
Iron Deficiency Caused by Intestinal Iron Loss—Novel Candidate Genes for Severe Anemia

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
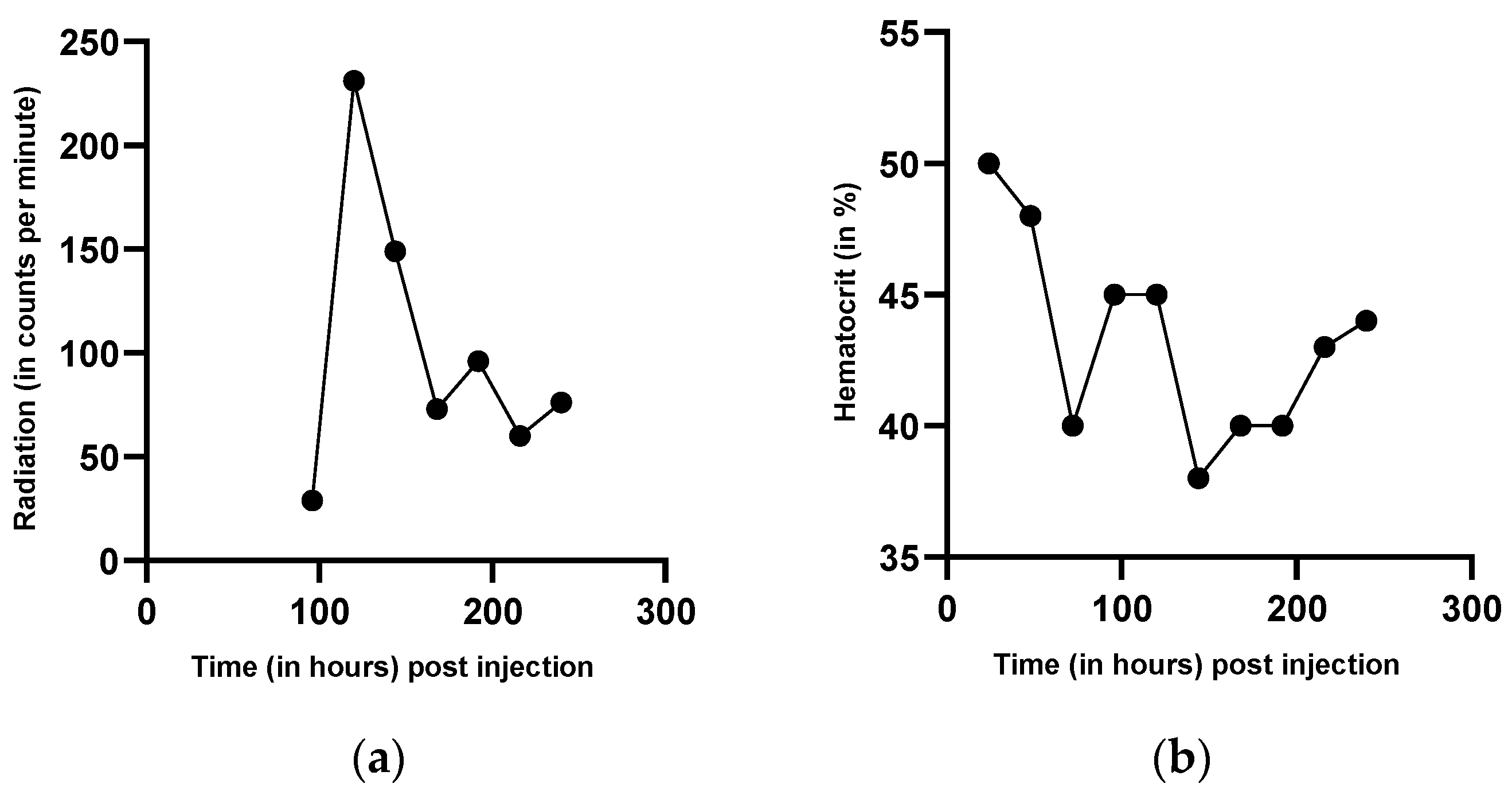
2.1. Measurement of Red Blood Cell Production by Intravenous Injection of Radioactively Marked Autologous Red Blood Cells
2.2. Measurement of Iron Excretion in the Urine and Intestinal Iron Excretion
2.3. Statistical Methods
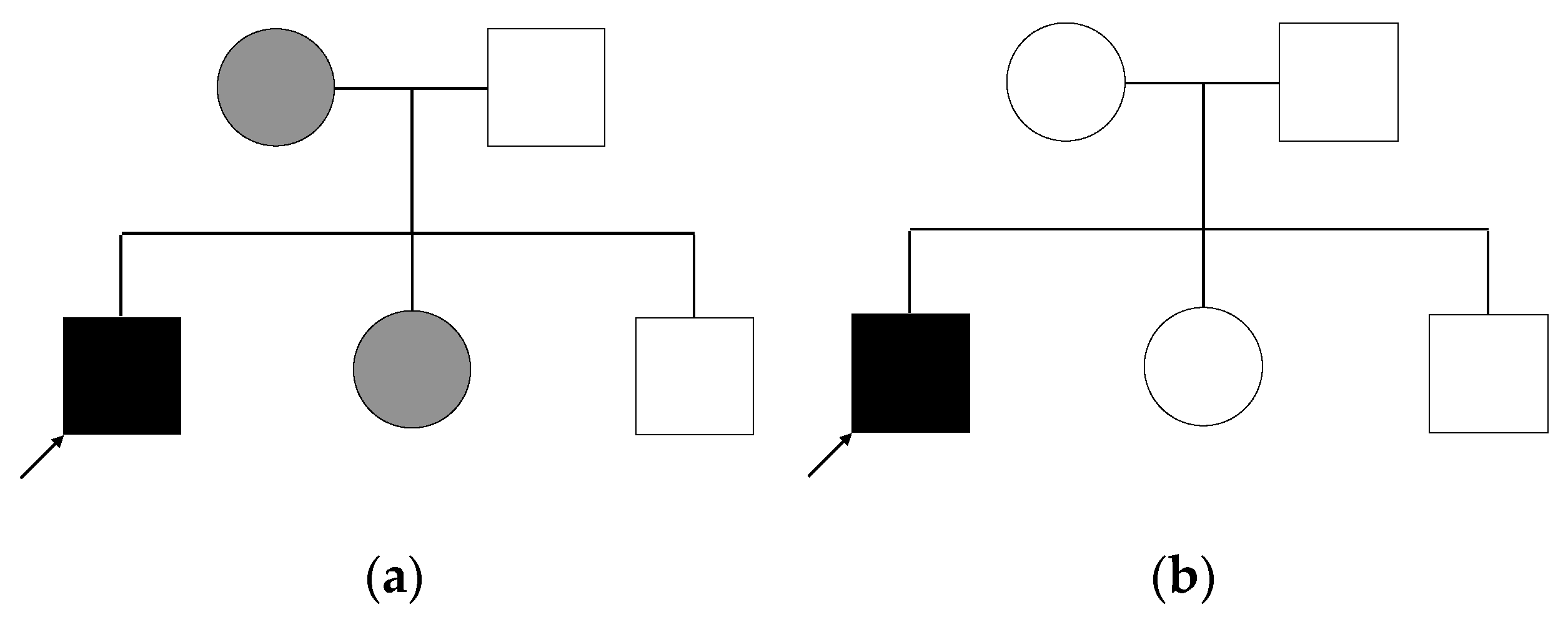
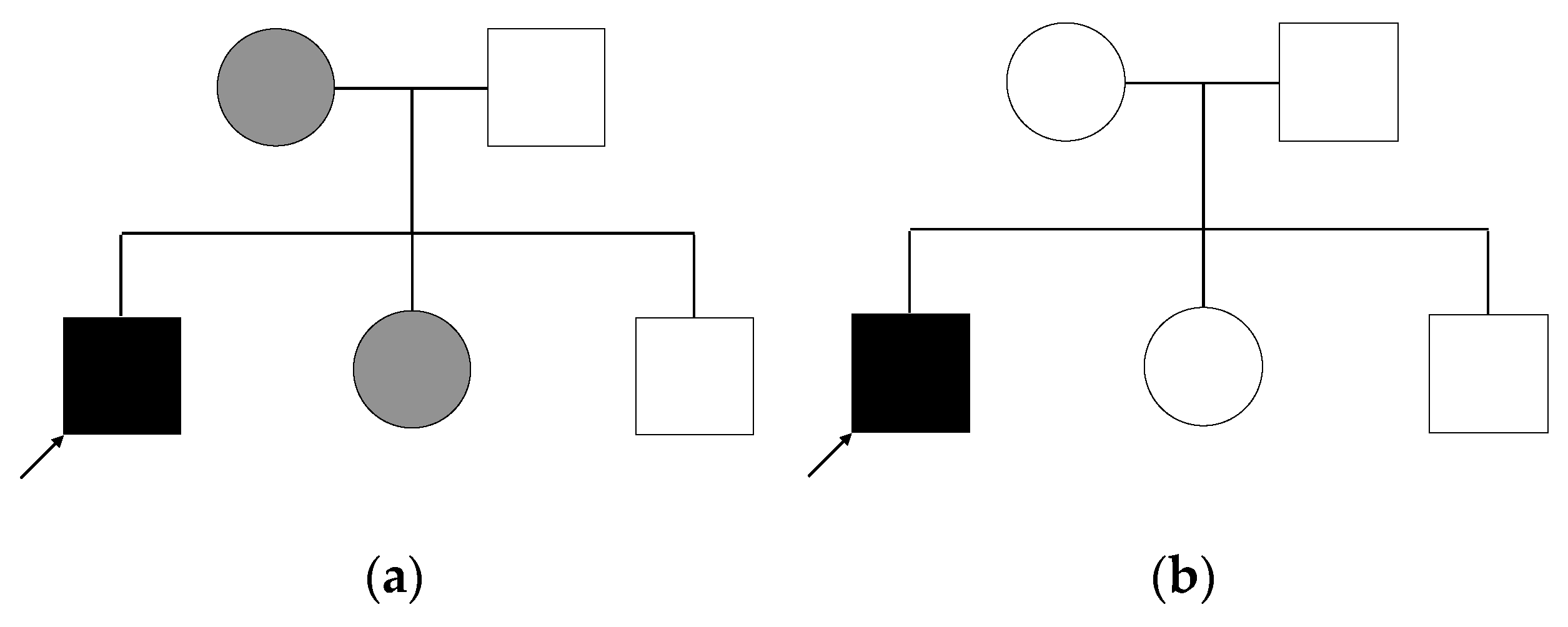
2.4. Genetic Analysis of Patient and Relatives
- Autosomal dominant inheritance with variable expression of the phenotype. In this case, each affected individual carries the variant heterozygously. We do not consider variants for which the mother is homozygous, as this is unlikely given the expected rarity of the variant.
- X-linked inheritance: the patient is a hemizygous carrier of the variant, while mother and sister are heterozygous.
- Autosomal dominant inheritance with a second hit in the patient: as mentioned above, patient, mother, and sister all carry the same variant, but in addition to that, the patient carries a second variant that the others do not have, and which would explain the increased severity of the patient.
- De novo mutation: the patient carries a variant in a heterozygous state, that he shares with no other individual in the family.
- Autosomal recessive inheritance: the patient is a homozygous carrier of the variant, while both parents are heterozygous. His siblings either do not carry the variant or are heterozygous.
- Compound heterozygosity: in case of compound heterozygosity, the patient carries two different heterozygous variants in the same gene, each inherited by one parent. The not affected father as well as both siblings could each be carriers of one of the variants. The mother and sister could also each be carriers of one variant.
