Real-Time Culture-Independent Microbial Profiling Onboard the International Space Station Using Nanopore Sequencing

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
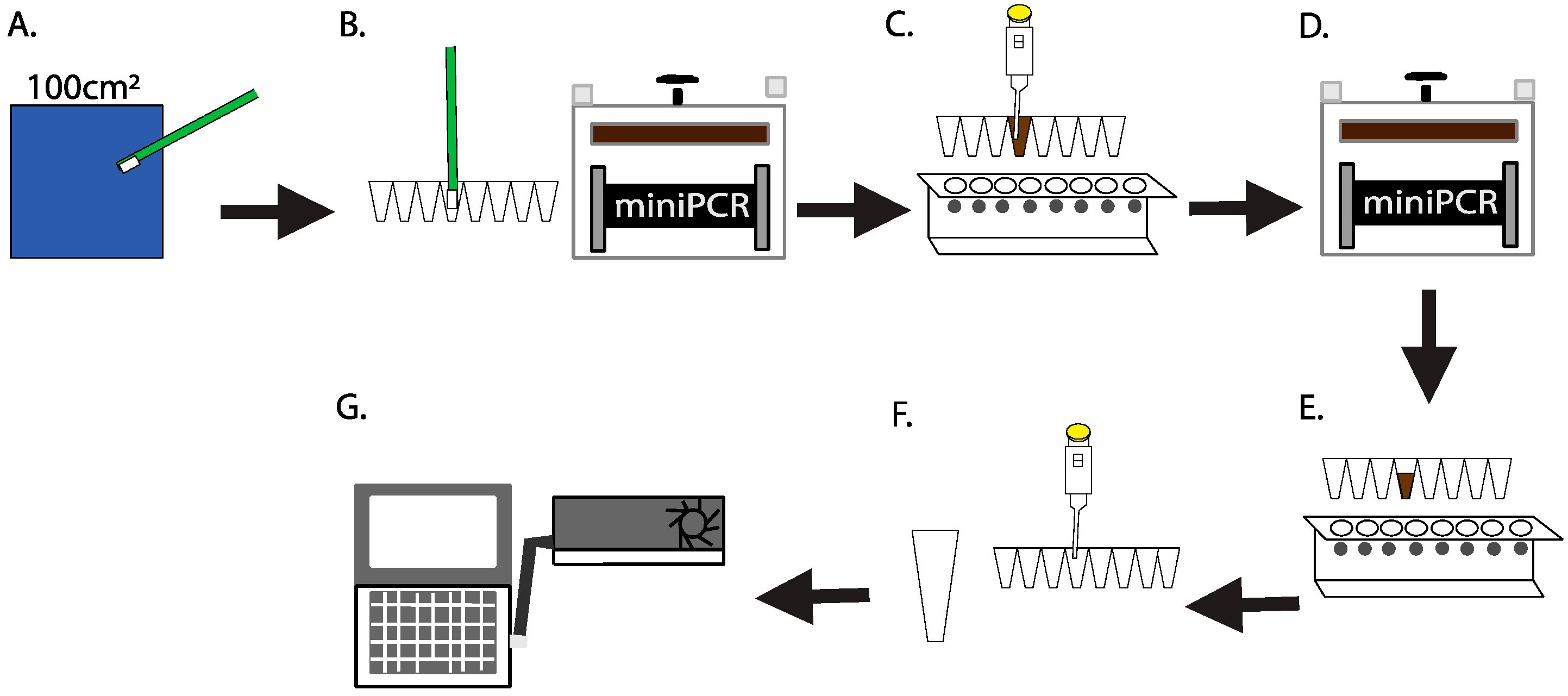
2.1. Culture-Independent, Swab-to-Sequencer Method
2.2. Comparison of Current Culture-Dependent to Swab-to-Sequencer, Culture-Independent Methods
2.3. NASA Analog Operations
2.4. Spaceflight Hardware
2.5. Swab-to-Sequencer Operations Onboard the International Space Station
2.6. Ground Control Testing of Identical Spaceflight Reagents
2.7. Nanopore Data Analysis
3. Results and Discussion
3.1. Swab-to-Sequencer Method for Rapid, In-Situ Microbial Profiling in Extreme Environments
3.2. Ground Validation: Comparison of Current Culture-Dependent to Swab-to-Sequencer, Culture-Independent Methods
3.3. Extreme Environment Method Validation during a NASA Analog Mission
3.4. Swab-to-Sequencer Processing Onboard the International Space Station
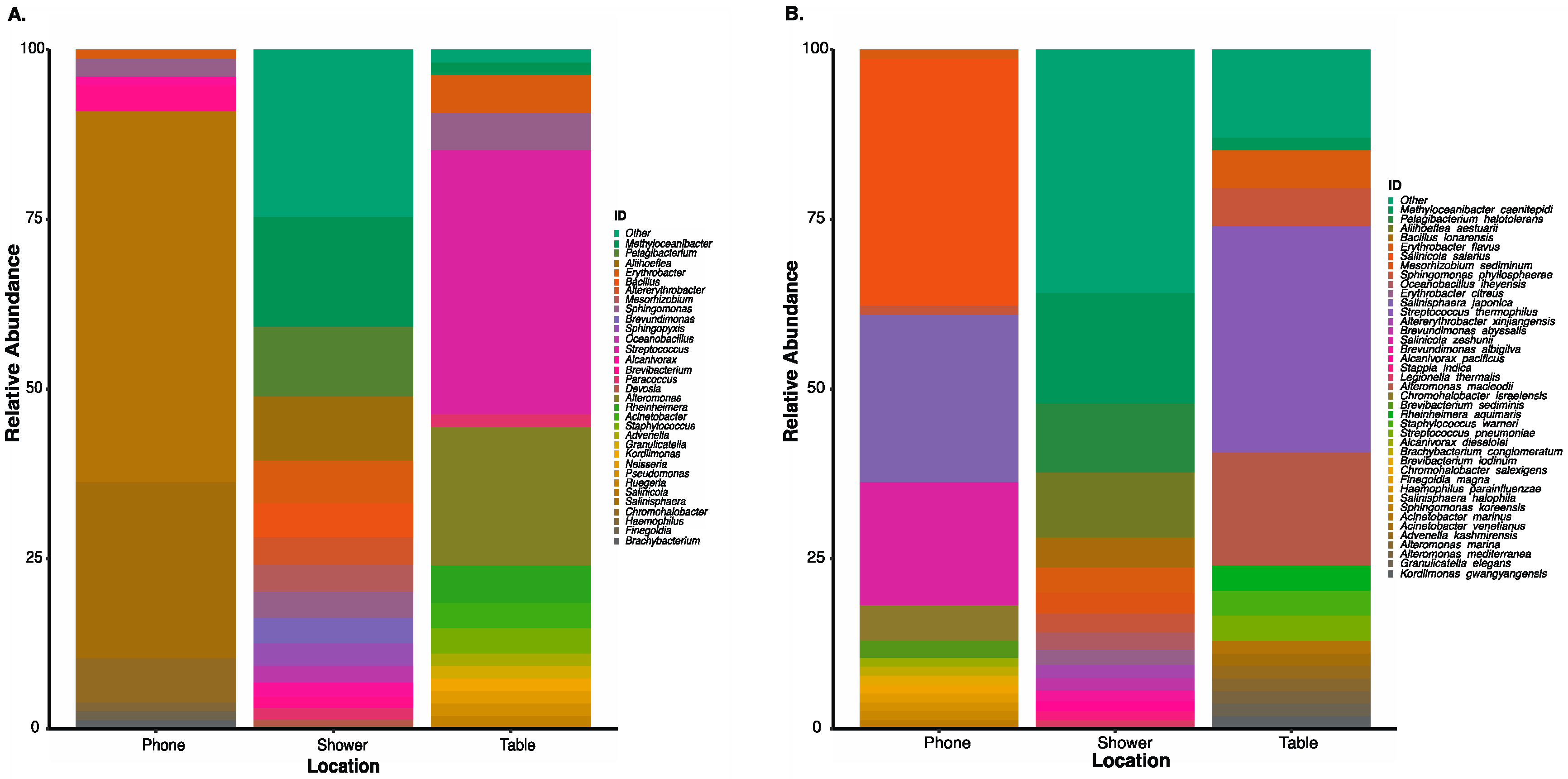
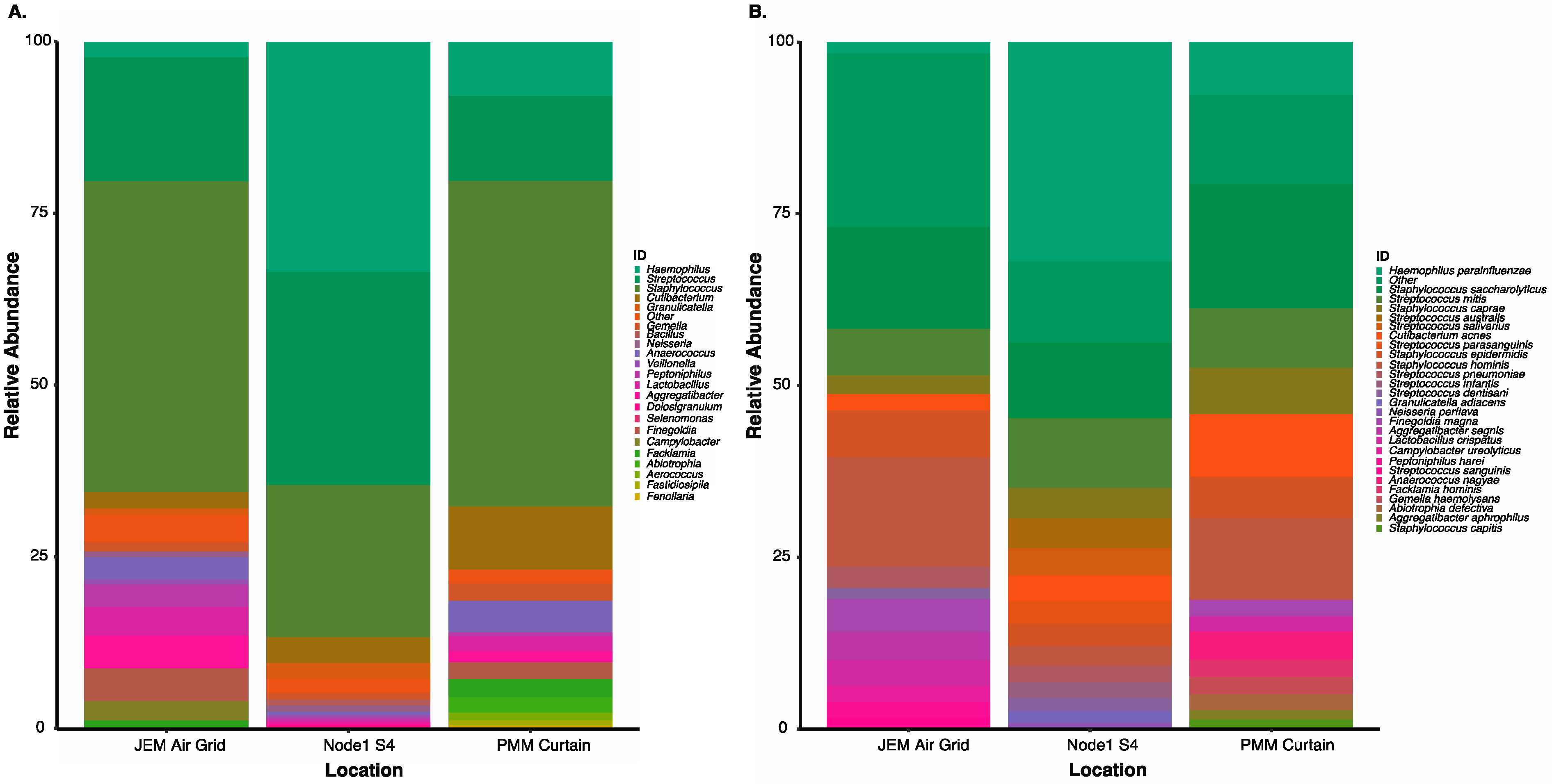
3.5. Culture-Independent Microbial Profiles of International Space Station Locations
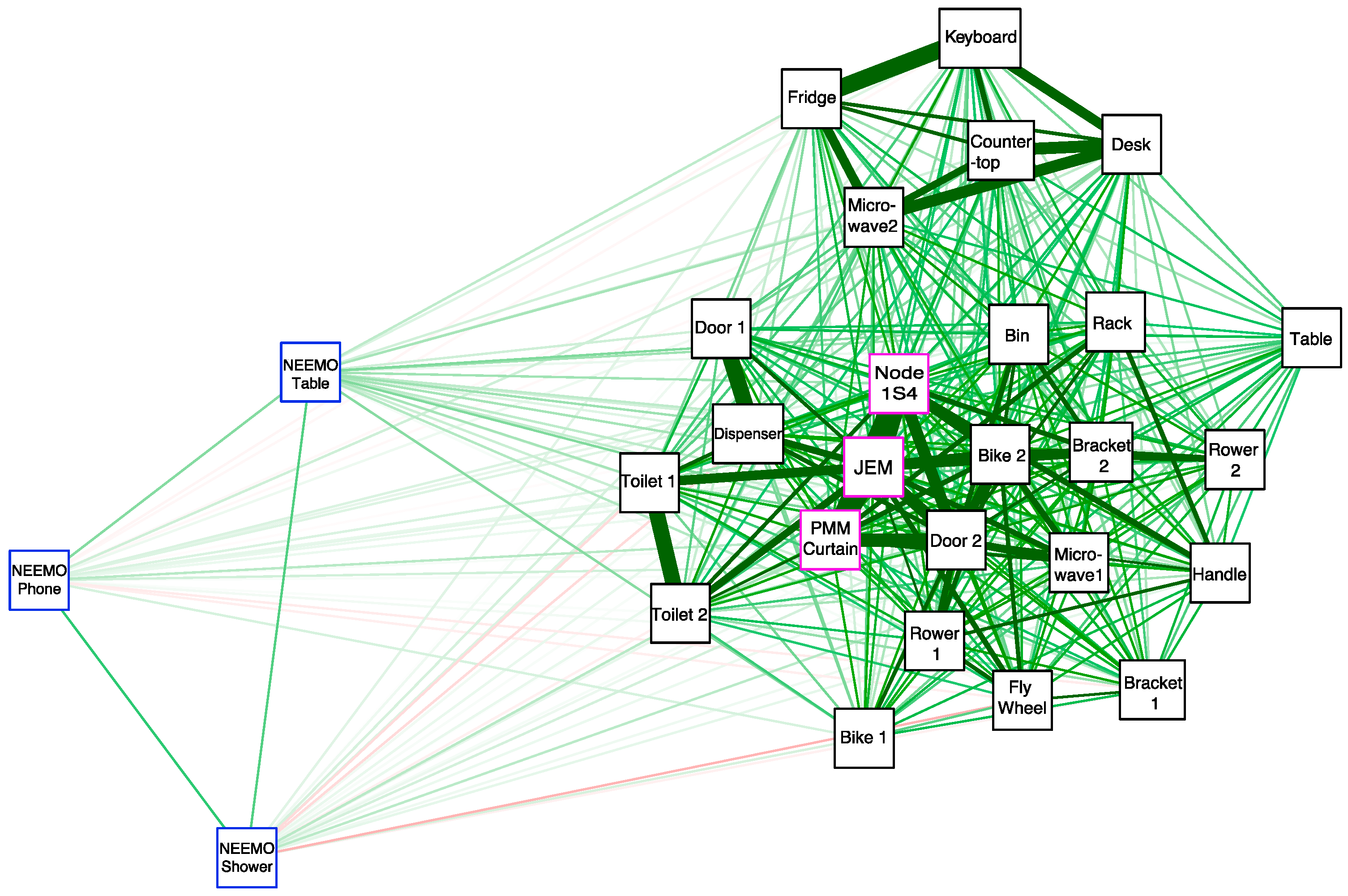
3.6. Correlation of Microbial Profiles from the Seafloor to Space Station
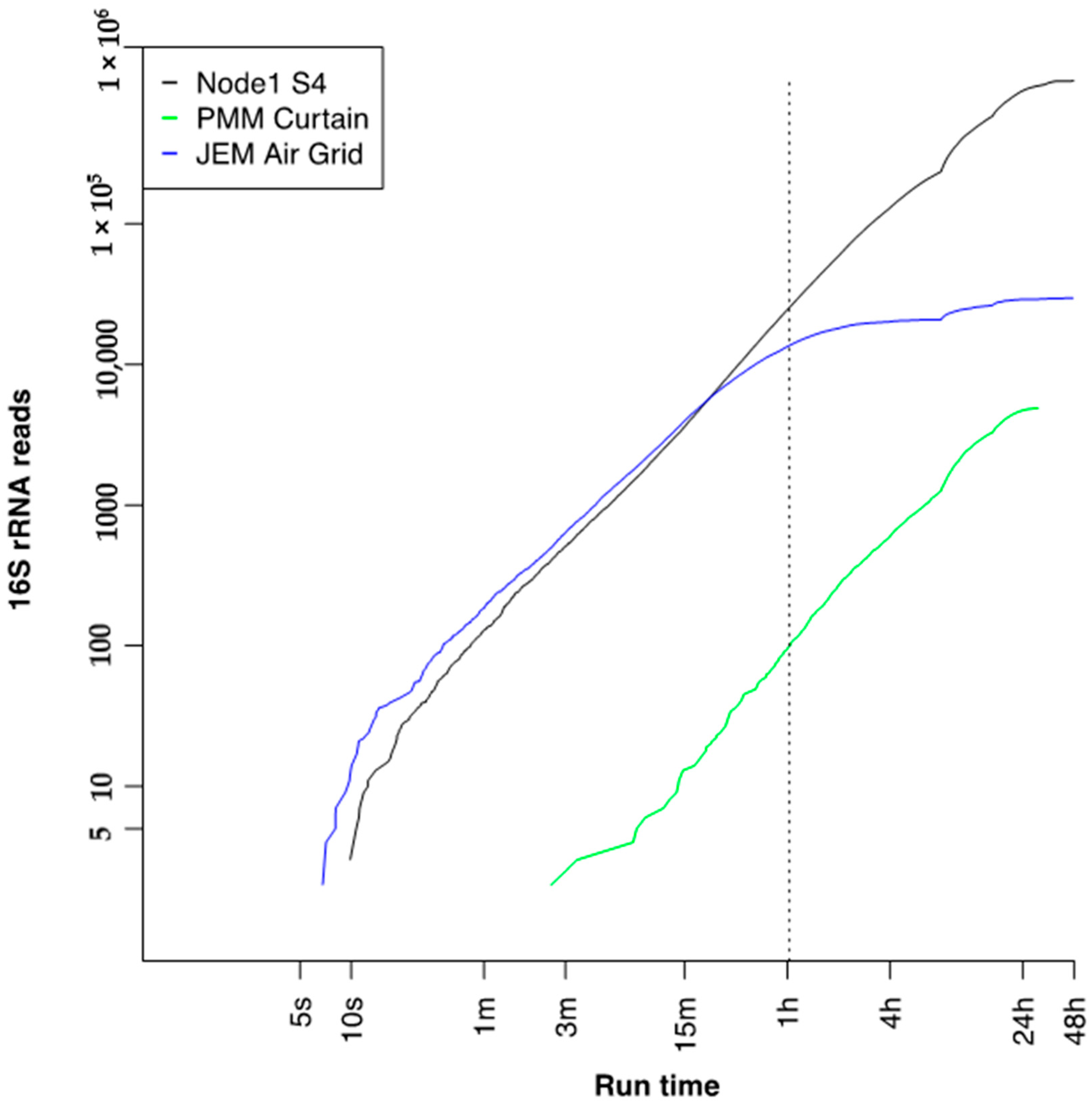
