Slow Adaptive Response of Budding Yeast Cells to Stable Conditions of Continuous Culture Can Occur without Genome Modifications

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Strain and Media
2.2. Continuous Culture Conditions
2.3. Preparation of the Total RNA and Microarray Analysis
2.4. Bioinformatics Analysis
2.5. Quantitative Real-Time RT-PCR Analyses
3. Results
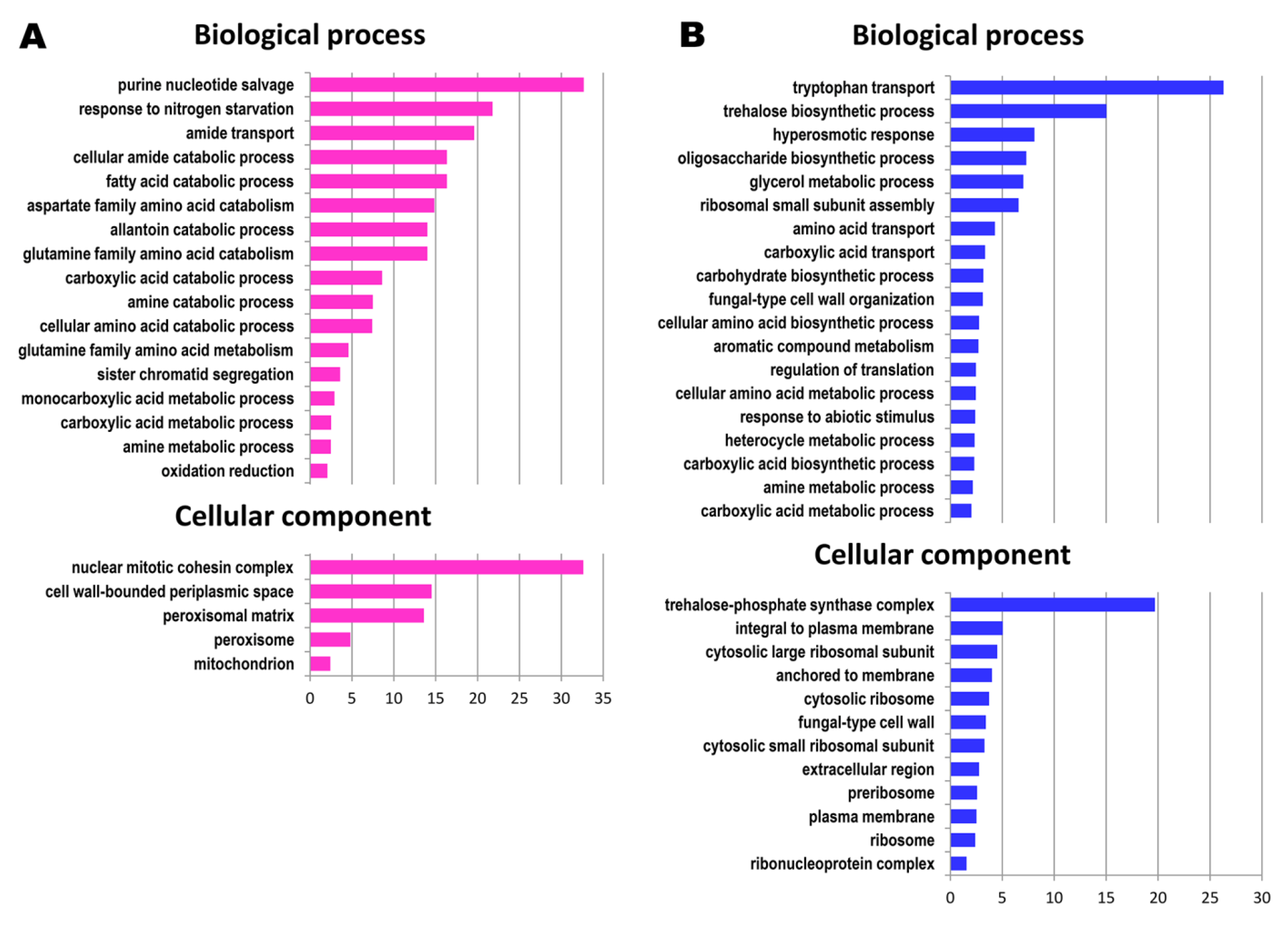
3.1. Functional Categories of Genes Upregulated during the Prolonged Continuous Culture
3.2. Functional Categories of Genes Downregulated during Prolonged Continuous Culture
3.3. Dubious ORFs
3.4. Search for Common Attributes Affecting the Expression of Selected Genes
3.5. Are There Other Explanations for the Observed Changes in Gene Expression?
4. Discussion
4.1. Regulatory Network Responsible for the Cellular Switch from a Variable to a Stable Environment
4.2. How Could a Constant Cell Environment Induce a Drift in Gene Expression Patterns, and Why Would It Make Biological Sense for the Drift to Be Slow?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hoskisson, P.A.; Hobbs, G. Continuous culture—Making a comeback? Microbiol. Read. 2005, 151, 3153–3159. [Google Scholar] [CrossRef] [PubMed]
- Daran-Lapujade, P.; Daran, J.-M.; van Maris, A.J.A.; de Winde, J.H.; Pronk, J.T. Chemostat-based micro-array analysis in baker’s yeast. Adv. Microb. Physiol. 2009, 54, 257–311. [Google Scholar] [CrossRef] [PubMed]
- Kayikci, Ö.; Nielsen, J. Glucose repression in Saccharomyces cerevisiae. FEMS Yeast Res. 2015, 15. [Google Scholar] [CrossRef]
- DeRisi, J.L.; Iyer, V.R.; Brown, P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 1997, 278, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, B.; Liang, X.B.; Robert, F.; Soontorngun, N. Transcriptional regulation of nonfermentable carbon utilization in budding yeast. FEMS Yeast Res. 2010, 10, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, T. A cultural divide on the use of chemostats. Microbiol. Read. 2006, 152, 1247–1248. [Google Scholar] [CrossRef]
- Ferea, T.L.; Botstein, D.; Brown, P.O.; Rosenzweig, R.F. Systematic changes in gene expression patterns following adaptive evolution in yeast. Proc. Natl. Acad. Sci. USA 1999, 96, 9721–9726. [Google Scholar] [CrossRef]
- Wenger, J.W.; Piotrowski, J.; Nagarajan, S.; Chiotti, K.; Sherlock, G.; Rosenzweig, F. Hunger artists: Yeast adapted to carbon limitation show trade-offs under carbon sufficiency. PLoS Genet. 2011, 7, e1002202. [Google Scholar] [CrossRef]
- Voordeckers, K.; Verstrepen, K.J. Experimental evolution of the model eukaryote Saccharomyces cerevisiae yields insight into the molecular mechanisms underlying adaptation. Curr. Opin. Microbiol. 2015, 28, 1–9. [Google Scholar] [CrossRef]
- Kvitek, D.J.; Sherlock, G. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet. 2013, 9, e1003972. [Google Scholar] [CrossRef]
