

Estimation of Metabolic Effects upon Cadmium Exposure during Pregnancy Using Tensor Decomposition



Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of the Dataset
2.2. Model Selection
2.2.1. Significance of Using Tensor Decomposition
2.2.2. Inappropriate Variable Selection with Multiple Regression Models
2.3. Data Processing
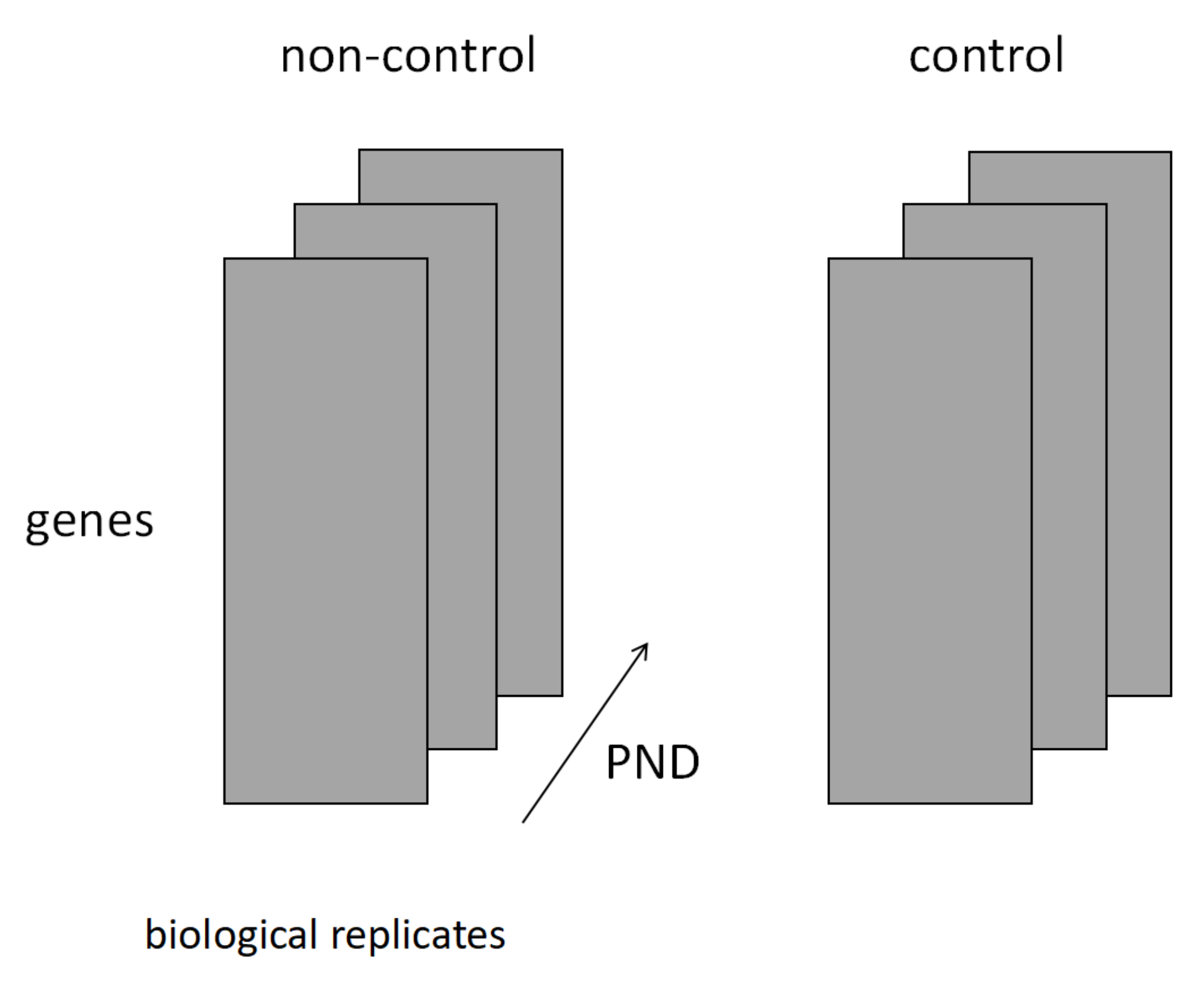
2.4. Structure of the Tensor Used in the Analysis
2.5. How to Analysis
2.5.1. Unsupervised Learning Using Tensor Decomposition
2.5.2. Variable Selection Using -Square Test
3. Results
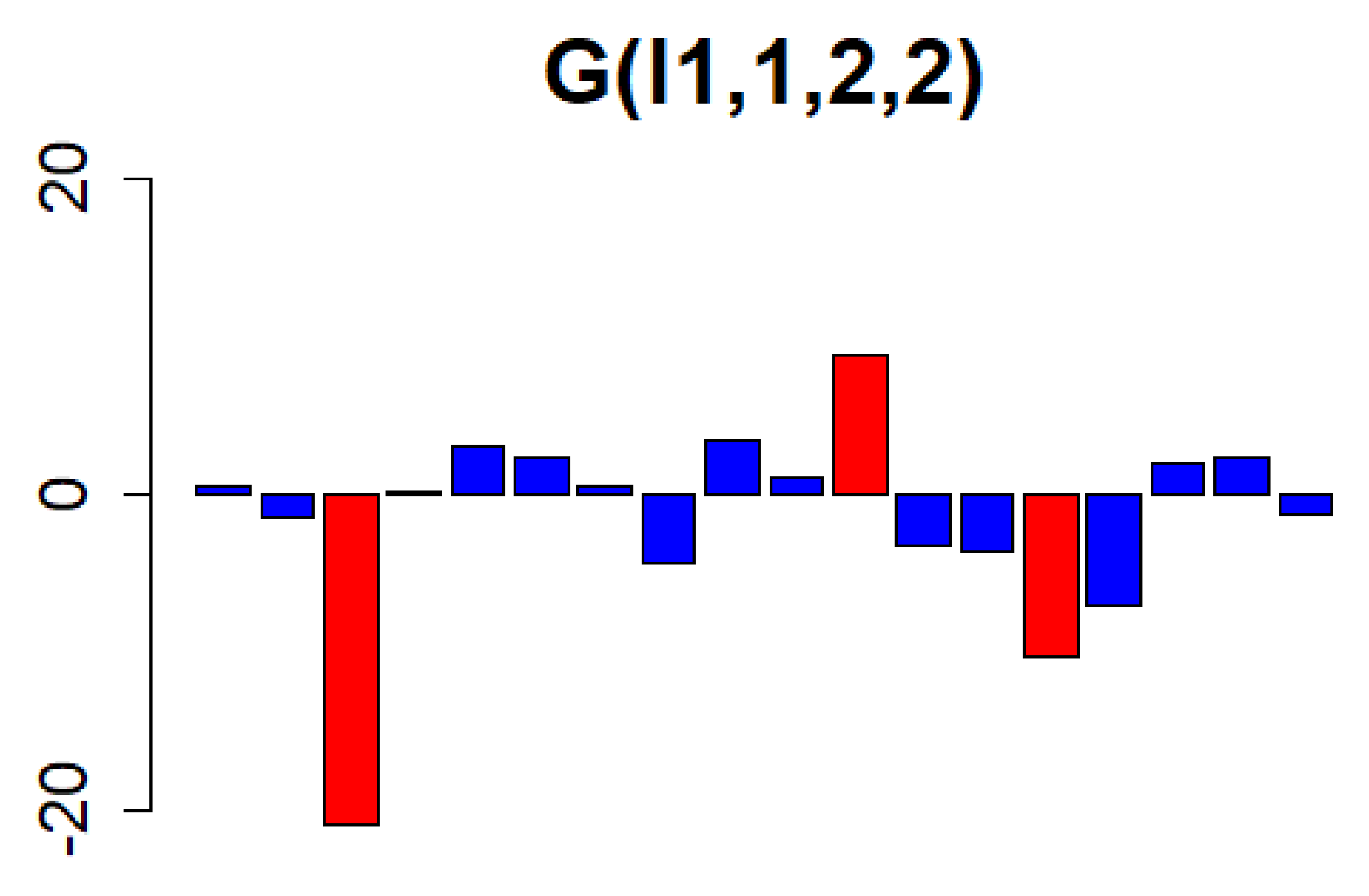
3.1. Identification of Genes with High Expression Levels
3.2. Checking the Ontology with gProfiler
3.3. Evaluation of Analysis Results
4. Discussion
4.1. Functions of the Identified Gene
4.2. Elucidation of the Mechanism of Insulin Metabolism Inhibition from the Obtained Genes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohba, K.I. Transport and Toxicity of Cadmium. Nichieishi 2006, 73, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, Z.A.; Vu, T.T.; Zaman, K. Oxidative stress as a mechanism of chronic cadmium-induced hepatotoxicity and renal toxicity and protection by antioxidants. Toxicol. Appl. Pharmacol. 1999, 154, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Takashige, K.; Hiroyuki, S.; Rie, F.; Yoshito, K.; Masahisa, I.; Kojun, S.; Shinya, S.; Masao, S. Cadmium reduces adipocyte size and expression levels of adiponectin and Peg1/Mest in adipose tissue. Toxicology 2010, 267, 20–26. [Google Scholar]
- Heindel, J.J.; Bruce, B.; Mathew, C.; Ronit, M.; Alberto, M.; Michelle, A.M.; Angel, N.; Paola, P.; Giancarlo, P. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef]
- Luo, Y.; McCullough, L.E.; Tzeng, J.Y.; Darrah, T.; Vengosh, A.; Maguire, R.L.; Maity, A.; Samuel-Hodge, C.; Murphy, S.K.; Mendez, M.A.; et al. Maternal blood cadmium, lead and arsenic levels, nutrient combinations, and offspring birth weight. BMC Public Health 2017, 17, 354. [Google Scholar]
- Thomas, W.J.; Garret, L.R.; Chris, M.S.; Aubrey, L.S.; Guillette, T.C.; Scott, M.B. Gestational Cd Exposure in the CD-1 Mouse Induces Sex-Specific Hepatic Insulin Insensitivity, Obesity, and Metabolic Syndrome in Adult Female Offspring. Toxicol. Sci. 2020, 178, 264–280. [Google Scholar]