3.7. Run Time to 16S Bacterial Identification on International Space Station
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamaguchi, N.; Roberts, M.; Castro, S.; Oubre, C.; Makimura, K.; Leys, N.; Grohmann, E.; Sugita, T.; Ichijo, T.; Nasu, M. Microbial Monitoring of Crewed Habitats in Space—Current Status and Future Perspectives. Microbes Environ. 2014, 29, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Pierson, D.L. Microbial contamination of spacecraft. Gravitat. Space Biol. Bull. 2001, 14, 1–6. [Google Scholar]
- Ferguson, J.K.; Taylor, G.R.; Mieszkuc, B.J. Microbiological Investigations. In Biomedical Results of Apollo; Johnston, R.S., Dietlein, L.F., Berry, C.A., Eds.; National Aeronautics and Space Administration: Washington, DC, USA, 1975. [Google Scholar]
- Taylor, G.R.; Graves, R.C.; Brockett, R.M.; Ferguson, J.K.; Mieszuc, B.J. Skylab Environmental and Crew Microbiology Studies. In Biomedical Results from Skylab; Johnston, R.S., Dietlein, L.F., Eds.; National Aeronautics and Space Administration: Washington, DC, USA, 1977; ISBN 9781503344945. [Google Scholar]
- Pierson, D.B.; Ott, C.M.; Bruce, R.; Castro, A.V.; Mehta, S.K. Microbiological Lessons Learned From the Space Shuttle. In American Institute of Aeronautics and Astronautics Meeting Papers, Proceedings of the 41st International Conference on Environmental Systems, International Conference on Environmental Systems (ICES), Portland, OR, USA, 17 July 2011; American Institute of Aeronautics and Astronautics: Reston, VA, USA, 2012; p. 5266. [Google Scholar]
- Castro, V.A.; Thrasher, A.N.; Healy, M.; Ott, C.M.; Pierson, D.L. Microbial Characterization during the Early Habitation of the International Space Station. Microb. Ecol. 2004, 47, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Bruce, R.J.; Ott, C.M.; Skuratov, V.M.; Pierson, D.L. Microbial Surveillance of Potable Water Sources of the International Space Station. SAE Trans. J. Mater. Manuf. 2005, 114, 283–292. [Google Scholar]
- Burton, A.S.; Stahl, S.E.; John, K.K.; Jain, M.; Juul, S.; Turner, D.J.; Harrington, E.; Stoddart, D.; Paten, B.; Akeson, M.; et al. Off Earth Identification of Bacterial Populations Using 16S rDNA Nanopore Sequencing. Genes 2020, 11, 76. [Google Scholar] [CrossRef]
- Boguraev, A.-S.; Christensen, H.C.; Bonneau, A.R.; Pezza, J.A.; Nichols, N.M.; Giraldez, A.J.; Gray, M.M.; Wagner, B.M.; Aken, J.T.; Foley, K.D.; et al. Successful amplification of DNA aboard the International Space Station. npj Microgravity 2017, 3, 1–4. [Google Scholar] [CrossRef]
