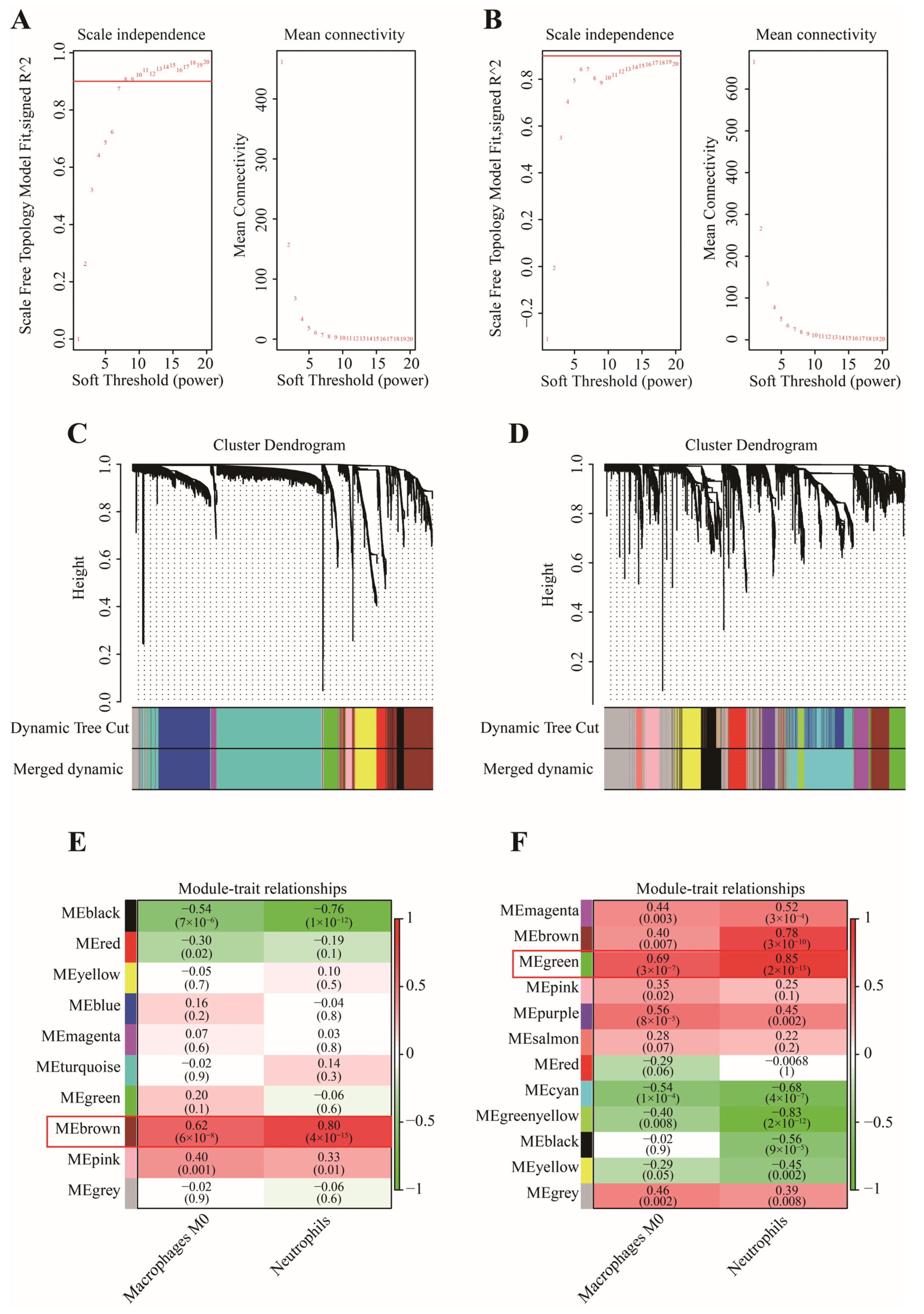
2.4. WGCNA Analysis
2.5. GO Enrichment Analysis
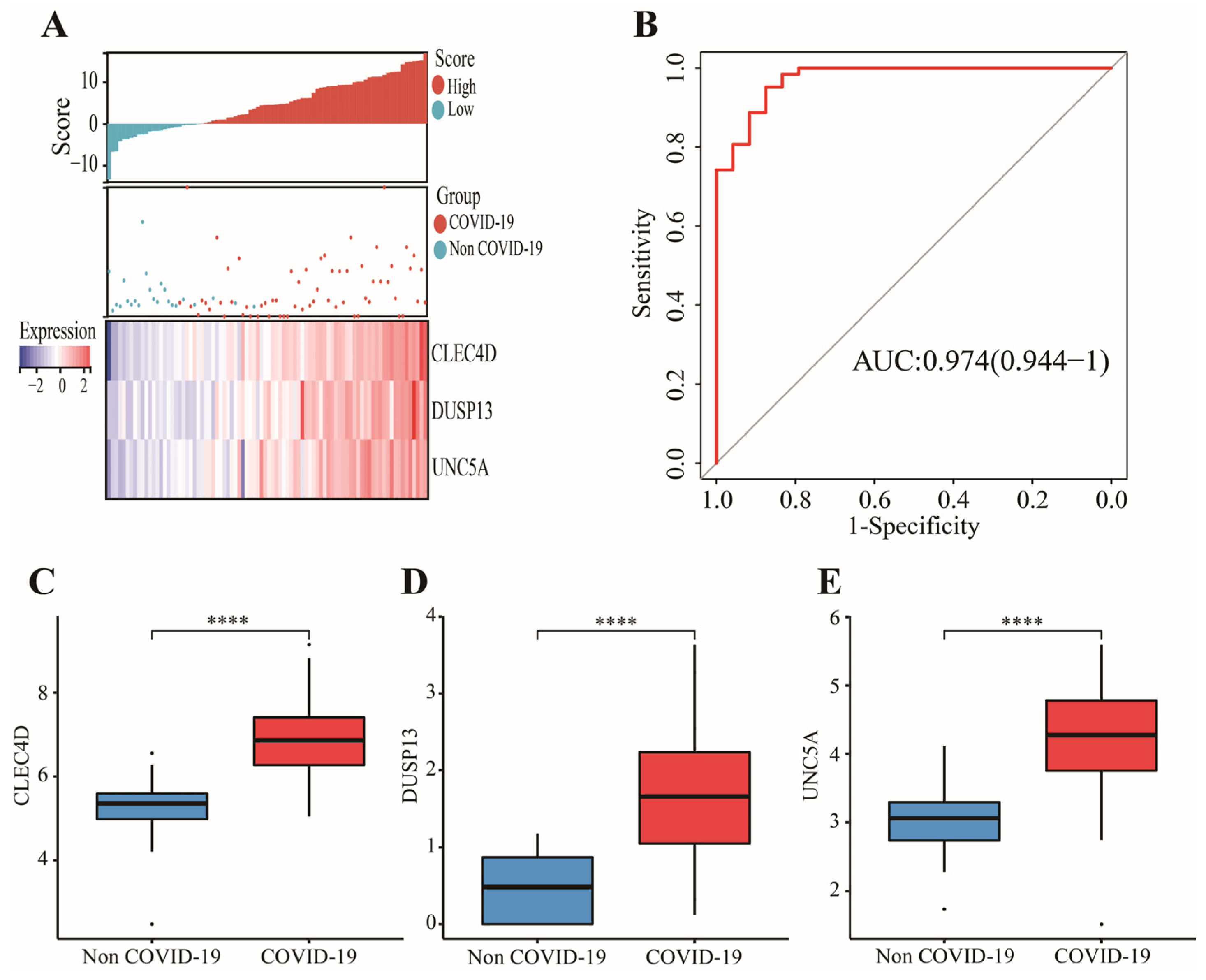
2.6. Construction and Validation of a Novel Gene Signature
2.7. Statistical Analysis
3. Results
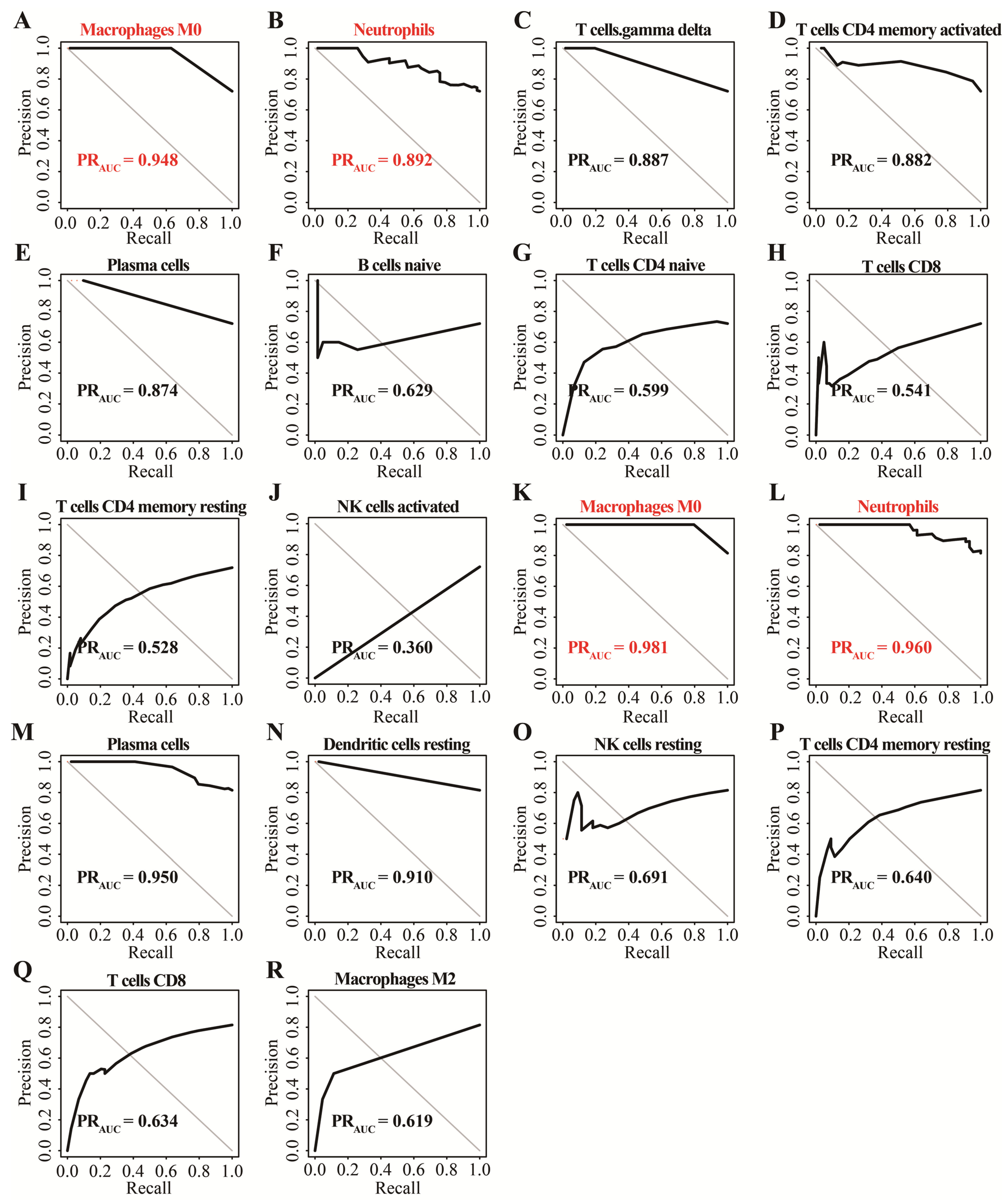
3.1. Identification of Two Important Immune Cells
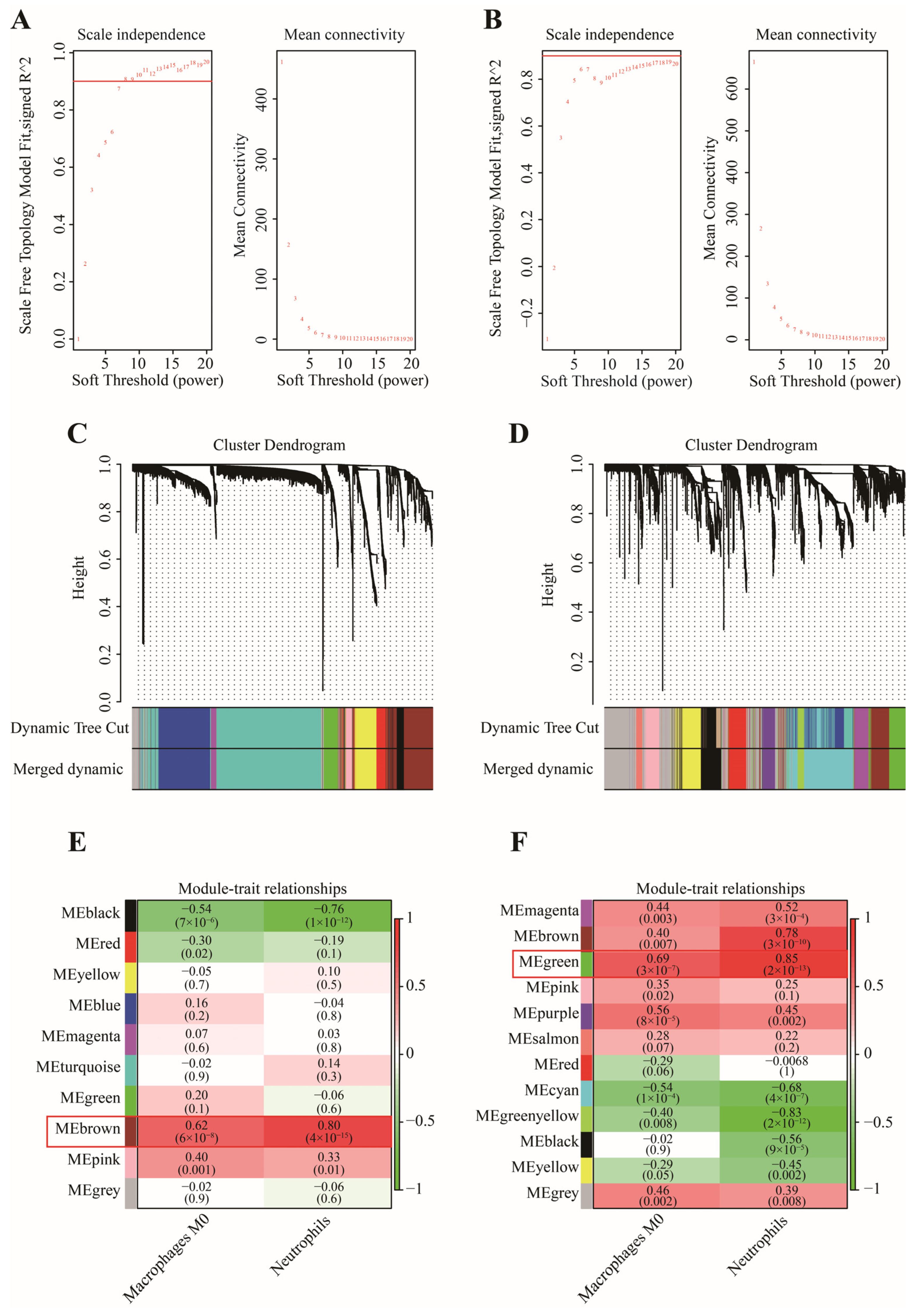
3.2. Determination of Key Modules
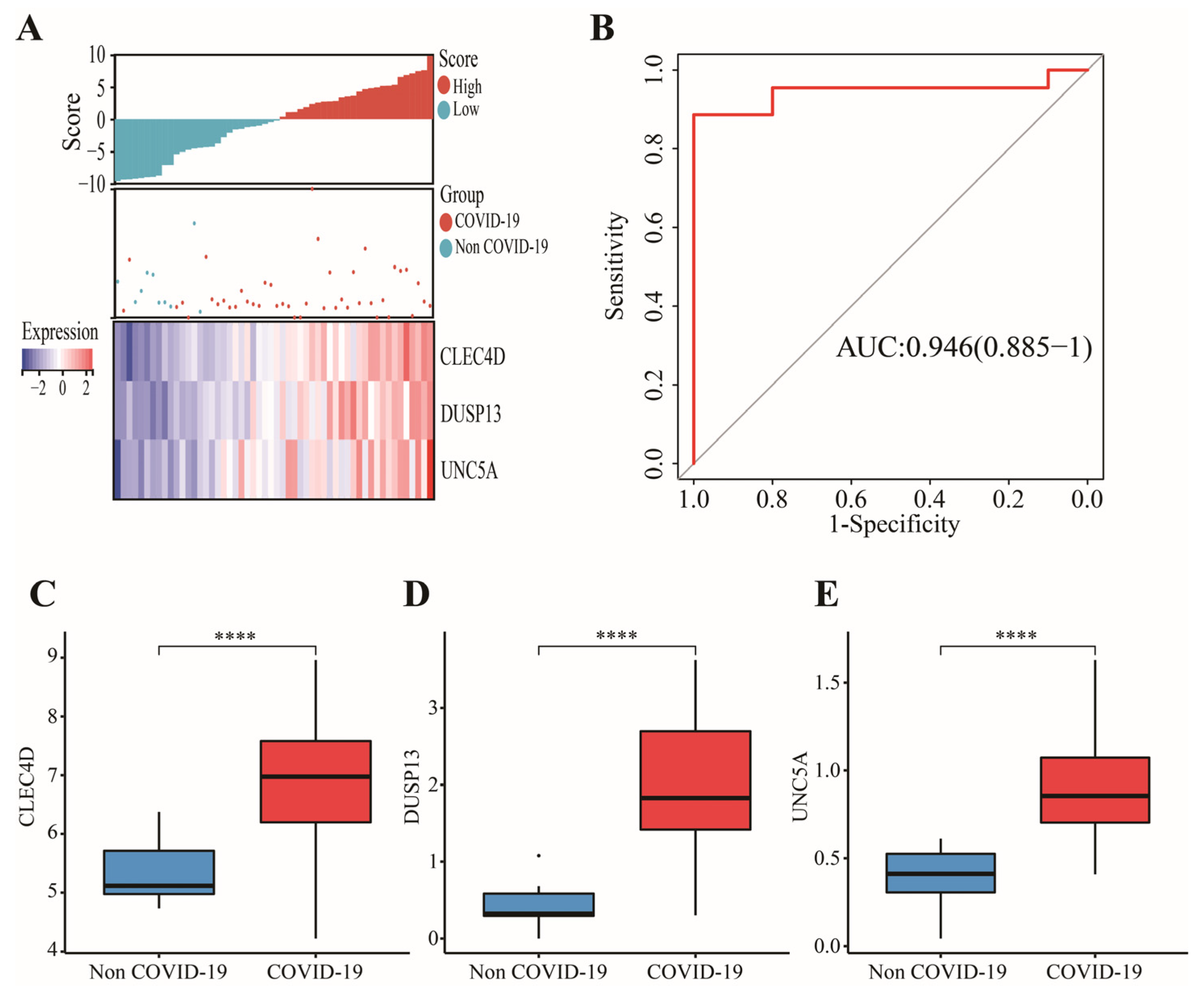
3.3. Construction and Validation of a 3-Gene Signature
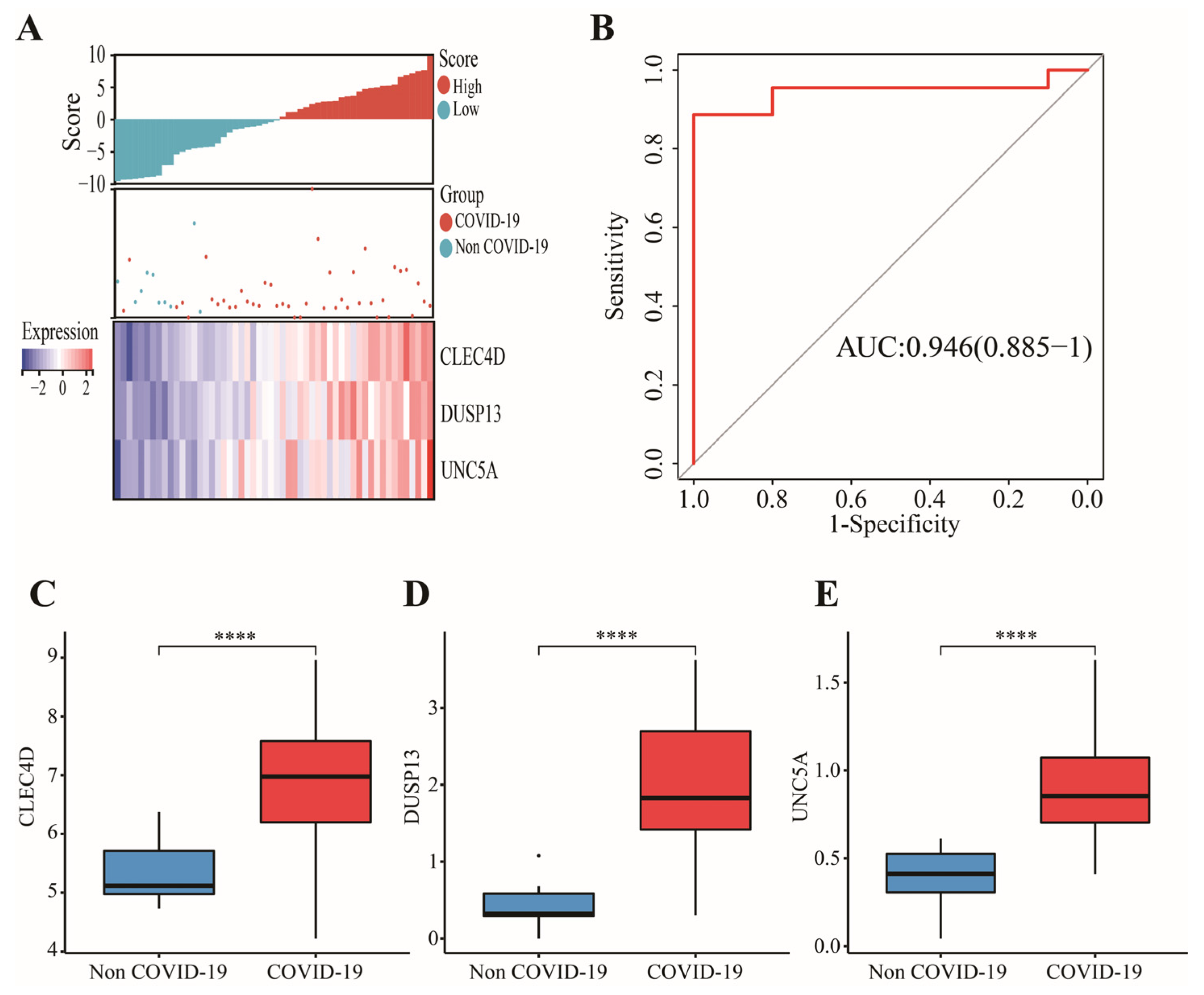
3.4. External Validation of Candidate Immune Cells and Genes in GSE196822
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Tomalka, J.A.; Suthar, M.S.; Deeks, S.G.; Sekaly, R.P. Fighting the SARS-CoV-2 pandemic requires a global approach to understanding the heterogeneity of vaccine responses. Nat. Immunol. 2022, 23, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef]
- Nguyen-Contant, P.; Embong, A.K.; Kanagaiah, P.; Chaves, F.A.; Yang, H.; Branche, A.R.; Topham, D.J.; Sangster, M.Y.; Ellebedy, A.; Schultz-Cherry, S.S. Protein-Reactive IgG and Memory B Cell Production after Human SARS-CoV-2 Infection Includes Broad Reactivity to the S2 Subunit. mBio 2020, 11, e01991-20. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Wauters, E.; Van Mol, P.; Garg, A.D.; Jansen, S.; Van Herck, Y.; Vanderbeke, L.; Bassez, A.; Boeckx, B.; Malengier-Devlies, B.; Timmerman, A.; et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021, 31, 272–290. [Google Scholar] [CrossRef]