- Hong, J.; Brandt, N.; Abdul-Rahman, F.; Yang, A.; Hughes, T.; Gresham, D. An incoherent feedforward loop facilitates adaptive tuning of gene expression. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Gresham, D. Molecular specificity, convergence and constraint shape adaptive evolution in nutrient-poor environments. PLoS Genet. 2014, 10, e1004041. [Google Scholar] [CrossRef] [PubMed]
- Ter Linde, J.J.; Liang, H.; Davis, R.W.; Steensma, H.Y.; van Dijken, J.P.; Pronk, J.T. Genome-wide transcriptional analysis of aerobic and anaerobic chemostat cultures of Saccharomyces cerevisiae. J. Bacteriol. 1999, 181, 7409–7413. [Google Scholar] [CrossRef] [PubMed]
- Kazemi Seresht, A.; Cruz, A.L.; de Hulster, E.; Hebly, M.; Palmqvist, E.A.; van Gulik, W.; Daran, J.-M.; Pronk, J.; Olsson, L. Long-term adaptation of Saccharomyces cerevisiae to the burden of recombinant insulin production. Biotechnol. Bioeng. 2013, 110, 2749–2763. [Google Scholar] [CrossRef]
- Jansen, M.L.A.; Diderich, J.A.; Mashego, M.; Hassane, A.; de Winde, J.H.; Daran-Lapujade, P.; Pronk, J.T. Prolonged selection in aerobic, glucose-limited chemostat cultures of Saccharomyces cerevisiae causes a partial loss of glycolytic capacity. Microbiol. Read. 2005, 151, 1657–1669. [Google Scholar] [CrossRef]
- Miller, A.W.; Befort, C.; Kerr, E.O.; Dunham, M.J. Design and use of multiplexed chemostat arrays. J. Vis. Exp. 2013, e50262. [Google Scholar] [CrossRef]
- Collart, M.A.; Oliviero, S. Preparation of Yeast RNA. Curr. Protoc. Mol. Biol. 1993, 23. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Monteiro, P.T.; Oliveira, J.; Pais, P.; Antunes, M.; Palma, M.; Cavalheiro, M.; Galocha, M.; Godinho, C.P.; Martins, L.C.; Bourbon, N.; et al. YEASTRACT+: A portal for cross-species comparative genomics of transcription regulation in yeasts. Nucleic Acids Res. 2020, 48, D642–D649. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Fink, G.R. The histidine permease gene (HIP1) of Saccharomyces cerevisiae. Gene 1985, 38, 205–214. [Google Scholar] [CrossRef]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Curcio, M.J.; Garfinkel, D.J. Single-step selection for Ty1 element retrotransposition. Proc. Natl. Acad. Sci. USA 1991, 88, 936–940. [Google Scholar] [CrossRef]
- Jack, C.V.; Cruz, C.; Hull, R.M.; Keller, M.A.; Ralser, M.; Houseley, J. Regulation of ribosomal DNA amplification by the TOR pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 9674–9679. [Google Scholar] [CrossRef]
- Brown, C.J.; Todd, K.M.; Rosenzweig, R.F. Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol. Biol. Evol. 1998, 15, 931–942. [Google Scholar] [CrossRef]
- Novick, A.; Szilard, L. Experiments with the Chemostat on spontaneous mutations of bacteria. Proc. Natl. Acad. Sci. USA 1950, 36, 708–719. [Google Scholar] [CrossRef]