3. Results
3.1. Patient History
3.2. Family History
3.3. Measurement of Red Blood Cell Production by Intravenous Injection of Radioactively Marked Autologous Red Blood Cells
3.4. Iron Levels in Urine
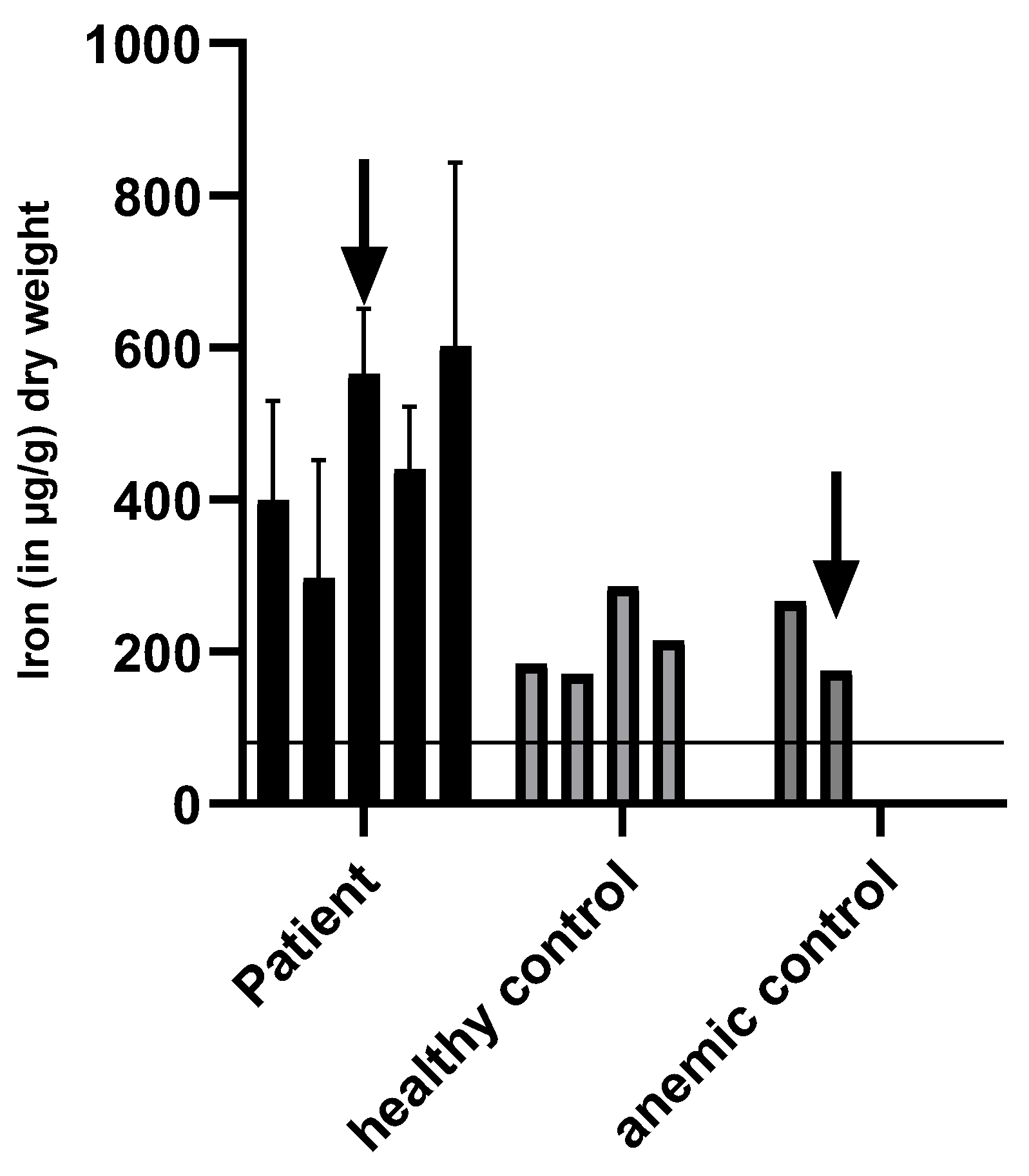
3.5. Iron Levels in Feces
3.6. Results of Whole Exome Sequencing
4. Discussion
4.1. Discussion of Anemia Causes
4.2. Discussion of Clinical Observations from a Hematological Point of View
4.3. Discussion of Radioactive Measurement
4.4. Discussion of Intestinal and Urinary Iron Excretion
4.5. Discussion of WES
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kassebaum, N.J.; Jasrasaria, R.; Naghavi, M.; Wulf, S.K.; Johns, N.; Lozano, R.; Regan, M.; Weatherall, D.; Chou, D.P.; Eisele, T.P.; et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood 2014, 123, 615–624. [Google Scholar] [CrossRef]
- Steinbicker, A.U.; Muckenthaler, M.U. Out of balance—Systemic iron homeostasis in iron-related disorders. Nutrients 2013, 5, 3034–3061. [Google Scholar] [CrossRef]
- Gulbis, B.; Eleftheriou, A.; Angastiniotis, M.; Ball, S.; Surrallés, J.; Castella, M.; Heimpel, H.; Hill, A.; Corrons, J.-L.V. Epidemiology of rare anaemias in Europe. Adv. Exp. Med. Biol. 2010, 686, 375–396. [Google Scholar] [CrossRef]
- European Network for Rare and Congenital Anaemias. Available online: https://rarediseases.info.nih.gov/organizations/872 (accessed on 23 November 2021).
