Bridging the Gap between Vertebrate Cytogenetics and Genomics with Single-Chromosome Sequencing (ChromSeq)

 , , ,
, , , 
{kind=link}
Abstract
1. Introduction
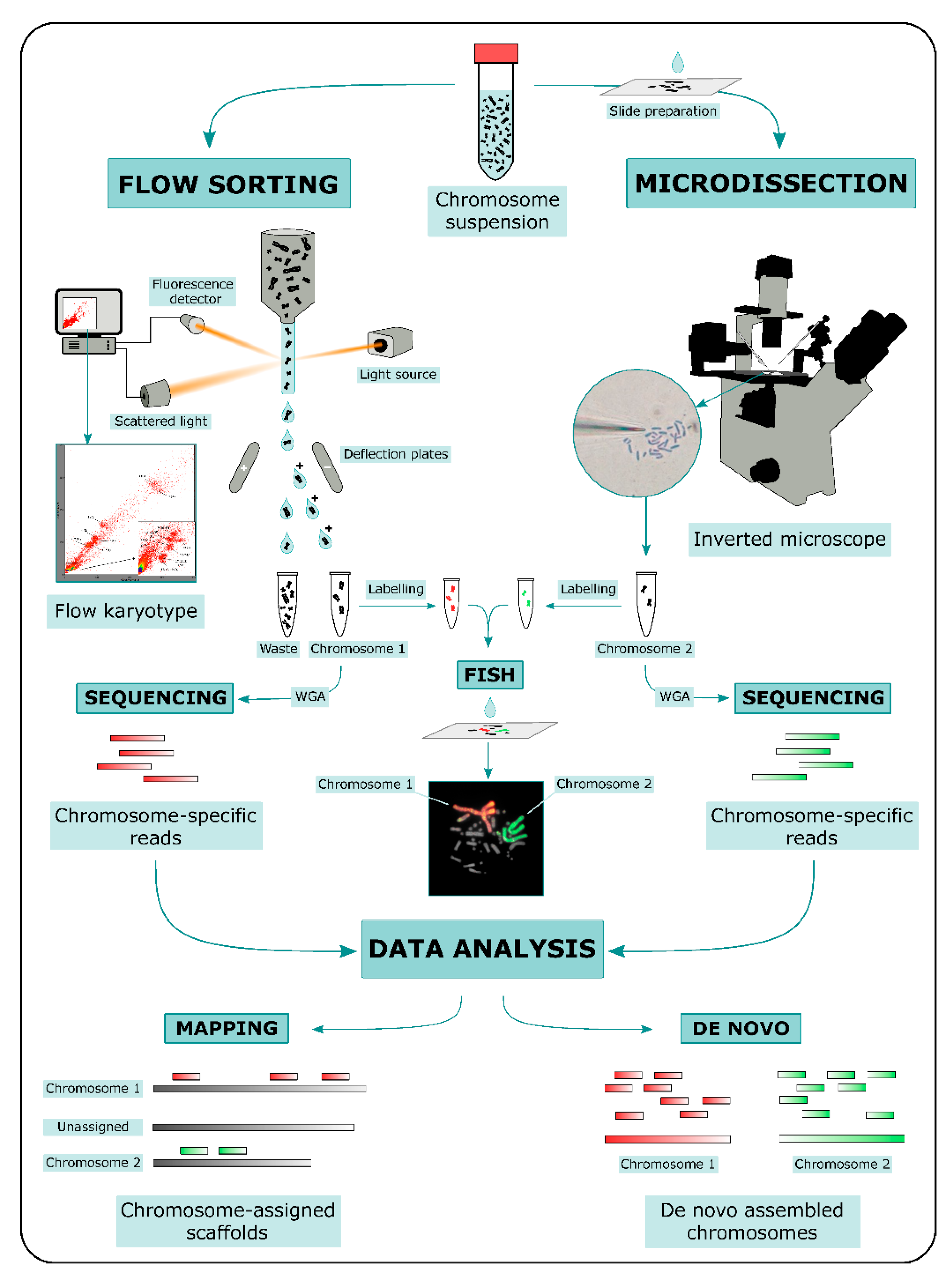
2. ChromSeq Workflow
3. Application in Vertebrate Genome Projects
4. Application in Vertebrate Karyotype Evolution Studies
5. ChromSeq to Refine Anolis carolinensis Genome Assembly
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Editorial. A reference standard for genome biology. Nat. Biotechnol. 2018, 36, 1121. Available online: https://www.nature.com/articles/nbt.4318 (accessed on 6 December 2018). [CrossRef] [PubMed]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, Q.; Wang, Y.; Luo, L.; Yang, J.; Yang, L.; Liu, M.; Li, Y.; Qian, T.; Zheng, Y.; et al. Gekko japonicus genome reveals evolution of adhesive toe pads and tail regeneration. Nat. Commun. 2015, 6, 10033. [Google Scholar] [CrossRef] [PubMed]
- Marlétaz, F.; Firbas, P.N.; Maeso, I.; Tena, J.J.; Bogdanovic, O.; Perry, M.; Wyatt, C.D.R.; de la Calle-Mustienes, E.; Bertrand, S.; Burguera, D.; et al. Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature 2018, 564, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Sacerdot, C.; Louis, A.; Bon, C.; Berthelot, C.; Crollius, H.R. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 2018, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Timoshevskaya, N.; Ye, C.; Holt, C.; Keinath, M.C.; Parker, H.J.; Cook, M.E.; Hess, J.E.; Narum, S.R.; Lamanna, F.; et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 2018, 50, 270–277. [Google Scholar] [CrossRef]
- Worley, K.C. A golden goat genome. Nat. Genet. 2017, 49, 485–486. [Google Scholar] [CrossRef]
- Garner, B.A.; Hand, B.K.; Amish, S.J.; Bernatchez, L.; Foster, J.T.; Miller, K.M.; Morin, P.A.; Narum, S.R.; O’Brien, S.J.; Roffler, G.; et al. Genomics in conservation: Case studies and bridging the gap between data and application. Trends Ecol. Evol. 2016, 31, 81–83. [Google Scholar] [CrossRef]
- Supple, M.A.; Shapiro, B. Conservation of biodiversity in the genomics era. Genome Biol. 2018, 19, 131. [Google Scholar] [CrossRef]
- Giani, A.M.; Gallo, G.R.; Gianfranceschi, L.; Formenti, G. Long walk to genomics: History and current approaches to genome sequencing and assembly. Comput. Struct. Biotechnol. J. 2020, 18, 9–19. [Google Scholar] [CrossRef]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION sequencing and genome assembly. GPB 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. GPB 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.T.; Hastie, A.; Lin, C.; Ehrlich, D.; Das, S.K.; Austin, M.D.; Deshpande, P.; Cao, H.; Nagarajan, N.; Xiao, M.; et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012, 30, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Gedman, G.L.; et al. Towards complete and error-free genome assemblies of all vertebrate species. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lewin, H.A.; Graves, J.A.M.; Ryder, O.A.; Graphodatsky, A.S.; O’Brien, S.J. Precision nomenclature for the new genomics. GigaScience 2019, 8, giz086. [Google Scholar] [CrossRef]
- Claussen, U. Chromosomics. Cytogenet. Genome Res. 2005, 111, 101–106. [Google Scholar] [CrossRef]