- Ter Linde, J.J.M.; Steensma, H.Y. A microarray-assisted screen for potential Hap1 and Rox1 target genes in Saccharomyces cerevisiae. Yeast 2002, 19, 825–840. [Google Scholar] [CrossRef]
- Kolkman, A.; Olsthoorn, M.M.A.; Heeremans, C.E.M.; Heck, A.J.R.; Slijper, M. Comparative proteome analysis of Saccharomyces cerevisiae grown in chemostat cultures limited for glucose or ethanol. Mol. Cell Proteom. 2005, 4, 1–11. [Google Scholar] [CrossRef]
- Wright, N.R.; Wulff, T.; Palmqvist, E.A.; Jørgensen, T.R.; Workman, C.T.; Sonnenschein, N.; Rønnest, N.P.; Herrgård, M.J. Fluctuations in glucose availability prevent global proteome changes and physiological transition during prolonged chemostat cultivations of Saccharomyces cerevisiae. Biotechnol. Bioeng. 2020, 117, 2074–2088. [Google Scholar] [CrossRef]
- Mashego, M.R.; Jansen, M.L.A.; Vinke, J.L.; van Gulik, W.M.; Heijnen, J.J. Changes in the metabolome of Saccharomyces cerevisiae associated with evolution in aerobic glucose-limited chemostats. FEMS Yeast Res. 2005, 5, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.I.; Desai, M.M. The spectrum of adaptive mutations in experimental evolution. Genomics 2014, 104, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, D.E.; Dean, A.M.; Hartl, D.L. Metabolic flux and fitness. Genetics 1987, 115, 25–31. [Google Scholar] [PubMed]
- Van den Brink, J.; Canelas, A.B.; van Gulik, W.M.; Pronk, J.T.; Heijnen, J.J.; de Winde, J.H.; Daran-Lapujade, P. Dynamics of glycolytic regulation during adaptation of Saccharomyces cerevisiae to fermentative metabolism. Appl. Environ. Microbiol. 2008, 74, 5710–5723. [Google Scholar] [CrossRef]
- De Groot, D.H.; van Boxtel, C.; Planqué, R.; Bruggeman, F.J.; Teusink, B. The number of active metabolic pathways is bounded by the number of cellular constraints at maximal metabolic rates. PLoS Comput. Biol. 2019, 15, e1006858. [Google Scholar] [CrossRef]
- Yu, R.; Campbell, K.; Pereira, R.; Björkeroth, J.; Qi, Q.; Vorontsov, E.; Sihlbom, C.; Nielsen, J. Nitrogen limitation reveals large reserves in metabolic and translational capacities of yeast. Nat. Commun. 2020, 11, 1881. [Google Scholar] [CrossRef]
- Newman, J.R.S.; Ghaemmaghami, S.; Ihmels, J.; Breslow, D.K.; Noble, M.; DeRisi, J.L.; Weissman, J.S. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 2006, 441, 840–846. [Google Scholar] [CrossRef]
- Venturelli, O.S.; Zuleta, I.; Murray, R.M.; El-Samad, H. Population diversification in a yeast metabolic program promotes anticipation of environmental shifts. PLoS Biol. 2015, 13, e1002042. [Google Scholar] [CrossRef]
- Wang, J.; Atolia, E.; Hua, B.; Savir, Y.; Escalante-Chong, R.; Springer, M. Natural variation in preparation for nutrient depletion reveals a cost-benefit tradeoff. PLoS Biol. 2015, 13, e1002041. [Google Scholar] [CrossRef]
- Smith, M.C.A.; Sumner, E.R.; Avery, S.V. Glutathione and Gts1p drive beneficial variability in the cadmium resistances of individual yeast cells. Mol. Microbiol. 2007, 66, 699–712. [Google Scholar] [CrossRef]