- Besson-Fournier, C.; Latour, C.; Kautz, L.; Bertrand, J.; Ganz, T.; Roth, M.-P.; Coppin, H. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 2012, 120, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, M.D. The regulation of hepcidin and its effects on systemic and cellular iron metabolism. Am. Soc. Hematol. Educ. Program 2008, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, N.K.N.; Ellenbogen, S.; Trebicka, E.; Wang, L.; Mukhopadhyay, S.; Lacy-Hulbert, A.; Gallini, C.A.; Garrett, W.S.; Cherayil, B.J. Tumor necrosis factor α inhibits expression of the iron regulating hormone hepcidin in murine models of innate colitis. PLoS ONE 2012, 7, e38136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchi, C.; Montosi, G.; Zhang, K.; Lamberti, I.; Duncan, S.A.; Kaufman, R.J.; Pietrangelo, A. ER stress controls iron metabolism through induction of hepcidin. Science 2009, 325, 877–880. [Google Scholar] [CrossRef] [Green Version]
- Pixberg, H.U. Isotopenmethoden in der Anämiediagnostik. Med. Welt 1975, 26, 1349–1353. [Google Scholar]
- Pollycove, M.; Mortimer, R. The quantitative determination of iron kinetics and hemoglobin synthesis in human subjects. J. Clin. Investig. 1961, 40, 753–782. [Google Scholar] [CrossRef] [Green Version]
- Ricketts, C.; Cavill, I. Ferrokinetics: Methods and interpretation. Clin. Nucl. Med. 1978, 3, 159–164. [Google Scholar] [CrossRef]
- Tapernon, K.; Zander, R.; Niehoff, D.; Sibrowski, W. Qualitätskontrolle der Hämolyserate von Erythrozytenkonzentraten: Ringversuch zur Bestimmung von freiem Hämoglobin [Quality control of hemolysis rate of erythrocyte concentrates: A proficiency test for determination of free hemoglobin]. Anasthesiol. Intensivmed. Notfallmed. Schmerzther 2000, 36, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org (accessed on 23 November 2021).
- Zhu, H.; Xia, W.; Mo, X.-B.; Lin, X.; Qiu, Y.-H.; Yi, N.-J.; Zhang, Y.-H.; Deng, F.-Y.; Lei, S.-F. Gene-Based Genome-Wide Association Analysis in European and Asian Populations Identified Novel Genes for Rheumatoid Arthritis. PLoS ONE 2016, 11, e0167212. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Geng, S.; Weng, J.; Lu, Z.; Zeng, L.; Li, M.; Deng, C.; Wu, X.; Li, Y.; Du, X. Analysis of the expression of PHTF1 and related genes in acute lymphoblastic leukemia. Cancer Cell Int. 2015, 15, 93. [Google Scholar] [CrossRef] [Green Version]
- Andjelkovic, M.; Minic, P.; Vreca, M.; Stojiljkovic, M.; Skakic, A.; Sovtic, A.; Rodic, M.; Skodric-Trifunovic, V.; Maric, N.; Visekruna, J.; et al. Genomic profiling supports the diagnosis of primary ciliary dyskinesia and reveals novel candidate genes and genetic variants. PLoS ONE 2018, 13, e0205422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Quereda, L.; Rodriguez, M.J.; Diaz-Manera, J.; Alonso-Perez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Natera-de Benito, D.; Olive, M.; Gonzalez-Mera, L.; et al. Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes 2020, 11, 539. [Google Scholar] [CrossRef]
- Pesesse, L.; Sanchez, C.; Walsh, D.A.; Delcour, J.-P.; Baudouin, C.; Msika, P.; Henrotin, Y. Bone sialoprotein as a potential key factor implicated in the pathophysiology of osteoarthritis. Osteoarthr. Cartil. 2014, 22, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Karmi, O.; Marjault, H.-B.; Pesce, L.; Carloni, P.; Onuchic, J.N.; Jennings, P.A.; Mittler, R.; Nechushtai, R. The unique fold and lability of the 2Fe-2S clusters of NEET proteins mediate their key functions in health and disease. J. Biol. Inorg. Chem. JBIC A Publ. Soc. Biol. Inorg. Chem. 2018, 23, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Tamir, S.; Paddock, M.L.; Darash-Yahana-Baram, M.; Holt, S.H.; Sohn, Y.S.; Agranat, L.; Michaeli, D.; Stofleth, J.T.; Lipper, C.H.; Morcos, F.; et al. Structure–Function Analysis of NEET Proteins Uncovers Their Role as Key Regulators of Iron and ROS Homeostasis in Health and Disease: SI: Fe/S Proteins. 2015. Available online: http://www.sciencedirect.com/science/article/pii/S0167488914003735 (accessed on 23 November 2021).
