
Identification of Novel Mutations by Targeted NGS Panel in Patients with Hyperferritinemia

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Panel Design
2.3. Library Preparation and Sequencing
2.4. Analysis
2.5. Multiplex Ligation-Dependent Probe Amplification (MLPA) Assay
2.6. Generation of SLC40A1 Mutant Construct
2.7. Cell Culture and Dual-Luciferase Reporter Assay
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | Alanine Aminotransferase |
| ESE | Exonic Splicing Enhancer |
| FTL | L-Ferritin gene |
| HAMP | Hepcidin gene |
| HFE | HFE gene |
| HDL | High Density Lipoprotein Cholesterol |
| HGMD | Human Gene Mutation Database |
| HGVS | Human Genome Variation Society |
| HH | Hereditary Hemochromatosis |
| HHCS | Hereditary Hyperferritinemia-Cataract Syndrome |
| HJV | Hemojuvelin gene |
| gnomAD | genome Aggregation Database |
| IRE | Iron Responsive Element |
| IRP | Iron Regulatory Protein |
| LIC | Liver Iron Concentration |
| MLPA | Multiplex Ligation-dependent Probe Amplification |
| MRI | Magnetic Resonance Imaging |
| NGS | Next Generation Sequencing |
| SLC40A1 | Ferroportin gene |
| TFR2 | Transferrin Receptor 2 gene |
| TSAT | Transferrin Saturation |
| WT | Wild-Type |
References
- Piperno, A.; Pelucchi, S. Mariani, Inherited iron overload disorders. Transl. Gastroenterol. Hepatol. 2020, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Poggiali, E. Inherited disorders of iron metabolism. Curr. Opin. Pediatr. 2011, 23, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Martinez, P.; Schved, J.F.; Brissot, P. The evaluation of hyperferritinemia: An updated strategy based on advances in detecting genetic abnormalities. Am. J. Gastroenterol. 2005, 100, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Mariani, R.; Pelucchi, S.; Arosio, C.; Coletti, S.; Pozzi, M.; Paolini, V.; Trombini, P.; Piperno, A. Genetic and metabolic factors are associated with increased hepatic iron stores in a selected population of p.Cys282Tyr heterozygotes. Blood Cells Mol. Dis. 2010, 44, 159–163. [Google Scholar] [CrossRef]
- Merryweather-Clarke, A.T.; Cadet, E.; Bomford, A.; Capron, D.; Viprakasit, V.; Miller, A.; McHugh, P.J.; Chapman, R.W.; Pointon, J.J.; Wimhurst, V.L.; et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet. 2003, 12, 2241–2247. [Google Scholar] [CrossRef]
- Pietrangelo, A. Ferroportin disease: Pathogenesis, diagnosis and treatment. Haematologica 2017, 102, 1972–1984. [Google Scholar] [CrossRef]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Primers 2018, 4, 18016. [Google Scholar] [CrossRef]
- Piperno, A. Molecular diagnosis of hemochromatosis. Expert Opin. Med. Diagn. 2013, 7, 161–177. [Google Scholar] [CrossRef]