- Deakin, J.E.; Ezaz, T. Understanding the evolution of reptile chromosomes through applications of combined cytogenetics and genomics approaches. Cytogenet. Genome Res. 2019, 157, 7–20. [Google Scholar] [CrossRef]
- Deakin, J.E.; Potter, S. Marsupial chromosomics: Bridging the gap between genomes and chromosomes. Reprod. Fertil. Dev. 2019, 31, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.E.; Potter, S.; O’Neill, R.; Ruiz-Herrera, A.; Cioffi, M.B.; Eldridge, M.D.; Fukui, K.; Graves, J.A.M.; Griffin, D.; Grutzner, F.; et al. Chromosomics: Bridging the gap between genomes and chromosomes. Genes 2019, 10, 627. [Google Scholar] [CrossRef] [PubMed]
- Graphodatsky, A.S. Comparative chromosomics. Mol. Biol. 2007, 41, 361–375. [Google Scholar] [CrossRef]
- Damas, J.; O’connor, R.; Farré, M.; Lenis, V.P.E.; Martell, H.J.; Mandawala, A.; Fowler, K.; Joseph, S.; Swain, M.T.; Griffin, D.K.; et al. Upgrading short-read animal genome assemblies to chromosome level using comparative genomics and a universal probe set. Genome Res. 2017, 27, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Kidd, C.; Tomasino, E.; Davis, J.T.; Wishon, C.; Stern, J.E.; Carleton, K.L.; Howe, A.E.; Kocher, T.D. A BAC-based physical map of the Nile tilapia genome. BMC Genom. 2005, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Lee, M.-K.; Yan, B.; Ding, K.; Cox, B.; Romanov, M.N.; Price, J.A.; Dodgson, J.B.; Zhang, H.-B. A BAC-based physical map of the chicken genome. Genome Res. 2003, 13, 2754–2758. [Google Scholar] [CrossRef]
- Snelling, W.M.; Chiu, R.; Schein, J.E.; Hobbs, M.; Abbey, C.A.; Adelson, D.L.; Aerts, J.; Bennett, G.L.; Bosdet, I.E.; Boussaha, M.; et al. A physical map of the bovine genome. Genome Biol. 2007, 8, R165. [Google Scholar] [CrossRef]
- Wells, D.E.; Gutierrez, L.; Xu, Z.; Krylov, V.; Macha, J.; Blankenburg, K.P.; Hitchens, M.; Bellot, L.J.; Spivey, M.; Stemple, D.L.; et al. A genetic map of Xenopus tropicalis. Dev. Biol. 2011, 354, 1–8. [Google Scholar] [CrossRef][Green Version]
- Chen, W.; Kalscheuer, V.; Tzschach, A.; Menzel, C.; Ullmann, R.; Schulz, M.H.; Erdogan, F.; Li, N.; Kijas, Z.; Arkesteijn, G.; et al. Mapping translocation breakpoints by next-generation sequencing. Genome Res. 2008, 18, 1143–1149. [Google Scholar] [CrossRef]
- Cápal, P.; Blavet, N.; Vrána, J.; Kubaláková, M.; Doležel, J. Multiple displacement amplification of the DNA from single flow–sorted plant chromosome. Plant J. 2015, 84, 838–844. [Google Scholar] [CrossRef]
- Doležel, J.; Vrána, J.; Cápal, P.; Kubaláková, M.; Burešová, V.; Šimková, H. Advances in plant chromosome genomics. Biotechnol. Adv. 2014, 32, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Doležel, J.; Vrána, J.; Šafář, J.; Bartoš, J.; Kubaláková, M.; Šimková, H. Chromosomes in the flow to simplify genome analysis. Funct. Integr. Genom. 2012, 12, 397–416. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.; Martis, M.; Dorado, G.; Pfeifer, M.; Gálvez, S.; Schaaf, S.; Jouve, N.; Šimková, H.; Valárik, M.; Doležel, J.; et al. Next-generation sequencing and syntenic integration of flow-sorted arms of wheat chromosome 4A exposes the chromosome structure and gene content. Plant J. 2012, 69, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.F.X.; Martis, M.; Hedley, P.E.; Šimková, H.; Liu, H.; Morris, J.A.; Steuernagel, B.; Taudien, S.; Roessner, S.; Gundlach, H.; et al. Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 2011, 23, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, J.; Steuernagel, B.; Ghosh, S.; Herren, G.; Hurni, S.; Adamski, N.; Vrána, J.; Kubaláková, M.; Krattinger, S.G.; Wicker, T.; et al. Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol. 2016, 17, 221. [Google Scholar] [CrossRef] [PubMed]
- Šimková, H.; Svensson, J.T.; Condamine, P.; Hřibová, E.; Suchánková, P.; Bhat, P.R.; Bartoš, J.; Šafář, J.; Close, T.J.; Doležel, J. Coupling amplified DNA from flow-sorted chromosomes to high-density SNP mapping in barley. BMC Genom. 2008, 9, 1–9. [Google Scholar] [CrossRef]
- Zwyrtková, J.; Šimková, H.; Doležel, J. Chromosome genomics uncovers plant genome organization and function. Biotechnol. Adv. 2020, 46, 107659. [Google Scholar] [CrossRef]
- Yang, F.; Trifonov, V.; Ng, B.L.; Kosyakova, N.; Carter, N.P. Generation of paint probes from flow-sorted and microdissected chromosomes. In Fluorescence In Situ Hybridization (FISH); Liehr, T., Ed.; Springer: Berlin, Germany, 2017; pp. 63–79. [Google Scholar]
- Benítez, J.J.; Topolancik, J.; Tian, H.C.; Wallin, C.B.; Latulippe, D.R.; Szeto, K.; Murphy, P.J.; Cipriany, B.R.; Levy, S.L.; Soloway, P.D.; et al. Microfluidic extraction, stretching and analysis of human chromosomal DNA from single cells. Lab Chip 2012, 12, 4848–4854. [Google Scholar] [CrossRef]
- Fan, H.C.; Wang, J.; Potanina, A.; Quake, S.R. Whole-genome molecular haplotyping of single cells. Nat. Biotechnol. 2011, 29, 51–57. [Google Scholar] [CrossRef]
- Takahashi, T.; Okeyo, K.O.; Ueda, J.; Yamagata, K.; Washizu, M.; Oana, H. A microfluidic device for isolating intact chromosomes from single mammalian cells and probing their folding stability by controlling solution conditions. Sci. Rep. 2018, 8, 13684. [Google Scholar] [CrossRef]
- Ibrahim, S.F.; van den Engh, G. High-speed chromosome sorting. Chrom. Res. 2004, 12, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Stanyon, R.; Stone, G. Phylogenomic analysis by chromosome sorting and painting. In Phylogenomics. Methods in Molecular Biology; William, J.M., Ed.; Springer: Berlin, Germany, 2008; Volume 422, pp. 13–29. [Google Scholar]
- Zhou, R.-N.; Hu, Z.-M. The development of chromosome microdissection and microcloning technique and its applications in genomic research. Curr. Genom. 2007, 8, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Weise, A.; Timmermann, B.; Grabherr, M.; Werber, M.; Heyn, P.; Kosyakova, N.; Liehr, T.; Neitzel, H.; Konrat, K.; Bommer, C.; et al. High-throughput sequencing of microdissected chromosomal regions. Eur. J. Hum. Genet. 2010, 18, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Zlotina, A.; Maslova, A.; Pavlova, O.; Kosyakova, N.; Al-Rikabi, A.; Liehr, T.; Krasikova, A. New Insights Into Chromomere Organization Provided by Lampbrush Chromosome Microdissection and High-Throughput Sequencing. Front. Genet. 2020, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Telenius, H.; Ponder, B.A.J.; Tunnacliffe, A.; Pelmear, A.H.; Carter, N.P.; Ferguson-Smith, M.A.; Behmel, A.; Nordenskjöld, M.; Pfragner, R. Cytogenetic analysis by chromosome painting using DOP-PCR amplified flow-sorted chromosomes. Genes Chromosom. Cancer 1992, 4, 257–263. [Google Scholar] [CrossRef]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef]