- Taguchi, Y. Unsupervised Feature Extraction Applied to Bioinformatics: A PCA Based and TD Based Approach; Springer International Publishing AG: Cham, Switzerland, 2019; 329p. [Google Scholar]
- Ito, K.; Kato, S.; Matsuda, Y.; Kimura, M.; Okano, Y. cDNA cloning, characterization, and chromosome mapping of UBE2E3(alias UbcH9), encoding an N-terminally extended human ubiqutin-conjugating enzyme. Cytogenet. Cell Genet. 1999, 84, 99–104. [Google Scholar]
- Marilyn, R.; Jacky, G.G.; Pascal, S.P.; Hao, P.; Monique, L.; Michel, B.; Phuong, U.L.; Ivan, R.N. The gene product of the gp78/AMFR ubiquitin E3 ligase cDNA is selectively recognized by the 3F3A antibody within a subdomain of the endoplasmic reticulum. Biochem. Biophys. Res. Commun. 2004, 320, 1316–1322. [Google Scholar]
- Laurent, L.C.; Laëtitia, K.L.; Conception, P.; Eric, J.; Matthieu, L.; Elodie, H.; Robinson, T.; Guillaume, B.; Ayelet, S.; Manuel, S.R.; et al. E4F1 Is an Atypical Ubiquitin Ligase that Modulates p53 Effector Functions Independently of Degradation. Cell 2006, 127, 775–788. [Google Scholar]
- Annie, P.C.; John, S.B.; Hsan-Jan, Y.; Marwan, K.T.; Todd, E.S.; Ruth, E.S.; Darryl, Y.N.; Terry, A.B.; Kwang-Youn, A.K.; Jian, H.; et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet–Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 2006, 103, 6287–6292. [Google Scholar]
- Zhan, W.; Wang, W.; Han, T.; Xie, C.; Zhang, T.; Gan, M.; Wang, J.B. COMMD9 promotes TFDP1/E2F1 transcriptional activity via interaction with TFDP1 in non-small cell lung cancer. Cell Signal. 2017, 30, 59–66. [Google Scholar] [CrossRef]
- Tanahashi, N.; Suzuki, M.; Fujiwara, T.; Takahashi, E.I.; Shimbara, N.; Chung, C.H.; Tanaka, K. Chromosomal Localization and Immunological Analysis of a Family of Human 26S Proteasomal ATPases. Biochem. Biophys. Res. Commun. 1998, 243, 229–232. [Google Scholar] [CrossRef]
- Ma, C.P.; Slaughter, C.A.; DeMartino, G.N. Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). J. Biol. Chem. 1992, 267, 10515–10523. [Google Scholar] [CrossRef]