- Jing, Y.; Luo, L.; Chen, Y.; Westerberg, L.S.; Zhou, P.; Xu, Z.; Herrada, A.A.; Park, C.S.; Kubo, M.; Mei, H.; et al. SARS-CoV-2 infection causes immunodeficiency in recovered patients by downregulating CD19 expression in B cells via enhancing B-cell metabolism. Signal Transduct. Target. Ther. 2021, 6, 345. [Google Scholar] [CrossRef]
- Rebillard, R.M.; Charabati, M.; Grasmuck, C.; Filali-Mouhim, A.; Tastet, O.; Brassard, N.; Daigneault, A.; Bourbonniere, L.; Anand, S.P.; Balthazard, R.; et al. Identification of SARS-CoV-2-specific immune alterations in acutely ill patients. J. Clin. Investig. 2021, 131, e145853. [Google Scholar] [CrossRef]
- Qiu, X.; Hua, X.; Li, Q.; Zhou, Q.; Chen, J. m6A Regulator-Mediated Methylation Modification Patterns and Characteristics of Immunity in Blood Leukocytes of COVID-19 Patients. Front. Immunol. 2021, 12, 774776. [Google Scholar] [CrossRef]
- Song, X.; Zhu, J.; Tan, X.; Yu, W.; Wang, Q.; Shen, D.; Chen, W. XGBoost-Based Feature Learning Method for Mining COVID-19 Novel Diagnostic Markers. Front. Public Health 2022, 10, 926069. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.; Chen, Q.; Wang, K. m6A Regulator-Mediated Methylation Modification Patterns and Characteristics in COVID-19 Patients. Front. Public Health 2022, 10, 914193. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Kumar, P. Integrated COVID-19 Predictor: Differential expression analysis to reveal potential biomarkers and prediction of coronavirus using RNA-Seq profile data. Comput. Biol. Med. 2022, 147, 105684. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Mo, S.; Dai, L.; Wang, Y.; Song, B.; Yang, Z.; Gu, W. Comprehensive Analysis of the Systemic Transcriptomic Alternations and Inflammatory Response during the Occurrence and Progress of COVID-19. Oxid. Med. Cell Longev. 2021, 2021, 9998697. [Google Scholar] [CrossRef] [PubMed]
- Lian, Q.; Zhang, K.; Zhang, Z.; Duan, F.; Guo, L.; Luo, W.; Mok, B.W.; Thakur, A.; Ke, X.; Motallebnejad, P.; et al. Differential effects of macrophage subtypes on SARS-CoV-2 infection in a human pluripotent stem cell-derived model. Nat. Commun. 2022, 13, 2028. [Google Scholar] [CrossRef]
- Masso-Silva, J.A.; Moshensky, A.; Lam, M.; Odish, M.F.; Patel, A.; Xu, L.; Hansen, E.; Trescott, S.; Nguyen, C.; Kim, R.; et al. Increased Peripheral Blood Neutrophil Activation Phenotypes and Neutrophil Extracellular Trap Formation in Critically Ill Coronavirus Disease 2019 (COVID-19) Patients: A Case Series and Review of the Literature. Clin. Infect. Dis. 2022, 74, 479–489. [Google Scholar] [CrossRef]
- Shi, S.; Nie, B.; Chen, X.; Cai, Q.; Lin, C.; Zhao, G.; Zhang, X. Clinical and laboratory characteristics of severe and non-severe patients with COVID-19: A retrospective cohort study in China. J. Clin. Lab. Anal. 2021, 35, e23692. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Lord, J.M. Neutrophils and COVID-19: Active Participants and Rational Therapeutic Targets. Front. Immunol. 2021, 12, 680134. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.C.; Korinek, M.; Cheng, W.J.; Hwang, T.L. Targeting Neutrophils to Treat Acute Respiratory Distress Syndrome in Coronavirus Disease. Front. Pharmacol. 2020, 11, 572009. [Google Scholar] [CrossRef] [PubMed]
- Bibert, S.; Guex, N.; Lourenco, J.; Brahier, T.; Papadimitriou-Olivgeris, M.; Damonti, L.; Manuel, O.; Liechti, R.; Gotz, L.; Tschopp, J.; et al. Transcriptomic Signature Differences Between SARS-CoV-2 and Influenza Virus Infected Patients. Front. Immunol. 2021, 12, 666163. [Google Scholar] [CrossRef]
- Bankar, R.; Suvarna, K.; Ghantasala, S.; Banerjee, A.; Biswas, D.; Choudhury, M.; Palanivel, V.; Salkar, A.; Verma, A.; Singh, A.; et al. Proteomic investigation reveals dominant alterations of neutrophil degranulation and mRNA translation pathways in patients with COVID-19. iScience 2021, 24, 102135. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Reyes, A.Z.; Hu, K.A.; Teperman, J.; Wampler Muskardin, T.L.; Tardif, J.C.; Shah, B.; Pillinger, M.H. Anti-inflammatory therapy for COVID-19 infection: The case for colchicine. Ann. Rheum. Dis. 2021, 80, 550–557. [Google Scholar] [CrossRef]
- Mudd, P.A.; Crawford, J.C.; Turner, J.S.; Souquette, A.; Reynolds, D.; Bender, D.; Bosanquet, J.P.; Anand, N.J.; Striker, D.A.; Martin, R.S.; et al. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci. Adv. 2020, 6, eabe3024. [Google Scholar] [CrossRef]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkeni, M.; Gharbi, J. RAGE receptor: May be a potential inflammatory mediator for SARS-CoV-2 infection? Med. Hypotheses 2020, 144, 109950. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.; Miele, M.C.; Di Timoteo, F.; De Angelis, M.; Mauro, V.; Aronica, R.; Al Ismail, D.; Ceccarelli, G.; Pinacchio, C.; d’Ettorre, G.; et al. Persistent Systemic Microbial Translocation and Intestinal Damage During Coronavirus Disease-19. Front. Immunol. 2021, 12, 708149. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, I.; Giroux, N.; Olson, L.; Morrison, S.A.; Llanga, T.; Akinade, T.O.; Zhu, Y.; Zhong, Y.; Bose, S.; Arvai, S.; et al. DAMPs/PAMPs induce monocytic TLR activation and tolerance in COVID-19 patients; nucleic acid binding scavengers can counteract such TLR agonists. Biomaterials 2022, 283, 121393. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, H.; Fan, L.; Fang, M.; He, X.; Lu, B.; Pang, Z. CLEC4s as Potential Therapeutic Targets in Hepatocellular Carcinoma Microenvironment. Front. Cell Dev. Biol. 2021, 9, 681372. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, X.; Zhou, D.; Li, T.; Tang, L.; Gong, T.; Su, J.; Liang, P. Modelling cadmium-induced cardiotoxicity using human pluripotent stem cell-derived cardiomyocytes. J. Cell Mol. Med. 2018, 22, 4221–4235. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.; Dupuis, J. Mapping gene and gene pathways associated with coronary artery disease: aCARDIoGRAM exome and multi-ancestry UK biobank analysis. Sci. Rep. 2021, 11, 16461. [Google Scholar] [CrossRef]