- Zhang, K.; Martiny, A.C.; Reppas, N.B.; Barry, K.W.; Malek, J.; Chisholm, S.W.; Church, G.M. Sequencing genomes from single cells by polymerase cloning. Nat. Biotechnol. 2006, 24, 680–686. [Google Scholar] [CrossRef]
- Ried, T.; Schröck, E.; Ning, Y.; Wienberg, J. Chromosome painting: A useful art. Hum. Mol. Genet. 1998, 7, 1619–1626. [Google Scholar] [CrossRef]
- Teer, J.K.; Johnston, J.J.; Anzick, S.L.; Pineda, M.; Stone, G.; Meltzer, P.S.; Mullikin, J.C.; Biesecker, L.G. Massively-parallel sequencing of genes on a single chromosome: A comparison of solution hybrid selection and flow sorting. BMC Genom. 2013, 14, 1–10. [Google Scholar] [CrossRef]
- Andreyushkova, D.A.; Makunin, A.I.; Beklemisheva, V.R.; Romanenko, S.A.; Druzhkova, A.S.; Biltueva, L.B.; Serdyukova, N.A.; Graphodatsky, A.S.; Trifonov, V.A. Next generation sequencing of chromosome-specific libraries sheds light on genome evolution in paleotetraploid sterlet (Acipenser ruthenus). Genes 2017, 8, 318. [Google Scholar] [CrossRef]
- Kichigin, I.G.; Giovannotti, M.; Makunin, A.I.; Ng, B.L.; Kabilov, M.R.; Tupikin, A.E.; Barucchi, V.C.; Splendiani, A.; Ruggeri, P.; Rens, W.; et al. Evolutionary dynamics of Anolis sex chromosomes revealed by sequencing of flow sorting-derived microchromosome-specific DNA. Mol. Genet. Genom. 2016, 291, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Lind, A.L.; Lai, Y.Y.Y.; Mostovoy, Y.; Holloway, A.K.; Iannucci, A.; Mak, A.C.Y.; Fondi, M.; Orlandini, V.; Eckalbar, W.L.; Milan, M.; et al. Genome of the Komodo dragon reveals adaptations in the cardiovascular and chemosensory systems of monitor lizards. Nat. Ecol. Evol. 2019, 3, 1241–1252. [Google Scholar] [CrossRef]
- Lisachov, A.P.; Makunin, A.I.; Giovannotti, M.; Pereira, J.C.; Druzhkova, A.S.; Barucchi, V.C.; Ferguson-Smith, M.A.; Trifonov, V.A. Genetic content of the neo-sex chromosomes in Ctenonotus and Norops (Squamata, Dactyloidae) and degeneration of the Y chromosome as revealed by high-throughput sequencing of individual chromosomes. Cytogenet. Genome Res. 2019, 157, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Makunin, A.I.; Kichigin, I.G.; Larkin, D.M.; O’Brien, P.C.; Ferguson-Smith, M.A.; Yang, F.; Proskuryakova, A.A.; Vorobieva, N.V.; Chernyaeva, E.N.; O’Brien, S.J.; et al. Contrasting origin of B chromosomes in two cervids (Siberian roe deer and grey brocket deer) unravelled by chromosome-specific DNA sequencing. BMC Genom. 2016, 17, 618. [Google Scholar] [CrossRef] [PubMed]