- Piperno, A.; Alessio, M. Aceruloplasminemia: Waiting for an Efficient Therapy. Front. Neurosci. 2018, 12, 903. [Google Scholar] [CrossRef]
- Girelli, D.; Bozzini, C.; Zecchina, G.; Tinazzi, E.; Bosio, S.; Piperno, A.; Ramenghi, U.; Peters, J.; Levi, S.; Camaschella, C.; et al. Clinical, biochemical and molecular findings in a series of families with hereditary hyperferritinaemia-cataract syndrome. Br. J. Haematol. 2001, 115, 334–340. [Google Scholar] [CrossRef]
- Kannengiesser, C.; Jouanolle, A.M.; Hetet, G.; Mosser, A.; Muzeau, F.; Henry, D.; Bardou-Jacquet, E.; Mornet, M.; Brissot, P.; Deugnier, Y.; et al. A new missense mutation in the L ferritin coding sequence associated with elevated levels of glycosylated ferritin in serum and absence of iron overload. Haematologica 2009, 94, 335–339. [Google Scholar] [CrossRef]
- Camaschella, C.; Poggiali, E. Towards explaining “unexplained hyperferritinemia”. Haematologica 2009, 94, 307–309. [Google Scholar] [CrossRef]
- Trombini, P.; Piperno, A. Ferritin, metabolic syndrome and NAFLD: Elective attractions and dangerous liaisons. J. Hepatol. 2007, 46, 549–552. [Google Scholar] [CrossRef]
- McDonald, C.J.; Ostini, L.; Wallace, D.F.; Lyons, A.; Crawford, D.H.; Subramaniam, V.N. Next-generation sequencing: Application of a novel platform to analyze atypical iron disorders. J. Hepatol. 2015, 63, 1288–1293. [Google Scholar] [CrossRef]
- Galimberti, S.; Trombini, P.; Bernasconi, D.P.; Redaelli, I.; Pelucchi, S.; Bovo, G.; di Gennaro, F.; Zucchini, N.; Paruccini, N.; Piperno, A. Simultaneous liver iron and fat measures by magnetic resonance imaging in patients with hyperferritinemia. Scand. J. Gastroenterol. 2015, 50, 429–438. [Google Scholar] [CrossRef]
- Piperno, A.; Sampietro, M.; Pietrangelo, A.; Arosio, C.; Lupica, L.; Montosi, G.; Vergani, A.; Fraquelli, M.; Girelli, D.; Pasquero, P.; et al. Heterogeneity of hemochromatosis in Italy. Gastroenterology 1998, 114, 996–1002. [Google Scholar] [CrossRef]
- Hofmann, W.K.; Tong, X.J.; Ajioka, R.S.; Kushner, J.P.; Koeffler, H.P. Mutation analysis of transferrin-receptor 2 in patients with atypical hemochromatosis. Blood 2002, 100, 1099–1100. [Google Scholar] [CrossRef][Green Version]
- Deugnier, Y.M.; Loréal, O.; Turlin, B.; Guyader, D.; Jouanolle, H.; Moirand, R.; Jacquelinet, C.; Brissot, P. Liver pathology in genetic hemochromatosis: A review of 135 homozygous cases and their bioclinical correlations. Gastroenterology 1992, 102, 2050–2059. [Google Scholar] [CrossRef]
- Ishak, K.; Baptista, A.; Bianchi, L.; Callea, F.; de Groote, J.; Gudat, F.; Denk, H.; Desmet, V.; Korb, G.; MacSween, R.N. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995, 22, 696–699. [Google Scholar] [CrossRef]