- Bohm, M.; Frey, N.; Giannitsis, E.; Sliwa, K.; Zeiher, A.M. Coronavirus Disease 2019 (COVID-19) and its implications for cardiovascular care: Expert document from the German Cardiac Society and the World Heart Federation. Clin. Res. Cardiol. 2020, 109, 1446–1459. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Nakagawara, A. UNC5 dependence receptor family in human cancer: A controllable double-edged sword. Cancer. Cancer Lett. 2021, 516, 28–35. [Google Scholar] [CrossRef]
- Plissonnier, M.L.; Lahlali, T.; Raab, M.; Michelet, M.; Romero-Lopez, C.; Rivoire, M.; Strebhardt, K.; Durantel, D.; Levrero, M.; Mehlen, P.; et al. Reciprocal antagonism between the netrin-1 receptor uncoordinated-phenotype-5A (UNC5A) and the hepatitis C virus. Oncogene 2017, 36, 6712–6724. [Google Scholar] [CrossRef]
- Gao, K.; Wang, R.; Chen, J.; Cheng, L.; Frishcosy, J.; Huzumi, Y.; Qiu, Y.; Schluckbier, T.; Wei, X.; Wei, G.W. Methodology-Centered Review of Molecular Modeling, Simulation, and Prediction of SARS-CoV-2. Chem. Rev. 2022, 122, 11287–11368. [Google Scholar] [CrossRef]
- Bok, K.; Sitar, S.; Graham, B.S.; Mascola, J.R. Accelerated COVID-19 vaccine development: Milestones, lessons, and prospects. Immunity 2021, 54, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; McNicholas, B.; Rezoagli, E.; Pham, T.; Curley, G.; McAuley, D.; O’Kane, C.; Nichol, A.; Dos Santos, C.; Rocco, P.; et al. Emerging pharmacological therapies for ARDS: COVID-19 and beyond. Intensive Care Med. 2020, 46, 2265–2283. [Google Scholar] [CrossRef] [PubMed]