- Kumar, P.L.; James, P.F. Identification and characterization of methylation-dependent/independent DNA regulatory elements in the human SLC9B1 gene. Gene 2015, 561, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Jia, Y.; Cooper, J.J.; Hale, T.; Zhang, Z.; Elbein, S.C. Common variants in glutamine: Fructose-6-phosphate amidotransferase 2 (GFPT2) gene are associated with type 2 diabetes, diabetic nephropathy, and increased GFPT2 mRNA levels. J. Clin. Endocrinol. Metab. 2004, 89, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Gao, J.; Yang, Y.; Qiu, R.; Zheng, Y.; Huang, W.; Zeng, Y.; Hou, Y.; Wang, S.; Leng, S.; et al. PHD finger protein 1 (PHF1) is a novel reader for histone H4R3 symmetric dimethylation and coordinates with PRMT5-WDR77/CRL4B complex to promote tumorigenesis. Nucleic Acids Res. 2018, 46, 6608–6626. [Google Scholar] [CrossRef] [Green Version]
- Hurt, J.A.; Obar, R.A.; Zhai, B.; Farny, N.G.; Gygi, S.P.; Silver, P.A. A conserved CCCH-type zinc finger protein regulates mRNA nuclear adenylation and export. J. Cell Biol. 2009, 185, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, M.C.; Johnstone, A.; Eccles, D.; Harmon, B.; Hayes, M.T.; Lea, R.A.; Griffiths, L.; Hoffman, E.P.; Stubbs, R.S.; Macartney-Coxson, D. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 2015, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.E.; Ma, K.; Xu, T.; Gao, L.; Wu, S.; Fu, C.; Zhang, W.; Wang, Z.; Liu, K.; Dong, M.; et al. Mutation of kri1l causes definitive hematopoiesis failure via PERK-dependent excessive autophagy induction. Cell Res. 2015, 25, 946–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, G.; Ghosheh, N.; Zeng, X.; Bogestål, Y.; Sartipy, P.; Synnergren, J. Identification of stable reference genes in differentiating human pluripotent stem cells. Physiol. Genom. 2015, 47, 232–239. [Google Scholar] [CrossRef]
- Barbashina, V.; Salazar, P.; Holland, E.C.; Rosenblum, M.K.; Ladanyi, M. Allelic losses at 1p36 and 19q13 in gliomas: Correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 1119–1128. [Google Scholar]
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H.; et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015, 75, 1516–1526. [Google Scholar] [CrossRef] [Green Version]
- Frezzato, F.; Gattazzo, C.; Martini, V.; Trimarco, V.; Teramo, A.; Carraro, S.; Cabrelle, A.; Ave, E.; Facco, M.; Zambello, R.; et al. HS1, a Lyn kinase substrate, is abnormally expressed in B-chronic lymphocytic leukemia and correlates with response to fludarabine-based regimen. PLoS ONE 2012, 7, e39902. [Google Scholar] [CrossRef] [PubMed]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [Green Version]
- Iolascon, A.; de Falco, L. Mutations in the gene encoding DMT1: Clinical presentation and treatment. Semin. Hematol. 2009, 46, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Island, M.-L.; Jouanolle, A.-M.; Détivaud, L.; Fatih, N.; Ropert, M.; Brissot, E.; Mosser, A.; Maisonneuve, H.; Brissot, P.; et al. A novel N491S mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol. Dis. 2011, 47, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Sendamarai, A.K.; Ohgami, R.S.; Fleming, M.D.; Lawrence, C.M. Structure of the membrane proximal oxidoreductase domain of human Steap3, the dominant ferrireductase of the erythroid transferrin cycle. Proc. Natl. Acad. Sci. USA 2008, 105, 7410–7415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.-S.; Sheftel, A.D.; Ponka, P. The anemia of “haemoglobin-deficit” (hbd/hbd) mice is caused by a defect in transferrin cycling. Exp. Hematol. 2006, 34, 593–598. [Google Scholar] [CrossRef]
- Troadec, M.-B.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.H.; Ward, D.M.; Kaplan, J. Targeted deletion of the mouse Mitoferrin1 gene: From anemia to protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Investig. 2010, 120, 1749–1761. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, A.K.; Campagna, D.R.; McLoughlin, E.M.; Agarwal, S.; Fleming, M.D.; Bottomley, S.S.; Neufeld, E.J. Systematic molecular genetic analysis of congenital sideroblastic anemia: Evidence for genetic heterogeneity and identification of novel mutations. Pediatric Blood Cancer 2010, 54, 273–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allikmets, R.; Raskind, W.H.; Hutchinson, A.; Schueck, N.D.; Dean, M.; Koeller, D.M. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum. Mol. Genet. 1999, 8, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Savary, S.; Allikmets, R.; Denizot, F.; Luciani, M.F.; Mattei, M.G.; Dean, M.; Chimini, G. Isolation and chromosomal mapping of a novel ATP-binding cassette transporter conserved in mouse and human. Genomics 1997, 41, 275–278. [Google Scholar] [CrossRef]
- Camaschella, C.; Campanella, A.; de Falco, L.; Boschetto, L.; Merlini, R.; Silvestri, L.; Levi, S.; Iolascon, A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 2007, 110, 1353–1358. [Google Scholar] [CrossRef]