- Ravasi, G.; Pelucchi, S.; Mariani, R.; Silvestri, L.; Camaschella, C.; Piperno, A. A severe hemojuvelin mutation leading to late onset of HFE2-hemochromatosis. Dig. Liver Dis. 2018, 50, 859–862. [Google Scholar] [CrossRef]
- Piubelli, C.; Castagna, A.; Marchi, G.; Rizzi, M.; Busti, F.; Badar, S.; Marchetti, M.; de Gobbi, M.; Roetto, A.; Xumerle, L.; et al. Identification of new BMP6 pro-peptide mutations in patients with iron overload. Am. J. Hematol. 2017, 92, 562–568. [Google Scholar] [CrossRef]
- Li, S.; Xue, J.; Chen, B.; Wang, Q.; Shi, M.; Xie, X.; Zhang, L. Two middle-age-onset hemochromatosis patients with heterozygous mutations in the hemojuvelin gene in a Chinese family. Int. J. Hematol. 2014, 99, 487–492. [Google Scholar] [CrossRef]
- Ravasi, G.; Rausa, M.; Pelucchi, S.; Arosio, C.; Greni, F.; Mariani, R.; Pelloni, I.; Silvestri, L.; Pineda, P.; Camaschella, C.; et al. Transferrin receptor 2 mutations in patients with juvenile hemochromatosis phenotype. Am. J. Hematol. 2015, 90, E226–E227. [Google Scholar] [CrossRef]
- Badar, S.; Busti, F.; Ferrarini, A.; Xumerle, L.; Bozzini, P.; Capelli, P.; Pozzi-Mucelli, R.; Campostrini, N.; de Matteis, G.; Vargas, S.M.; et al. Identification of novel mutations in hemochromatosis genes by targeted next generation sequencing in Italian patients with unexplained iron overload. Am. J. Hematol. 2016, 91, 420–425. [Google Scholar] [CrossRef]
- Faria, R.; Silva, B.; Silva, C.; Loureiro, P.; Queiroz, A.; Fraga, S.; Esteves, J.; Mendes, D.; Fleming, R.; Vieira, L.; et al. Next-generation sequencing of hereditary hemochromatosis-related genes: Novel likely pathogenic variants found in the Portuguese population. Blood Cells Mol. Dis. 2016, 61, 10–15. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Sadikovic, B.; Waye, J.S.; Levstik, A.; Lanktree, B.B.; Yudin, J.; Crowther, M.A.; Pare, G.; Adams, P.C. Clinical evaluation of a hemochromatosis next-generation sequencing gene panel. Eur. J. Haematol. 2017, 98, 228–234. [Google Scholar] [CrossRef]
- Vlasveld, L.T.; Janssen, R.; Bardou-Jacquet, E.; Venselaar, H.; Hamdi-Roze, H.; Drakesmith, H.; Swinkels, D.W. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals 2019, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Mayr, R.; Janecke, A.R.; Schranz, M.; Griffiths, W.J.; Vogel, W.; Pietrangelo, A.; Zoller, H. Ferroportin disease: A systematic meta-analysis of clinical and molecular findings. J. Hepatol. 2010, 53, 941–949. [Google Scholar] [CrossRef]
- Liu, W.; Shimomura, S.; Imanishi, H.; Iwamoto, Y.; Ikeda, N.; Saito, M.; Ohno, M.; Hara, N.; Yamamoto, T.; Nakamura, H.; et al. Hemochromatosis with mutation of the ferroportin 1 (IREG1) gene. Intern. Med. 2005, 44, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Lymboussaki, A.; Pignatti, E.; Montosi, G.; Garuti, C.; Haile, D.J.; Pietrangelo, A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J. Hepatol. 2003, 39, 710–715. [Google Scholar] [CrossRef]