- Castro-Wallace, S.L.; Chiu, C.Y.; John, K.K.; Stahl, S.E.; Rubins, K.H.; McIntyre, A.B.R.; Dworkin, J.P.; Lupisella, M.L.; Smith, L.M.; Botkin, D.J.; et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Nygaard, A.B.; Tunsjø, H.S.; Meisal, R.; Charnock, C. A preliminary study on the potential of Nanopore MinION and Illumina MiSeq 16S rRNA gene sequencing to characterize building-dust microbiomes. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Voorhies, A.A.; Ott, C.M.; Mehta, S.; Pierson, D.L.; Crucian, B.E.; Feiveson, A.; Oubre, C.M.; Torralba, M.; Moncera, K.; Zhang, Y.; et al. Study of the impact of long-duration space missions at the International Space Station on the astronaut microbiome. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]
- Minich, J.J.; Zhu, Q.; Janssen, S.; Hendrickson, R.; Amir, A.; Vetter, R.; Hyde, J.; Doty, M.M.; Stillwell, K.; Benardini, J.; et al. KatharoSeq Enables High-Throughput Microbiome Analysis from Low-Biomass Samples. mSystems 2018, 3, e00218-17. [Google Scholar] [CrossRef]
- Avila-Herrera, A.; Thissen, J.; Urbaniak, C.; Be, N.A.; Smith, D.J.; Karouia, F.; Mehta, S.; Venkateswaran, K.; Jaing, C. Crewmember microbiome may influence microbial composition of ISS habitable surfaces. PLoS ONE 2020, 15, e0231838. [Google Scholar] [CrossRef] [PubMed]
- Fontana, C.; Favaro, M.; Pelliccioni, M.; Pistoia, E.S.; Favalli, C. Use of the MicroSeq 500 16S rRNA Gene-Based Sequencing for Identification of Bacterial Isolates That Commercial Automated Systems Failed To Identify Correctly. J. Clin. Microbiol. 2005, 43, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Gimmon, Y.; Sorathia, S.; Beaton, K.H.; Schubert, M.C. Exposure to an extreme environment comes at a sensorimotor cost. npj Microgravity 2018, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Todd, W.; Reagan, M. The NEEMO Undersea Analog: Another Type of Deep Space Exploration. Space 2005, 2005, 6754. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Project, G.; et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef]
- R Core Team. R: Foundation for Statistical Computing; Version 2.6.2; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; p. 213. [Google Scholar]
- Epskamp, S.; Cramer, A.O.J.; Waldorp, L.J.; Schmittmann, V.D.; Borsboom, D. qgraph: Network Visualizations of Relationships in Psychometric Data. J. Stat. Softw. 2012, 48, 1–18. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package, Version 1.0.2. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 4 January 2019).