- Tomaszkiewicz, M.; Rangavittal, S.; Cechova, M.; Campos Sanchez, R.; Fescemyer, H.W.; Harris, R.; Ye, D.; O’Brien, P.C.M.; Chikhi, R.; Ryder, O.A.; et al. A time-and cost-effective strategy to sequence mammalian Y Chromosomes: An application to the de novo assembly of gorilla Y. Genome Res. 2016, 26, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ullmann, R.; Langnick, C.; Menzel, C.; Wotschofsky, Z.; Hu, H.; Döring, A.; Hu, Y.; Kang, H.; Tzschach, A.; et al. Breakpoint analysis of balanced chromosome rearrangements by next-generation paired-end sequencing. Eur. J. Hum. Genet. 2010, 18, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Komissarov, A.; Vij, S.; Yurchenko, A.; Trifonov, V.; Thevasagayam, N.; Saju, J.; Sridatta, P.S.R.; Purushothaman, K.; Graphodatsky, A.; Orbán, L.; et al. B Chromosomes of the Asian seabass (Lates calcarifer) contribute to genome variations at the level of individuals and populations. Genes 2018, 9, 464. [Google Scholar] [CrossRef]
- Brinkrolf, K.; Rupp, O.; Laux, H.; Kollin, F.; Ernst, W.; Linke, B.; Kofler, R.; Romand, S.; Hesse, F.; Budach, W.E.; et al. Chinese hamster genome sequenced from sorted chromosomes. Nat. Biotechnol. 2013, 31, 694–695. [Google Scholar] [CrossRef]
- Luo, W.; Xia, Y.; Yue, B.; Zeng, X. Assigning the sex-specific markers via genotyping-by-sequencing onto the Y chromosome for a torrent frog Amolops mantzorum. Genes 2020, 11, 727. [Google Scholar] [CrossRef]
- Kuderna, L.F.K.; Solís-Moruno, M.; Batlle-Masó, L.; Julià, E.; Lizano, E.; Anglada, R.; Ramírez, E.; Bote, A.; Tormo, M.; Marquès-Bonet, T.; et al. Flow sorting enrichment and nanopore sequencing of chromosome 1 from a Chinese individual. Front. Genet. 2020, 10, 1315. [Google Scholar] [CrossRef]
- Murchison, E.P.; Schulz-Trieglaff, O.B.; Ning, Z.; Alexandrov, L.B.; Bauer, M.J.; Fu, B.; Hims, M.; Ding, Z.; Ivakhno, S.; Stewart, C.; et al. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell 2012, 148, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Iannucci, A.; Altmanová, M.; Ciofi, C.; Ferguson-Smith, M.; Pereira, J.C.; Rehák, I.; Stanyon, R.; Velenský, P.; Rovatsos, M.; Kratochvíl, L.; et al. Isolating chromosomes of the Komodo dragon: New tools for comparative mapping and sequence assembly. Cytogenet. Genome Res. 2019, 157, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Alföldi, J.; Di Palma, F.; Grabherr, M.; Williams, C.; Kong, L.; Mauceli, E.; Russell, P.; Lowe, C.B.; Glor, R.E.; Jaffe, J.D.; et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011, 477, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Stöck, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 2020, 4, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Seifertova, E.; Zimmerman, L.B.; Gilchrist, M.J.; Macha, J.; Kubickova, S.; Cernohorska, H.; Zarsky, V.; Owens, N.D.; Sesay, A.K.; Tlapakova, T.; et al. Efficient high-throughput sequencing of a laser microdissected chromosome arm. BMC Genom. 2013, 14, 357. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Traut, W.; Vogel, H.; Glöckner, G.; Hartmann, E.; Heckel, D.G. High-throughput sequencing of a single chromosome: A moth W chromosome. Chromosome Res. 2013, 21, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Iwasaki, Y.; Nishiki, I.; Nomura, K.; Fujiwara, A. Identification of genetic linkage group 1-linked sequences in Japanese eel (Anguilla japonica) by single chromosome sorting and sequencing. PLoS ONE 2018, 13, e0197040. [Google Scholar] [CrossRef]