- Pomerantz, J.L.; Baltimore, D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef]
- Bertin, J.; Wang, L.; Guo, Y.; Jacobson, M.D.; Poyet, J.L.; Srinivasula, S.M.; Merriam, S.; DiStefano, P.S.; Alnemri, E.S. CARD11 and CARD14 Are Novel Caspase Recruitment Domain (CARD)/Membrane-associated Guanylate Kinase (MAGUK) Family Members that Interact with BCL10 and Activate NF-κB. J. Biol. Chem. 2001, 276, 11877–11882. [Google Scholar] [CrossRef]
- Maine, G.N.; Burstein, E. COMMD proteins: COMMing to the scene. Cell. Mol. Life Sci. 2007, 64, 1997–2005. [Google Scholar] [CrossRef]
- Martha, L.S.; Abbie, L.; Kristina, L.B.; Roger, K.W. Tumor necrosis factor-related genes and colon and rectal cancer. Int. J. Mol. Epidemiol. Genet. 2011, 2, 328–338. [Google Scholar]
- Liu, Q.; Yang, X.; Long, G.; Hu, Y.; Gu, Z.; Boisclair, Y.R.; Long, Q. Endoplasmic Reticulum Associated Degradation(ERAD) Deficiency Promotes Mitochondrial Dysfunction and Transcriptional Rewiring in Human Hepatic Cells. J. Biol. Chem. 2020, 295, 16743–16753. [Google Scholar] [CrossRef]
- Olga, Z.R.; Lissiene, S.N.; Florin, S.; Eric, A.S. The arginine methyltransferase NDUFAF7 is essential for complex I assembly and early vertebrate embryogenesis. Hum. Mol. Genet. 2014, 23, 5159–5170. [Google Scholar]
- Małecki, J.; Jakobsson, M.E.; Ho, A.Y.; Moen, A.; Rustan, A.C.; Falnes, P.Ø. Uncovering human METTL12 as a mitochondrial methyltransferase that modulates citrate synthase activity through metabolite-sensitive lysine methylation. J. Biol. Chem. 2017, 292, 17950–17962. [Google Scholar] [CrossRef]
- Rommel, A.M.; Todd, M.G.; Adam, O.; Hanna, G.B.; Rumela, C.; Elizabeth, A.R.; Yibin, K.; Thomas, S.; Ileana, M.C. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar]
- Young, S.G.; Davies, B.S.J.; Fong, L.G.; Gin, P.; Weinstein, M.M.; Bensadoun, A.; Beigneux, A.P. GPIHBP1: An endothelial cell molecule important for the lipolytic processing of chylomicrons. Curr. Opin. Lipidol. 2007, 18, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Sander, K. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar]