- Cazzola, M.; Skoda, R.C. Translational pathophysiology: A novel molecular mechanism of human disease. Blood 2000, 95, 3280–3288. [Google Scholar] [CrossRef]
- Beutler, E. The significance of the 187G (H63D) mutation in hemochromatosis. Am. J. Hum. Genet. 1997, 61, 762–764. [Google Scholar] [CrossRef]
- Ritchie, D.; van Steen, K. The search for gene-gene interactions in genome-wide association studies: Challenges in abundance of methods, practical considerations, and biological interpretation. Ann. Transl. Med. 2018, 6, 157. [Google Scholar] [CrossRef]


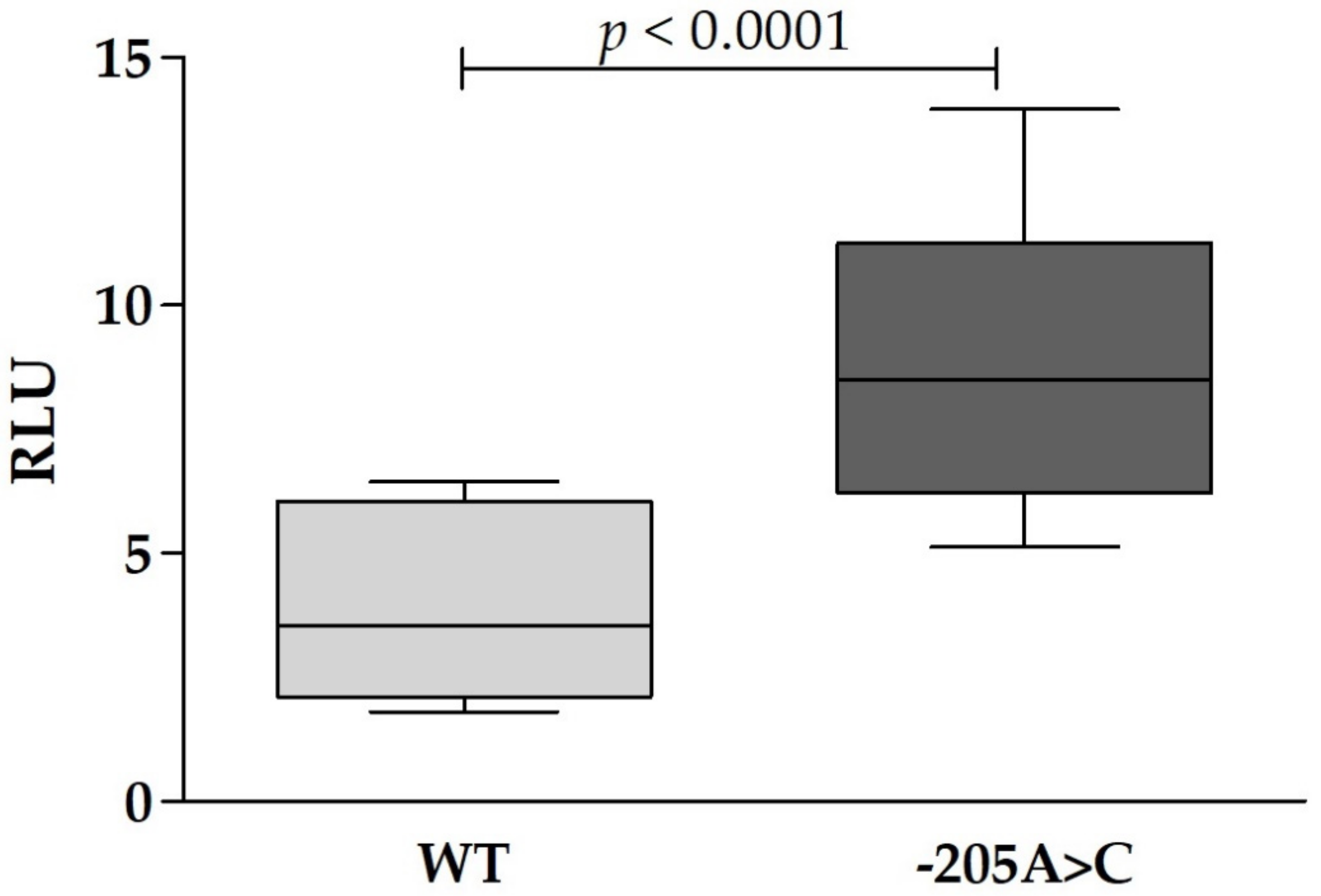{kind=link}
| Gene | Amino Acid Change | dbSNP | Allelic Frequency (All) | Allelic Frequency (TSAT < 45%) | Allelic Frequency (TSAT ≥ 45%) | Allelic Frequency (gnomAD) | p (All vs. gnomAD) |
|---|---|---|---|---|---|---|---|
| ACO1 | V498M | rs375879049 | 0.014 | 0.000 | 0.045 | 0.000 | ns |
| BMP6 | L96P | rs200573175 | 0.028 | 0.040 | 0.000 | 0.006 | 0.01 |
| CDAN1 | R891C | rs8023524 | 0.111 | 0.120 | 0.091 | 0.186 | ns |
| CP | P477L | rs35331711 | 0.014 | 0.020 | 0.000 | 0.004 | ns |
| CP | P876S | rs767188986 | 0.014 | 0.000 | 0.000 | 0.000 | ns |
| CP | T551I | rs61733458 | 0.042 | 0.060 | 0.000 | 0.031 | ns |
| ERFE | G196R | rs1247409936 | 0.014 | 0.020 | 0.000 | 0.000 | ns |
| HAMP | K83delinsLIYSCCPR | rs1159254691 | 0.014 | 0.020 | 0.000 | 0.000 | ns |
| HAMP | R59P | novel | 0.014 | 0.000 | 0.045 | // | // |
| HJV | C89R | novel | 0.014 | 0.000 | 0.045 | // | // |
| HJV | Deletion | novel | 0.014 | 0.000 | 0.045 | // | // |
| HIF1A | P582S | rs11549465 | 0.097 | 0.080 | 0.136 | 0.105 | ns |
| PCSK7 | T182M | rs150784623 | 0.014 | 0.000 | 0.045 | 0.000 | ns |
| SERPINA1 | E366K | rs28929474 | 0.014 | 0.000 | 0.045 | 0.000 | ns |
| SERPINA1 | D280V | rs121912714 | 0.014 | 0.020 | 0.000 | 0.001 | ns |
| SLC40A1 | V160A | novel | 0.014 | 0.020 | 0.000 | // | // |
| SLC40A1 | R179T | rs765023388 | 0.014 | 0.020 | 0.000 | 0.00001 | <0.001 |
| SLC40A1 | A350D | novel | 0.014 | 0.020 | 0.000 | // | // |
| SLC40A1 | G494D | novel | 0.014 | 0.020 | 0.000 | // | // |
| SLC40A1 | c.-205A>C | novel | 0.014 | 0.020 | 0.000 | // | // |
| SLC40A1 | V531A | novel | 0.014 | 0.020 | 0.000 | // | // |
| SMAD4 | N188D | novel | 0.014 | 0.020 | 0.000 | // | // |
| SMAD7 | G137A | rs757109163 | 0.014 | 0.000 | 0.045 | 0.000 | ns |
| SMAD7 | G39R | rs144204026 | 0.028 | 0.040 | 0.000 | 0.011 | ns |
| TF | G544E | rs121918677 | 0.014 | 0.020 | 0.045 | 0.000 | ns |
| TF | G150S | rs1799899 | 0.083 | 0.080 | 0.091 | 0.060 | ns |
| TFR2 | L224R | novel | 0.014 | 0.000 | 0.045 | // | // |
| TFR2 | D514Mfs12 | novel | 0.014 | 0.000 | 0.045 | // | // |