- Karaderi, T.; Bareke, H.; Kunter, I.; Seytanoglu, A.; Cagnan, I.; Balci, D.; Barin, B.; Hocaoglu, M.B.; Rahmioglu, N.; Asilmaz, E.; et al. Host Genetics at the Intersection of Autoimmunity and COVID-19: A Potential Key for Heterogeneous COVID-19 Severity. Front. Immunol. 2020, 11, 586111. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Siegel, E.; Shen, D. Deep Learning and Medical Image Analysis for COVID-19 Diagnosis and Prediction. Annu. Rev. Biomed. Eng. 2022, 24, 179–201. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Reddy Maddikunta, P.K.; Pham, Q.V.; Gadekallu, T.R.; Krishnan, S.S.R.; Chowdhary, C.L.; Alazab, M.; Jalil Piran, M. Deep learning and medical image processing for coronavirus (COVID-19) pandemic: A survey. Sustain. Cities Soc. 2021, 65, 102589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Boruta-Decision | LASSO-Coefficient |
|---|---|---|
| ADM | Rejected | |
| ALOX5AP | Confirmed | - |
| ANXA3 | Rejected | |
| ATP9A | Rejected | |
| BEND7 | Confirmed | 0.56877713 |
| BMX | Rejected | |
| CA4 | Confirmed | - |
| CD177 | Confirmed | - |
| CKAP4 | Rejected | |
| CLEC4D | Confirmed | 1.372927226 |
| CST7 | Rejected | |
| DDAH2 | Rejected | |
| DUSP13 | Confirmed | 0.985482029 |
| DYSF | Rejected | |
| FCAR | Confirmed | - |
| FFAR3 | Rejected | |
| FUT7 | Rejected | |
| GADD45A | Tentative | |
| GALNT14 | Rejected | |
| GPR84 | Rejected | |
| GYG1 | Confirmed | - |
| HK3 | Confirmed | - |