- Liu, J.; Lestrade, D.; Arabaciyan, S.; Cescut, J.; François, J.-M.; Capp, J.-P. A GRX1 Promoter Variant Confers Constitutive Noisy Bimodal Expression That Increases Oxidative Stress Resistance in Yeast. Front. Microbiol. 2018, 9, 2158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qian, W.; Zhang, J. Positive selection for elevated gene expression noise in yeast. Mol. Syst. Biol. 2009, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Acar, M. Mechanisms for the epigenetic inheritance of stress response in single cells. Curr. Genet. 2018, 64, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Brickner, J.H. Epigenetic transcriptional memory. Curr. Genet. 2017, 63, 435–439. [Google Scholar] [CrossRef]
- Choi, J.K.; Hwang, S.; Kim, Y.-J. Stochastic and regulatory role of chromatin silencing in genomic response to environmental changes. PLoS ONE 2008, 3, e3002. [Google Scholar] [CrossRef]
- Ahmed, S.; Brickner, J.H. Regulation and epigenetic control of transcription at the nuclear periphery. Trends Genet. 2007, 23, 396–402. [Google Scholar] [CrossRef]
- Fabrizio, P.; Garvis, S.; Palladino, F. Histone Methylation and Memory of Environmental Stress. Cells 2019, 8, 339. [Google Scholar] [CrossRef]
- Sorida, M.; Murakami, Y. Unprogrammed epigenetic variation mediated by stochastic formation of ectopic heterochromatin. Curr. Genet. 2020, 66, 319–325. [Google Scholar] [CrossRef]
- Choi, J.K.; Kim, Y.-J. Implications of the nucleosome code in regulatory variation, adaptation and evolution. Epigenetics 2009, 4, 291–295. [Google Scholar] [CrossRef][Green Version]
- Chakravarty, A.K.; Jarosz, D.F. More than Just a Phase: Prions at the Crossroads of Epigenetic Inheritance and Evolutionary Change. J. Mol. Biol. 2018, 430, 4607–4618. [Google Scholar] [CrossRef]
- Itakura, A.K.; Chakravarty, A.K.; Jakobson, C.M.; Jarosz, D.F. Widespread Prion-Based Control of Growth and Differentiation Strategies in Saccharomyces cerevisiae. Mol. Cell 2020, 77, 266–278.e6. [Google Scholar] [CrossRef]
- Palková, Z.; Devaux, F.; Icicová, M.; Mináriková, L.; Le Crom, S.; Jacq, C. Ammonia pulses and metabolic oscillations guide yeast colony development. Mol. Biol. Cell 2002, 13, 3901–3914. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Transcriptional Factors for Up-Regulated Genes | ||||||
| TF | Fraction | p-Value | Gene Count | Over-represen-tation | Rank | Description |
| Adr1p | 0.07 | 0 | 54 | 2.96 | 11.1 | Carbon source-responsive zinc-finger transcription factor; required for transcription of the glucose-repressed gene ADH2, of peroxisomal protein genes, and of genes required for ethanol, glycerol, and fatty acid utilization |
| Mga1p | 0.06 | 0 | 54 | 2.86 | 9.4 | Protein similar to heat shock transcription factor; multicopy suppressor of pseudohyphal growth defects of ammonium permease mutants |