- Sudbery, I.; Stalker, J.; Simpson, J.T.; Keane, T.; Rust, A.G.; Hurles, M.E.; Walter, K.; Lynch, D.; Teboul, L.; Brown, S.D.; et al. Deep short-read sequencing of chromosome 17 from the mouse strains A/J and CAST/Ei identifies significant germline variation and candidate genes that regulate liver triglyceride levels. Genome Biol. 2009, 10, R112. [Google Scholar] [CrossRef]
- Vij, S.; Kuhl, H.; Kuznetsova, I.S.; Komissarov, A.; Yurchenko, A.A.; Van Heusden, P.; Singh, S.; Thevasagayam, N.M.; Prakki, S.R.S.; Purushothaman, K.; et al. Chromosomal-level assembly of the Asian seabass genome using long sequence reads and multi-layered scaffolding. PLoS Genet. 2016, 12, e1005954. [Google Scholar]
- International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345. [Google Scholar] [CrossRef]
- Keinath, M.C.; Timoshevskiy, V.A.; Timoshevskaya, N.Y.; Tsonis, P.A.; Voss, S.R.; Smith, J.J. Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing. Sci. Rep. 2015, 5, 16413. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Timoshevskaya, N.; Timoshevskiy, V.A.; Keinath, M.C.; Hardy, D.; Voss, S.R. A chromosome-scale assembly of the axolotl genome. Genome Res. 2019, 29, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Chiatante, G.; Capozzi, O.; Svartman, M.; Perelman, P.; Centrone, L.; Romanenko, S.S.; Ishida, T.; Valeri, M.; Roelke-Parker, M.E.; Stanyon, R. Centromere repositioning explains fundamental number variability in the New World monkey genus Saimiri. Chromosoma 2017, 126, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.B.; Sánchez, A.; Marchal, J.A.; Kosyakova, N.; Liehr, T.; Trifonov, V.; Bertollo, L.A.C. Whole chromosome painting reveals independent origin of sex chromosomes in closely related forms of a fish species. Genetica 2011, 139, 1065. [Google Scholar] [CrossRef] [PubMed]
- Iannucci, A.; Altmanová, M.; Ciofi, C.; Ferguson-Smith, M.; Milan, M.; Pereira, J.C.; Pether, J.; Rehák, I.; Rovatsos, M.; Stanyon, R.; et al. Conserved sex chromosomes and karyotype evolution in monitor lizards (Varanidae). Heredity 2019, 123, 215–227. [Google Scholar] [CrossRef]
- Kretschmer, R.; de Oliveira Furo, I.; Gunski, R.J.; del Valle Garnero, A.; Pereira, J.C.; O’Brien, P.C.M.; Ferguson-Smith, M.A.; de Oliveira, E.H.C.; de Freitas, T.R.O. Comparative chromosome painting in Columbidae (Columbiformes) reinforces divergence in Passerea and Columbea. Chrom. Res. 2018, 26, 211–223. [Google Scholar] [CrossRef]
- Srikulnath, K.; Nishida, C.; Matsubara, K.; Uno, Y.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S.; Matsuda, Y. Karyotypic evolution in squamate reptiles: Comparative gene mapping revealed highly conserved linkage homology between the butterfly lizard (Leiolepis reevesii rubritaeniata, Agamidae, Lacertilia) and the Japanese four-striped rat snake (Elaphe quadrivirgata, Colubridae, Serpentes). Chromosome Res. 2009, 17, 975. [Google Scholar]
- Srikulnath, K.; Uno, Y.; Nishida, C.; Ota, H.; Matsuda, Y. Karyotype reorganization in the Hokou Gecko (Gekko hokouensis, Gekkonidae): The process of microchromosome disappearance in Gekkota. PLoS ONE 2015, 10, e0134829. [Google Scholar] [CrossRef]