| TFR2 | R455N | rs41303501 | 0.014 | 0.000 | 0.045 | 0.00342 | ns |
| TFR2 | Y547F | rs573769443 | 0.014 | 0.000 | 0.045 | 0.00009 | <0.001 |
| Transferrin Saturation < 45% | Transferrin Saturation ≥ 45% | p | |
|---|---|---|---|
| N° | 25 | 11 | |
| M/F | 21/4 | 9/2 | ns |
| Age (years) | 54 (34–58) | 45 (33.5–54) | ns |
| Hemoglobin (g/dL) | 15.2 ^ (14.3–15.7) | 14.5 ^^ (12.9–16) | ns |
| S-Iron (μg/dL) | 97 (92–110) | 199 (162.5–215.5) | <0.0001 |
| Transferrin Saturation (%) | 31.7 (27.1–36) | 62.8 (56.4–75) | <0.0001 |
| S-Ferritin (μg/L) | 945 (745–1105) | 1339 (844.5–2423.5) | 0.057 |
| LIC (μmol/g) | 51.2 (39.0–69.2) | 100.0 (52.9 –283.2) | <0.002 |
| Body Mass Index (kg/m2) | 22.6 (21.9–25.3) | 23.5 (20.9–24.3) | ns |
| Glycemia (mg/dL) | 87 (83.5–101) | 90 (80.5–103) | ns |
| Triglycerides (mg/dL) | 104 (77.5–122.8) | 85 (63.3–106.8) | ns |
| HDL (mg/dL) | 56 (49–64) | 57 (46.5–60.5) | ns |
| ALT (U/L) | 22 (16–27.5) | 28 (26–41.3) | 0.015 |
| Patient | Sex | Age (years) | Hb (g/dL) | S-Iron (μg/dL) | Transferrin Saturation (%) | S-Ferritin (μg/L) | RMN T2* (msec) |
|---|---|---|---|---|---|---|---|
| 1 | M | 25 | 15.2 | 116 | 31.7 | 1000 | 9 |
| 2 | M | 41 | 14.5 | 132 | 39.4 | 819 | 9.7 |
| 3 | M | 62 | 11.9 | 74 | 23.7 | 2600 | 14.29 |
| 4 | F | 21 | 12.1 | 108 | 38 | 1010 | -† |
| 5 | M | 59 | 13.6 | 143 | 36 | 1025 | 6.96 |
| 6 | M | 54 | 15.7 | 129 | 37 | 1908 | 6.5 ‡ |
| 7 | M | 12 | 15.8 | 297 | 90 | 151 | 19.5 |
| 8 | M | 46 | 16.1 | 183 | 83 | 3909 | 1.4 |
| 9 | M | 38 | 12.3 | 208 | 92 | 3510 | 1.02 |
| 10 | F | 19 | 14.4 | 272 | 87 | 1339 | 2.2 |
| Patient | Gene | cDNA— Protein Mutation | Protein Lenght | SIFT | Polyphen | Mutation Taster | Human Splicing Finder |
|---|---|---|---|---|---|---|---|
| 1 | SLC40A1 (exon 5) | c.479T>C p.Val160Ala | not affected | affect protein function, score = 0.00 | probably damaging score = 0.992 | disease causing | - |
| 2 | SLC40A1 (exon 6) | c.536G>C p. Arg179Thr s765023388 | not affected | tolerated score = 0.57 | probably damaging score = 0.996 | disease causing; might affect splice site | alteration of an exonic ESE site; potential splicing alteration |
| 3 | SLC40A1 (exon 7) | c.1049 C>A p.Ala350Asp | not affected | affect protein function, score = 0.00 | probably damaging, score = 0.999 | disease causing | - |
| 4 | SLC40A1 (exon 8) | c.1481 G>A p.Gly494Asp | not affected | affect protein function, score = 0.00 | probably damaging, score = 1.000 | disease causing; might affect of splice site | no splicing site alteration |
| 5 | SLC40A1 (5′ UTR) | c.-205A>C | not affected | - | - | disease causing | - |
| 6 | SLC40A1 (exon 8) | c.1592T>C p. Val531Ala | not affected | affect protein function, score = 0.00 | probably damaging, score = 1.000 | disease causing; splice site change | alteration of an exonic ESE site; potential splicing alteration |