- Camaschella, C.; Poggiali, E. Inherited disorders of iron metabolism. Curr. Opin. Pediatr. 2011, 23, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Meynard, D.; Vaja, V.; Sun, C.C.; Corradini, E.; Chen, S.; López-Otín, C.; Grgurevic, L.; Hong, C.C.; Stirnberg, M.; Gütschow, M.; et al. Regulation of TMPRSS6 by BMP6 and iron in human cells and mice. Blood 2011, 118, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Pagani, A.; Nai, A.; de Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of tissue iron homeostasis: The macrophage “ferrostat”. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Fomon, S.J.; Ziegler, E.E.; Serfass, R.E.; Nelson, S.E.; Rogers, R.R.; Frantz, J.A. Less than 80% of absorbed iron is promptly incorporated into erythrocytes of infants. J. Nutr. 2000, 130, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Charlton, R.; Seftel, H.; Bothwell, T.; Mayet, F.; Adams, B.; Finch, C.; Layrisse, M. Body iron excretion in man: A collaborative study. Am. J. Med. 1968, 45, 336–353. [Google Scholar] [CrossRef]
- Kreuzer, M.; Kirchgessner, M. Endogenous iron excretion. A quantitative means to control iron metabolism? Biol. Trace Elem. Res. 1991, 29, 77–92. [Google Scholar] [CrossRef]
- McCance, R.A.; Widdowson, E.M. The absorption and excretion of iron following oral and intravenous administration. J. Physiol. 1938, 94, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Finch, C.A.; Hegsted, M.; Kinney, T.D.; Thomas, E.D.; Rath, C.E.; Haskins, D.; Finch, S.; Fluharty, R.G. Iron metabolism: The pathophysiology of iron storage. Blood 1950, 5, 983–1008. [Google Scholar] [CrossRef] [Green Version]
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.E.; Weintraub, L.R.; Crosby, W.H. The role of the intestine in iron kinetics. J. Clin. Investig. 1964, 43, 963–974. [Google Scholar] [CrossRef]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Griffiths, T.A.M.; Mauk, A.G.; MacGillivray, R.T.A. Recombinant expression and functional characterization of human hephaestin: A multicopper oxidase with ferroxidase activity. Biochemistry 2005, 44, 14725–14731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Closuit, M.; Govaerts, J.; Lambrechts, A. Contribution à l’étude du métabolisme du fer à l’aide du radiofer 59/26 Fe. I. Quelques données nouvelles sur l’élimination rénale du fer. Arch. Int. Physiol. 1953, 61, 306–317. [Google Scholar] [CrossRef]
- Van Raaij, S.E.G.; Rennings, A.J.; Biemond, B.J.; Schols, S.E.M.; Wiegerinck, E.T.G.; Roelofs, H.M.J.; Hoorn, E.J.; Walsh, S.B.; Nijenhuis, T.; Swinkels, D.W.; et al. Iron handling by the human kidney: Glomerular filtration and tubular reabsorption both contribute to urinary iron excretion. Am. J. Physiol. Ren. Physiol. 2019, 316, F606–F614. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, C.; Moore, D.; Delorme, C.; Lacas-Gervais, S.; Ait-El-Mkadem, S.; Fragaki, K.; Burté, F.; Serre, V.; Bannwarth, S.; Chaussenot, A.; et al. A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Hum. Mol. Genet. 2017, 26, 1599–1611. [Google Scholar] [CrossRef]
- Tamir, S.; Zuris, J.A.; Agranat, L.; Lipper, C.H.; Conlan, A.R.; Michaeli, D.; Harir, Y.; Paddock, M.L.; Mittler, R.; Cabantchik, Z.I.; et al. Nutrient-deprivation autophagy factor-1 (NAF-1): Biochemical properties of a novel cellular target for anti-diabetic drugs. PLoS ONE 2013, 8, e61202. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Kao, C.-H.; Chen, Y.-T.; Wang, C.-H.; Wu, C.-Y.; Tsai, C.-Y.; Liu, F.-C.; Yang, C.-W.; Wei, Y.-H.; Hsu, M.-T.; et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Sankaran, V.G.; Weiss, M.J. Anemia: Progress in molecular mechanisms and therapies. Nat. Med. 2015, 21, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, H.; Okuno, Y.; Yoshida, K.; Shiraishi, Y.; Doisaki, S.; Narita, A.; Sakaguchi, H.; Kawashima, N.; Wang, X.; Xu, Y.; et al. Clinical utility of next-generation sequencing for inherited bone marrow failure syndromes. Genet. Med. 2017, 19, 796–802. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 06/12/ 10 | 07/07/ 11 | 20/06/ 12 | 20/12/ 12 | 25/04/ 13 | 29/08/ 13 | 19/12/ 13 | 08/05/ 14 | 05/03/ 15 | |
|---|---|---|---|---|---|---|---|---|---|
| RBC, (mcL) norm 4.44–5.61 | 3.15 | 4.8 | 3.48 | 3.91 | 3.98 | 4.33 | 4.45 | 3.58 | 4.32 |
| Hemoglobin (g/dL): 13.5–16.9 | 8.6 | 13.0 | 9.4 | 9.8 | 11.2 | 11.2 | 10.9 | 9.0 | 11.2 |
| (Hct, %): 40–49.4 | 27.7 | 39.1 | 28.8 | 31.1 | 33.7 | 36.0 | 34.9 | 29.4 | 35.2 |
| MCV (fL): 81.8–95.5 | 87.9 | 81.5 | 82.8 | 79.5 | 84.7 | 83.1 | 78.4 | 82.1 | 81.5 |
| MCH (pg): 27–32.3 | 27.3 | 27.1 | 27.0 | 25.1 | 28.1 | 25.9 | 24.5 | 25.1 | 25.9 |
| MCHC (g/dL): 32.4–35 | 31.0 | 33.2 | 32.6 | 31.5 | 33.2 | 31.1 | 31.2 | 30.6 | 31.8 |
| Reticulocyte count (%): 0.4–1.36 | 5.7 | 1.6 | 4.0 | 3.3 | 4.5 | 2.2 | 5.8 | ||
| Reticulocyte count (103/mcL): 23–70 | 180.2 | 75.4 | 138 | 129 | 179 | 93 | 208 | ||
| Ret He (pg): 32,1–38.8 | 16.7 | 22.7 | 22.1 | 16.9 | 24.6 | 24.0 | 17.9 | ||
| Reticulocyte production index: 2–3 | 2.0 | 1.5 | 1.5 | 2.4 | 1.3 | 2.3 | |||
| Free hemoglobin (mg/dL): 0–15 | 6.7 | ||||||||
| Ferritin (µg/L): 22–322 | 10 | 19 | 58 | 9 | 262 | 33 | 39 | 102 | 12 |