- Parra, M.; Jung, J.; Boone, T.D.; Tran, L.; Blaber, E.A.; Brown, M.; Chin, M.; Chinn, T.; Cohen, J.; Doebler, R.; et al. Microgravity validation of a novel system for RNA isolation and multiplex quantitative real time PCR analysis of gene expression on the International Space Station. PLoS ONE 2017, 12, e0183480. [Google Scholar] [CrossRef]
- Stortchevoi, A.; Kamelamela, N.; Levine, S.S. SPRI Beads-based Size Selection in the Range of 2-10kb. J. Biomol. Tech. 2020, 31, 7–10. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Xu, H.; Xiong, M.; Gu, H. Size-selective DNA separation: Recovery spectra help determine the sodium chloride (NaCl) and polyethylene glycol (PEG) concentrations required. Biotechnol. J. 2014, 9, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Carleo, M.A.; Del Giudice, A.; Viglietti, R.; Rosario, P.; Esposito, V. Aortic Valve Endocarditis Caused by Abiotrophia defectiva: Case Report and Literature Overview. In Vivo 2015, 29, 515–518. [Google Scholar] [PubMed]
- Gross, K.C.; Houghton, M.P.; Roberts, R.B. Evaluation of blood culture media for isolation of pyridoxal-dependent Streptococcus mitior (mitis). J. Clin. Microbiol. 1981, 13, 266–272. [Google Scholar] [CrossRef]
- González-Díaz, A.; Tubau, F.; Pinto, M.; Sierra, Y.; Cubero, M.; Càmara, J.; Ayats, J.; Bajanca-Lavado, M.P.; Ardanuy, C.; Martínez, J.A. Identification of polysaccharide capsules among extensively drug-resistant genitourinary Haemophilus parainfluenzae isolates. Sci. Rep. 2019, 9, 4481. [Google Scholar] [CrossRef]
- McManus, C.; Kelley, S.T. Molecular survey of aeroplane bacterial contamination. J. Appl. Microbiol. 2005, 99, 502–508. [Google Scholar] [CrossRef]
- Gibbons, S.M.; Schwartz, T.; Fouquier, J.; Mitchell, M.; Sangwan, N.; Gilbert, J.A.; Kelley, S.T. Ecological Succession and Viability of Human-Associated Microbiota on Restroom Surfaces. Appl. Environ. Microbiol. 2015, 81, 765–773. [Google Scholar] [CrossRef]
- Mukherjee, N.; Dowd, S.E.; Wise, A.; Kedia, S.; Vohra, V.; Banerjee, P. Diversity of Bacterial Communities of Fitness Center Surfaces in a U.S. Metropolitan Area. Int. J. Environ. Res. Public Health 2014, 11, 12544–12561. [Google Scholar] [CrossRef]
- Grice, E.A. The skin microbiome: Potential for novel diagnostic and therapeutic approaches to cutaneous disease. Semin. Cutan. Med. Surg. 2014, 33, 98–103. [Google Scholar] [CrossRef]
- Kai, S.; Matsuo, Y.; Nakagawa, S.; Kryukov, K.; Matsukawa, S.; Tanaka, H.; Iwai, T.; Imanishi, T.; Hirota, K. Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinION™ nanopore sequencer. FEBS Open Bio 2019, 9, 548–557. [Google Scholar] [CrossRef]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE 2019, 14, e0216709. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Páez, A.; Portune, K.J.; Sanz, Y. Species-level resolution of 16S rRNA gene amplicons sequenced through the MinION™ portable nanopore sequencer. GigaScience 2016, 5, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Spakowicz, D.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Van Aerle, R.; Barrientos, L.; Martinez-Urtaza, J. Computational methods for 16S metabarcoding studies using Nanopore sequencing data. Comput. Struct. Biotechnol. J. 2020, 18, 296–305. [Google Scholar] [CrossRef]
- Peker, N.; Garcia-Croes, S.; Dijkhuizen, B.; Wiersma, H.H.; Van Zanten, E.; Wisselink, G.; Friedrich, A.W.; Kooistra-Smid, M.; Sinha, B.; Rossen, J.W.A.; et al. A Comparison of Three Different Bioinformatics Analyses of the 16S–23S rRNA Encoding Region for Bacterial Identification. Front. Microbiol. 2019, 10, 620. [Google Scholar] [CrossRef]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef]
- Cuscó, A.; Catozzi, C.; Viñes, J.; Sanchez, A.; Francino, O. Microbiota profiling with long amplicons using Nanopore sequencing: Full-length 16S rRNA gene and the 16S-ITS-23S of the rrn operon. F1000Research 2019, 7, 1755. [Google Scholar] [CrossRef]
- Wee, Y.; Bhyan, S.B.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. The bioinformatics tools for the genome assembly and analysis based on third-generation sequencing. Briefings Funct. Genom. 2018, 18, 1–12. [Google Scholar] [CrossRef]