| 7 | TFR2 (exon 5 and 13) | c.671T>G p.Leu224Arg | not affected | tolerated score = 0.53 | possibly damaging, score = 0.8 | protein features (might be) affected; splice site changes | alteration of an exonic ESE site; potential splicing alteration |
| c.1540delG p.Asp514Metfs12 | affected | affect protein function, score = 0.05 | probably damaging, score = 0.987 | disease causing; frameshift-splice site change | alteration of an exonic ESE site; potential splicing alteration | ||
| 8 | TFR2 (exon 10 and 14) | c.1364G>A p.Arg455Gln [17] rs41303501 | not affected | tolerated score = 0.29 | possibly damaging, score = 0.837 | protein features (might be) affected; mught affect splice site changes | activation of an exonic cryptic acceptor site and of one or more cryptic branch point(s); potential splicing alteration |
| c.1640A>T p.Tyr547Phe rs573769443 | not affected | affect protein function, score = 0.00 | benign score = 0.238 | protein features (might be) affected; might affect splice site changes | alteration of an exonic ESE site; potential splicing alteration | ||
| 9 | HJV (exon 3) | c.265 T>C p.Cys89Arg | not affected | affect protein function, score = 0.00 | probably damaging score = 0.999 | disease causing | no splicing alteration |
| deletion: g.(?_144124721)_(144126704_?)del | - | - | - | - | - | ||
| 10 | HAMP (exon 3) | c.176 G>C p.Arg59Pro | not affected | tolerated score = 0.88 | probably damaging score = 1.0 | protein features (might be) affected; might affect splice site changes | activation of a cryptic acceptor site; potential splicing alteration |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravasi, G.; Pelucchi, S.; Bertola, F.; Capelletti, M.M.; Mariani, R.; Piperno, A. Identification of Novel Mutations by Targeted NGS Panel in Patients with Hyperferritinemia. Genes 2021, 12, 1778. https://doi.org/10.3390/genes12111778
Ravasi G, Pelucchi S, Bertola F, Capelletti MM, Mariani R, Piperno A. Identification of Novel Mutations by Targeted NGS Panel in Patients with Hyperferritinemia. Genes. 2021; 12(11):1778. https://doi.org/10.3390/genes12111778
Chicago/Turabian StyleRavasi, Giulia, Sara Pelucchi, Francesca Bertola, Martina Maria Capelletti, Raffaella Mariani, and Alberto Piperno. 2021. "Identification of Novel Mutations by Targeted NGS Panel in Patients with Hyperferritinemia" Genes 12, no. 11: 1778. https://doi.org/10.3390/genes12111778
APA StyleRavasi, G., Pelucchi, S., Bertola, F., Capelletti, M. M., Mariani, R., & Piperno, A. (2021). Identification of Novel Mutations by Targeted NGS Panel in Patients with Hyperferritinemia. Genes, 12(11), 1778. https://doi.org/10.3390/genes12111778