| HP | Confirmed | - |
| IFITM10 | Confirmed | 0.700047504 |
| KREMEN1 | Rejected | |
| LRRN1 | Confirmed | - |
| LTB4R | Confirmed | 0.512153962 |
| MCEMP1 | Tentative | |
| MMP9 | Rejected | |
| NR2E1 | Rejected | |
| OPLAH | Rejected | |
| OSM | Confirmed | - |
| PADI4 | Tentative | |
| PFKFB3 | Rejected | |
| RAB19 | Rejected | |
| ROPN1L | Confirmed | - |
| S100A12 | Rejected | |
| S100A8 | Confirmed | - |
| S100A9 | Confirmed | - |
| S100P | Rejected | |
| UNC5A | Confirmed | 0.539946778 |
| UPP1 | Confirmed | 0.321891042 |
| ZDHHC19 | Rejected |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, G.; Liu, H.; Deng, J.; Li, K.; Xie, B. A Novel 3-Gene Signature for Identifying COVID-19 Patients Based on Bioinformatics and Machine Learning. Genes 2022, 13, 1602. https://doi.org/10.3390/genes13091602
Lai G, Liu H, Deng J, Li K, Xie B. A Novel 3-Gene Signature for Identifying COVID-19 Patients Based on Bioinformatics and Machine Learning. Genes. 2022; 13(9):1602. https://doi.org/10.3390/genes13091602
Chicago/Turabian StyleLai, Guichuan, Hui Liu, Jielian Deng, Kangjie Li, and Biao Xie. 2022. "A Novel 3-Gene Signature for Identifying COVID-19 Patients Based on Bioinformatics and Machine Learning" Genes 13, no. 9: 1602. https://doi.org/10.3390/genes13091602
APA StyleLai, G., Liu, H., Deng, J., Li, K., & Xie, B. (2022). A Novel 3-Gene Signature for Identifying COVID-19 Patients Based on Bioinformatics and Machine Learning. Genes, 13(9), 1602. https://doi.org/10.3390/genes13091602