- Targueta, C.P.; Krylov, V.; Nondilo, T.E.; Lima, J.; Lourenço, L.B. Sex chromosome evolution in frogs—Helpful insights from chromosome painting in the genus Engystomops. Heredity 2020, 1–14. [Google Scholar] [CrossRef]
- Lisachov, A.; Tishakova, K.; Romanenko, S.; Molodtseva, A.; Prokopov, D.; Pereira, J.; Ferguson-Smith, M.A.; Borodin, P.; Trifonov, V. Whole-chromosome fusions in the karyotype evolution of Sceloporus (Iguania, Reptilia) are more intense in sex chromosomes than autosomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kichigin, I.G.; Lisachov, A.P.; Giovannotti, M.; Makunin, A.I.; Kabilov, M.R.; O’Brien, P.C.M.; Ferguson-Smith, M.A.; Graphodatsky, A.S.; Trifonov, V.A. First report on B chromosome content in a reptilian species: The case of Anolis carolinensis. Mol. Genet. Genom. 2019, 294, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Makunin, A.I.; Rajičić, M.; Karamysheva, T.V.; Romanenko, S.A.; Druzhkova, A.S.; Blagojević, J.; Vujošević, M.; Rubtsov, N.B.; Graphodatsky, A.S.; Trifonov, V.A. Low-pass single-chromosome sequencing of human small supernumerary marker chromosomes (sSMCs) and Apodemus B chromosomes. Chromosoma 2018, 127, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.T.; Conte, M.A.; Fantinatti, B.E.A.; Cabral-de-Mello, D.C.; Carvalho, R.F.; Vicari, M.R.; Kocher, T.D.; Martins, C. Origin and evolution of B chromosomes in the cichlid fish Astatotilapia latifasciata based on integrated genomic analyses. Mol. Biol. Evol. 2014, 31, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Torgasheva, A.A.; Malinovskaya, L.P.; Zadesenets, K.S.; Karamysheva, T.V.; Kizilova, E.A.; Akberdina, E.A.; Pristyazhnyuk, I.E.; Shnaider, E.P.; Volodkina, V.A.; Saifitdinova, A.F.; et al. Germline-restricted chromosome (GRC) is widespread among songbirds. Proc. Natl. Acad. Sci. USA 2019, 116, 11845–11850. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Damas, J.; Corbo, M.; Lewin, H.A. Vertebrate Chromosome Evolution. Annu. Rev. Anim. Biosci. 2020, 9. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannucci, A.; Makunin, A.I.; Lisachov, A.P.; Ciofi, C.; Stanyon, R.; Svartman, M.; Trifonov, V.A. Bridging the Gap between Vertebrate Cytogenetics and Genomics with Single-Chromosome Sequencing (ChromSeq). Genes 2021, 12, 124. https://doi.org/10.3390/genes12010124
Iannucci A, Makunin AI, Lisachov AP, Ciofi C, Stanyon R, Svartman M, Trifonov VA. Bridging the Gap between Vertebrate Cytogenetics and Genomics with Single-Chromosome Sequencing (ChromSeq). Genes. 2021; 12(1):124. https://doi.org/10.3390/genes12010124
Chicago/Turabian StyleIannucci, Alessio, Alexey I. Makunin, Artem P. Lisachov, Claudio Ciofi, Roscoe Stanyon, Marta Svartman, and Vladimir A. Trifonov. 2021. "Bridging the Gap between Vertebrate Cytogenetics and Genomics with Single-Chromosome Sequencing (ChromSeq)" Genes 12, no. 1: 124. https://doi.org/10.3390/genes12010124
APA StyleIannucci, A., Makunin, A. I., Lisachov, A. P., Ciofi, C., Stanyon, R., Svartman, M., & Trifonov, V. A. (2021). Bridging the Gap between Vertebrate Cytogenetics and Genomics with Single-Chromosome Sequencing (ChromSeq). Genes, 12(1), 124. https://doi.org/10.3390/genes12010124