| Serum iron (µg/dL): 80–150 | 44 | 23 | 21 | 346 | 29 | 34 | |||
| Transferrin (mg/dL): 200–360 | 301 | 424 | 284 | 337 | |||||
| Transferrin saturation (%): 16–45 | 5 | 4 | 86 | 7 | |||||
| Haptoglobin (mg/dL) | 153 | 120 | |||||||
| Erythropoetin (mU/mL): 6–23 | 174 | ||||||||
| Coeruloplasmin (mg/dL): 20–60 | 21 | ||||||||
| alpha HBDH (U/L): 72–182 | 180 | 190 | |||||||
| Zinc (mc/L): 750–1400 | 662 | ||||||||
| Copper (mc/dL): 65–165 | 125 |
| Date | Type of Examination | Results |
|---|---|---|
| 19/07/2011 27/06/2012 17/05/2013 | Molecular markers of AML examination with PCR |
|
| 08/05/2014 | Hemostasis |
During Acetylsalicylic acid therapy sufficient prolongation of shutter speed, no evidence for aspirin resistance or thrombocytic hyperreactivity |
| 06/12/2010 | Blood typing | Blood group: 0 Rhesus factor: positive Rhesus system: CcD.ee Kell: negative Antibodies: negative Cytomegalovirus antibodies: IgG and IgM negative |
| 08/05/2014 | Blood typing | monospecific Coombs-test: IgG, IgA, IgM, C3d, C3c: negative AKS indirect Coombs Diamed: negative |
| Date | WBC (/mcL) | RBC (/mL) | HGB (g/dL) | HCT (%) | MCV (fL) | MCH (pg) | MCHC (g/dL) | PLT (mcL) | Ferritin (ng/mL) |
|---|---|---|---|---|---|---|---|---|---|
| 23/1/19 * | 5.5 | 4.5 | 12.7 | 38.1 | 84.7 | 28.2 | 33.3 | 188 | 167 |
| 30/1/19 | 4.5 | 4.51 | 12.7 | 38.3 | 84.9 | 28.2 | 33.2 | 186 | |
| 12/6/19 | 4.3 | 4.63 | 13.1 | 39.8 | 86.0 | 28.3 | 32.9 | 156 | |
| 26/6/19 | 4.3 | 3.78 | 10.6 | 33.0 | 87.3 | 28.0 | 32.1 | 163 | 551 |
| 3/7/19 | 4.6 | 4.81 | 13.6 | 41.3 | 85.9 | 28.3 | 32.9 | 162 | |
| 10/7/19 | 5.2 | 4.78 | 13.5 | 41.2 | 86.2 | 28.2 | 32.8 | 177 | |
| 20/11/19 | 5.9 | 4.96 | 14.2 | 42.2 | 85.1 | 28.6 | 33.6 | 174 | 418 |
| 4/12/19 | 5.2 | 4.83 | 13.8 | 41.6 | 86.1 | 28.6 | 33.2 | 144 | |
| 5/2/20 | 6.5 | 5.02 | 14.6 | 43.0 | 85.7 | 29.1 | 34.0 | 164 | |
| 22/2/20 | 5.5 | 4.88 | 14.0 | 42.2 | 86.5 | 28.7 | 33.2 | 164 | 605 |
| 4/3/20 | 5.4 | 5.15 | 14.9 | 44.3 | 86.0 | 28.9 | 33.6 | 156 | |
| 17/6/20 | 4.8 | 4.67 | 13.4 | 39.5 | 84.6 | 28.7 | 33.9 | 157 | 602 |
| 1/7/20 | 5.6 | 3.72 | 10.6 | 32.9 | 88.4 | 28.5 | 32.2 | 173 | 306 |
| 9/12/20 | 5.4 | 4.83 | 14.1 | 41.1 | 85.7 | 29.2 | 34.1 | 179 | |
| 16/3/21 | 9.2 | 2.70 | 7.8 | 25.1 | 93.0 | 28.9 | 31.1 | 166 | 573 |
| 9/6/21 | 5.7 | 4.95 | 13.7 | 41.8 | 84.4 | 27.7 | 32.8 | 159 | |
| 22/9/21 | 5.9 | 4.24 | 12.3 | 37.5 | 88.4 | 29.0 | 32.8 | 155 |
| Date | Type of Examination | Results |
|---|---|---|
| 27/07/2011 | Bone marrow examination | Hematopoetic bone marrow with discrete dysnuclear stigmata of erythropoesis and of megakaryocytes as well as microfocal abnormal lymphoid infitration. Taking the clinical information into account, the findings can be correlated with MDS. |
| 25/04/2013 | Bone marrow examination | Myelogramm: Myeloblasts 0–2; promyelocytes 2–5, myelocytes 9–17, metamyelocytes 7–25, banded neutrophils 9–15, segmented neutrophils 4–11, eosinophils 1–5, basophils 0–1, monocytes 0–1, proerythroblasts 1–3, macroblasts 2–5, normoblasts 12–28, lymphocytes 7–22, plasmacells 0–4, reticulum cells 0–1, tissuebasophile mastcells 0–1, other blasts <5%. Granulopoesis: quantification normal, hypogranulation Megakaryopoesis: increased, hyposegmentation 5q Erythropoese: quant normal, dyserythropoesis Iron in bone marrow shows severe depletion. FACS analysis: no significant evidence for the presence of blasts (<5%) Diagnosis: Mild erythropoetic hyperplasia of the hematopoetic bone marrow with reticulocytic iron depletion, no infiltrations of a malign tumor, iron defeciency. |
| 05/03/2015 | Bone marrow examination | Maturing hematopoesis without significant signs of dysplasia, hemoglobin deficiency and significant deficiency of stored iron, no increase in blasts FACS analysis: no significant population of blasts, maturing hematopoesis |
| 30/04/2013 | Skin biopsy | Microscopy shows skin with unobtrusive epidermis and normal skin appendage, periadnexal lymphocytic infiltration. Diagnosis: mostly age-appropriate skin with no evidence of cutaneous accumulation. |
| 08/05/2014 | Sctintigraphy (Tc-99m-Ultra-Tag) with SPECT/CT for the detection of a gastrointestinal hemorrhage | No evidence for a gastrointestinal bleeding. The constant presentation of the intestinal loop in the upper left abdomen cannot exclude a angiodysplasia (differential diagnosis: regional hyperemic intestinal loop). Comparison with morphologic imaging is recommended. |
| Chromosome | Location | Ref | Alt | Gene | Change of Amino Acid | CADD Score | rsID | gnomAD_ Exome |
|---|---|---|---|---|---|---|---|---|
| 1 | 114269137 | G | A | PHTF1 | p.P131S | 23.6 | - | - |
| 1 | 118584464 | C | T | SPAG17 | p.E1006K | 21 | rs200539422 | 0.00008064 |
| 2 | 179456326 | C | G | TTN | p.V11009L | 22.9 | - | - |
| 4 | 88731867 | C | T | IBSP | p.T119M | 25 | rs866752943 | 0.00003591 |
| 4 | 103806433 | C | T | CISD2 SLC9B1 * | p.C462Y p.A55V | 29.2 | rs866399747 | - |
| 5 | 179751867 | G | A | GFPT2 | p.R209W | 34 | rs753649376 | 0.00002686 |
| 6 | 33382090 | C | A | PHF1 | p.R275S | 23.7 | - | - |