- Akio, K. Various Metabolisms, Physiological Functions and Related Diseases of Fatty Acids. Biochemistry 2010, 82, 591–605. [Google Scholar]
- Fikadu, G.T.; Ana, M.V.; Elleke, F.B.; Martin, H.; Amrita, J.; Angelika, H.; Pentti, S.; Joost, C.M.H. Sphingomyelin synthase-related protein SMSr is a suppressor of ceramide-induced mitochondrial apoptosis. J. Cell Sci. 2014, 127, 445–454. [Google Scholar]
- Atsuhiro, N.; Masaji, O.; Yukiko, O.; Ichiro, W. Placental Transfer of Cadmium in Pregnant Women. Jpn. J. Hyg. 1989, 44, 579–586. [Google Scholar]
- Tsuruzoe, K.; Nishikawa, T.; Araki, E. Oxidative Stress and Insulin Resistance. Diabetes 2006, 49, 845–848. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Term Name | Term ID | p-Value |
|---|---|---|---|
| KEGG | Metabolic pathway | KEGG01100 | 1.661 × |
| KEGG | Activation of platelets | KEGG04611 | 2.294 × |
| REAC | Lipid metabolism | R-HSA-556833 | 1.107 × |
| GO BP | Ionic transport | GO0006811 | 2.741 × |
| GO BP | Cell adhesion | GO0007155 | 4.702 × |
| GO BP | Intracellular signaling | GO0035556 | 4.207 × |
| GO BP | Cellular protein modification process | GO0036211 | 5.519 × |
| GO BP | Regulation of gene expression | GO0010468 | 1.241 × |
| GO BP | RNA metabolism | GO0016070 | 9.895 × |
| GO CC | Mitochondria | GO0005739 | 1.990 × |
| GO CC | Endoplasmic reticulum | GO0005783 | 1.990 × |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amakura, Y.; Taguchi, Y.-h. Estimation of Metabolic Effects upon Cadmium Exposure during Pregnancy Using Tensor Decomposition. Genes 2022, 13, 1698. https://doi.org/10.3390/genes13101698
Amakura Y, Taguchi Y-h. Estimation of Metabolic Effects upon Cadmium Exposure during Pregnancy Using Tensor Decomposition. Genes. 2022; 13(10):1698. https://doi.org/10.3390/genes13101698
Chicago/Turabian StyleAmakura, Yuki, and Y-h. Taguchi. 2022. "Estimation of Metabolic Effects upon Cadmium Exposure during Pregnancy Using Tensor Decomposition" Genes 13, no. 10: 1698. https://doi.org/10.3390/genes13101698
APA StyleAmakura, Y., & Taguchi, Y.-h. (2022). Estimation of Metabolic Effects upon Cadmium Exposure during Pregnancy Using Tensor Decomposition. Genes, 13(10), 1698. https://doi.org/10.3390/genes13101698