- Brooks, J.P.; Edwards, D.J.; Harwich, M.D.; Rivera, M.C.; Fettweis, J.M.; Serrano, M.G.; Reris, R.A.; Sheth, N.U.; Huang, B.; Girerd, P.; et al. The truth about metagenomics: Quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015, 15, 1–14. [Google Scholar] [CrossRef]
- Browne, P.D.; Nielsen, T.K.; Kot, W.P.; Aggerholm, A.; Gilbert, M.T.P.; Puetz, L.; Rasmussen, M.; Zervas, A.; Hansen, L.H. GC bias affects genomic and metagenomic reconstructions, underrepresenting GC-poor organisms. GigaScience 2020, 9, 9. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- von Wintzingerode, F.; Gobel, U.B.; Stackebrandt, E. Determination of microbial diversity in environmental samples: Pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 1997, 21, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pérez, H.; Ciuffreda, L.; Flores, C. NanoCLUST: A species-level analysis of 16S rRNA nanopore sequencing data. Bioinformatics 2020. [Google Scholar] [CrossRef] [PubMed]
- Karst, S.M.; Ziels, R.M.; Kirkegaard, R.H.; Sørensen, E.A.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. Enabling high-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. bioRxiv 2020, 645903. [Google Scholar] [CrossRef]
- Weiser, J.N.; Ferreira, D.M.; Paton, J.C. Streptococcus pneumoniae: Transmission, colonization and invasion. Nat. Rev. Genet. 2018, 16, 355–367. [Google Scholar] [CrossRef]
- O’Sullivan, J.N.; Rea, M.C.; O’Connor, P.M.; Hill, C.; Ross, R.P. Human skin microbiota is a rich source of bacteriocin-producing staphylococci that kill human pathogens. FEMS Microbiol. Ecol. 2018, 95, 95. [Google Scholar] [CrossRef]
- Takeuchi, M.; Katayama, T.; Yamagishi, T.; Hanada, S.; Tamaki, H.; Kamagata, Y.; Oshima, K.; Hattori, M.; Marumo, K.; Nedachi, M.; et al. Methyloceanibacter caenitepidi gen. nov., sp. nov., a facultatively methylotrophic bacterium isolated from marine sediments near a hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2014, 64, 462–468. [Google Scholar] [CrossRef]
- Roh, S.W.; Kim, K.-H.; Nam, Y.-D.; Chang, H.-W.; Kim, M.-S.; Shin, K.-S.; Yoon, J.-H.; Oh, H.-M.; Bae, J.-W. Aliihoeflea aestuarii gen. nov., sp. nov., a novel bacterium isolated from tidal flat sediment. J. Microbiol. 2008, 46, 594–598. [Google Scholar] [CrossRef]
- Shimane, Y.; Tsuruwaka, Y.; Miyazaki, M.; Mori, K.; Minegishi, H.; Echigo, A.; Ohta, Y.; Maruyama, T.; Grant, W.D.; Hatada, Y. Salinisphaera japonica sp. nov., a moderately halophilic bacterium isolated from the surface of a deep-sea fish, Malacocottus gibber, and emended description of the genus Salinisphaera. Int. J. Syst. Evol. Microbiol. 2013, 63, 2180–2185. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Kim, H.; Kim, I.-G.; Kang, K.H.; Park, Y.-H. Erythrobacter flavus sp. nov., a slight halophile from the East Sea in Korea. Int. J. Syst. Evol. Microbiol. 2003, 53, 1169–1174. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.R.; Yu, W.-H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, T.; Yamaguchi, N.; Tanigaki, F.; Shirakawa, M.; Nasu, M. Four-year bacterial monitoring in the International Space Station—Japanese Experiment Module “Kibo” with culture-independent approach. npj Microgravity 2016, 2, 16007. [Google Scholar] [CrossRef] [PubMed]
- Sielaff, A.C.; Urbaniak, C.; Mohan, G.B.M.; Stepanov, V.G.; Tran, Q.; Wood, J.M.; Minich, J.; McDonald, D.; Mayer, T.; Knight, R.; et al. Characterization of the total and viable bacterial and fungal communities associated with the International Space Station surfaces. Microbiome 2019, 7, 1–21. [Google Scholar] [CrossRef]
- Lang, J.M.; Coil, D.A.; Neches, R.Y.; Brown, W.E.; Cavalier, D.; Severance, M.; Hampton-Marcell, J.T.; Gilbert, J.A.; Eisen, J.A. A microbial survey of the International Space Station (ISS). PeerJ 2017, 5, e4029. [Google Scholar] [CrossRef]