| 8 | 144550669 | C | T | ZC3H3 | p.R663Q | 23.1 | rs754727062 | - |
| 19 | 6467566 | C | A | DENND1C | p.Q741H | 23.6 | rs778032935 | - |
| 19 | 10670511 | C | T | KRI1 | p.R307H | 32 | rs1202991375 | 0.000008976 |
| 19 | 58967096 | G | A | ZNF324B | p.C262Y | 25.2 | rs774176207 | 0.00005501 |
| Chromosome | location | Ref | Alt | Gene | Change of Amino Acid | CADD Score | rsID | gnomAD Exome |
|---|---|---|---|---|---|---|---|---|
| 1 | 7792598 | C | A | CAMTA1 | p.A45E | 22.5 | rs367848023 | 0.000045 |
| 1 | 155240725 | C | T | CLK2 | p.R15Q | 24.2 | rs776199117 | 0.000008952 |
| Gene | Gene Name | Gene Function and Phenotypes of Genetic Mutations | Literature |
|---|---|---|---|
| PHTF1 | Putative homeodomain transcription factor 1 | Main expression in testes, associated with rheumatoid arthritis and type 1 diabetes, overexpression in acute lymphoblastic leukemia | [14] [15] |
| SPAG17 | Sperm associated antigen 17 | Organization of microtubuli and function of the axoneme; mutations cause primary ciliary dyskinesia, SNP cause skeletal malformations of the limbs in mice | [16] |
| TTN | Titin | Encodes protein of striated muscle, mutations cause neuromuscular diseases | [17] |
| IBSP | Integrin binding sialoprotein | Encodes a major structural protein of bone matrix, discussed as a factor in the development of osteoarthritis | [18] |
| CISD2 | CDGSH iron sulfur domain 2 | Encodes zinc finger protein NAF-1, a recently discovered member of the NEET protein family in the endoplasmatic reticulum, involved in iron and ROS homeostasis, as well as autophagy and apoptosis, cause of Wolfram syndrome 2 | [19] [20] |
| SLC9B1 | Solute carrier family 9 member B1 | Encoded protein is a sodium/hydrogen exchanger and transmembrane protein which is primarily expressed in testes, essential for sperm motility and fertility | [21] |
| GFPT2 | Glutamine-fructose-6-phosphate transaminase 2 | Controls flux of glucose into the hexamine pathway, common variants are associated with type 2 diabetes and diabetic nephropathy | [22] |
| PHF1 | PHD finger protein 1 | Encodes polycomb group protein, functions in transcriptional repression of homeotic genes, recruited to double streak breaks, promotes cell proliferation, invasion and tumorigenesis, key factor for tumor progression | [23] |
| ZC3H3 | Zinc finger CCCH domain-containing protein 3 | Relevant for export of polyadenylated mRNAs from the nucleus, highly expressed in bone marrow | [24] |
| DENND1C | Differentially expressed in normal and neoplastic cells domain 1C | Guanine nucleotide exchange factor for the early endosomal small GTPase, which regulates endosomal membrane trafficking, involved in actin polymerization, potential role in glucose transport and homeostasis | [25] |
| KRI1 | KRI1 homolog | DNA binding, importance in hematopoiesis | [26] |
| ZNF324B | Zinc finger protein 324B | Transcriptional regulation, discussed as a reference gene in human pluripotent stem cells | [27] |
| CAMTA1 | Calmodulin binding transcription activator 1 | Mainly expressed in adult brain tissue, encodes a transcription factor and is discussed as a tumor suppressor | [28] |
| CLK2 | Cdc-like kinase 2 | Phosphorylation of serine/threonine and tyrosine-containing substrates, importance in cell cycle and different malignancies, overexpression in breast cancer, modulates hepatic gluconeogenesis and fatty acid oxidation | [29] |
| HCLS1 | Hematopoietic lineage cell-specific substrate 1 | Antigen receptor signaling for clonal expansion and deletion in lymphoid cells, overexpression in B-chronic lymphocytic leukemia | [30] |
| RP1L1 | Retinitis pigmentosa 1-like 1 protein | Differentiation of photoreceptor cells, discussed as cause for occult macular dystrophy | [31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huettmann, C.; Stelljes, M.; Sivalingam, S.; Fobker, M.; Vrachimis, A.; Exler, A.; Wenning, C.; Wempe, C.; Penke, M.; Buness, A.; et al. Iron Deficiency Caused by Intestinal Iron Loss—Novel Candidate Genes for Severe Anemia. Genes 2021, 12, 1869. https://doi.org/10.3390/genes12121869
Huettmann C, Stelljes M, Sivalingam S, Fobker M, Vrachimis A, Exler A, Wenning C, Wempe C, Penke M, Buness A, et al. Iron Deficiency Caused by Intestinal Iron Loss—Novel Candidate Genes for Severe Anemia. Genes. 2021; 12(12):1869. https://doi.org/10.3390/genes12121869
Chicago/Turabian StyleHuettmann, Carolina, Matthias Stelljes, Sugirthan Sivalingam, Manfred Fobker, Alexis Vrachimis, Anne Exler, Christian Wenning, Carola Wempe, Matthias Penke, Andreas Buness, and et al. 2021. "Iron Deficiency Caused by Intestinal Iron Loss—Novel Candidate Genes for Severe Anemia" Genes 12, no. 12: 1869. https://doi.org/10.3390/genes12121869
APA StyleHuettmann, C., Stelljes, M., Sivalingam, S., Fobker, M., Vrachimis, A., Exler, A., Wenning, C., Wempe, C., Penke, M., Buness, A., Ludwig, K. U., Muckenthaler, M. U., & Steinbicker, A. U. (2021). Iron Deficiency Caused by Intestinal Iron Loss—Novel Candidate Genes for Severe Anemia. Genes, 12(12), 1869. https://doi.org/10.3390/genes12121869