- Mora, M.; Wink, L.; Kögler, I.; Mahnert, A.; Rettberg, P.; Schwendner, P.; DeMets, R.; Cockell, C.; Alekhova, T.; Klingl, A.; et al. Space Station conditions are selective but do not alter microbial characteristics relevant to human health. Nat. Commun. 2019, 10, 3990. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nat. Cell Biol. 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Holgerson, P.L.; Esberg, A.; Sjödin, A.; West, C.E.; Johansson, I. A longitudinal study of the development of the saliva microbiome in infants 2 days to 5 years compared to the microbiome in adolescents. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Gupta, S.; Garg, M.; Misra, S.; Singhal, S. Granulicatella adiacens abscess: Two rare cases and review. J. Lab. Physicians 2018, 10, 121–123. [Google Scholar] [CrossRef]
- Urbaniak, C.; Lorenzi, H.; Thissen, J.; Jaing, C.; Crucian, B.; Sams, C.; Pierson, D.; Venkateswaran, K.; Mehta, S. The influence of spaceflight on the astronaut salivary microbiome and the search for a microbiome biomarker for viral reactivation. Microbiome 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Pulliam, L.; Porschen, R.K.; Hadley, W.K. Biochemical properties of CO2-dependent streptococci. J. Clin. Microbiol. 1980, 12, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Wassef, N.; Rizkalla, E.; Shaukat, N.; Sluka, M. HACEK-induced endocarditis. BMJ Case Rep. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, H.; Poehlein, A.; Brzuszkiewicz, E.; Scavenius, C.; Enghild, J.J.; Al-Zeer, M.A.; Brinkmann, V.; Jensen, A.; Söderquist, B. Staphylococcus saccharolyticus Isolated From Blood Cultures and Prosthetic Joint Infections Exhibits Excessive Genome Decay. Front. Microbiol. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Matty, C.; Meyers, V.; Slipes, W.; Scully, R. Crew Health and Performance Improvements with Reduced Carbon Dioxide Levels and the Resource Impact to Accomplish Those Reductions. In American Institute of Aeronautics and Astronautics Meeting Papers, Proceedings of 41st International Conference on Environmental Systems, 17–21 July 2011, Portland, OR, USA; American Institute of Aeronautics and Astronautics: Reston, VA, USA, 2011. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date | Location | Total Reads (Fast5) | QC Pore Count | Sequencing Pore Count |
|---|---|---|---|---|
| 19 July 2018 | Node 1S4 Dining Table Wall | 1,660,150 | 1646 | 1635 |
| 17 September 2018 | Permanent Multipurpose Module (PMM) Curtain | 172,891 | 1610 | 1574 |
| 25 September 2018 | Japanese Experiment Module (JEM) air grid | 92,106 | 1530 | 1002 |
| 7 November 2018 | Flight Negative Swab | 42,880 | 1505 | 1486 |
| 23 October 2018 | Ground Negative Swab | 31,970 | 1613 | 1589 |
| 16 November 2018 | Ground Positive Control | 1,689,464 | 1613 | 1589 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stahl-Rommel, S.; Jain, M.; Nguyen, H.N.; Arnold, R.R.; Aunon-Chancellor, S.M.; Sharp, G.M.; Castro, C.L.; John, K.K.; Juul, S.; Turner, D.J.; et al. Real-Time Culture-Independent Microbial Profiling Onboard the International Space Station Using Nanopore Sequencing. Genes 2021, 12, 106. https://doi.org/10.3390/genes12010106
Stahl-Rommel S, Jain M, Nguyen HN, Arnold RR, Aunon-Chancellor SM, Sharp GM, Castro CL, John KK, Juul S, Turner DJ, et al. Real-Time Culture-Independent Microbial Profiling Onboard the International Space Station Using Nanopore Sequencing. Genes. 2021; 12(1):106. https://doi.org/10.3390/genes12010106
Chicago/Turabian StyleStahl-Rommel, Sarah, Miten Jain, Hang N. Nguyen, Richard R. Arnold, Serena M. Aunon-Chancellor, Gretta Marie Sharp, Christian L. Castro, Kristen K. John, Sissel Juul, Daniel J. Turner, and et al. 2021. "Real-Time Culture-Independent Microbial Profiling Onboard the International Space Station Using Nanopore Sequencing" Genes 12, no. 1: 106. https://doi.org/10.3390/genes12010106
APA StyleStahl-Rommel, S., Jain, M., Nguyen, H. N., Arnold, R. R., Aunon-Chancellor, S. M., Sharp, G. M., Castro, C. L., John, K. K., Juul, S., Turner, D. J., Stoddart, D., Paten, B., Akeson, M., Burton, A. S., & Castro-Wallace, S. L. (2021). Real-Time Culture-Independent Microbial Profiling Onboard the International Space Station Using Nanopore Sequencing. Genes, 12(1), 106. https://doi.org/10.3390/genes12010106