| Fzf1p | 0.09 | 0 | 18 | 4.31 | 6.8 | Transcription factor involved in sulfite metabolism; sole identified regulatory target is SSU1; overexpression suppresses sulfite-sensitivity of many unrelated mutants due to hyperactivation of SSU1 |
| Gzf3p | 0.07 | 0 | 31 | 2.51 | 5.1 | GATA zinc finger protein; negatively regulates nitrogen catabolic gene expression by competing with Gat1p for GATA site binding; function requires a repressive carbon source; dimerizes with Dal80p and binds to Tor1p |
| Dal81p | 0.06 | 0.0001 | 24 | 3.12 | 4.8 | Positive regulator of genes in multiple nitrogen degradation pathways |
| Cat8p | 0.09 | 0 | 16 | 3.16 | 4.6 | Zinc cluster transcriptional activator; necessary for derepression of a variety of genes under non-fermentative growth conditions, active after diauxic shift, binds carbon source responsive elements |
| Dal80p | 0.08 | 0.0003 | 15 | 3.18 | 3.6 | Negative regulator of genes in multiple nitrogen degradation pathways; expression is regulated by nitrogen levels and by Gln3p; member of the GATA-binding family, forms homodimers and heterodimers with Gzf3p |
| Tod6p | 0.1 | 0.0002 | 10 | 3.06 | 3.1 | PAC motif binding protein involved in rRNA and ribosome biogenesis; subunit of the RPD3L histone deacetylase complex; Myb-like HTH transcription factor |
| Sut2p | 0.07 | 0.0012 | 13 | 2.35 | 2.1 | Zn2Cys6 family transcription factor; positively regulates sterol uptake under anaerobic conditions with SUT1; represses filamentation-inducing genes during non-starvation conditions; positively regulates mating |
| Wtm2p | 0.07 | 0.0014 | 12 | 2.39 | 2 | Transcriptional modulator; involved in regulation of meiosis, silencing, and expression of RNR genes; involved in response to replication stress |
| Rpi1p | 0.06 | 0.0053 | 11 | 2.13 | 1.5 | Transcription factor, allelic differences between S288C and Sigma1278b; mediates fermentation stress tolerance by modulating cell wall integrity |
| Transcriptional Factors for Down-Regulated Genes | ||||||
| TF | Fraction | p-Value | Gene Count | Over-represen-tation | Rank | Description |
| Hot1p | 0.31 | 0 | 32 | 12.68 | 126 | Transcription factor for glycerol biosynthetic genes; required for the transient induction of glycerol biosynthetic genes GPD1 and GPP2 in response to high osmolarity; targets Hog1p to osmostress responsive promoters |
| Crz1p | 0.11 | 0 | 102 | 3.63 | 40 | Transcription factor, activates transcription of stress response genes; nuclear localization is positively regulated by calcineurin-mediated dephosphorylation |
| Gis1p | 0.12 | 0 | 73 | 3.7 | 33.5 | Histone demethylase and transcription factor; regulates genes during nutrient limitation |
| Rlm1p | 0.11 | 0 | 77 | 3.51 | 29.4 | MADS-box transcription factor; component of the protein kinase C-mediated MAP kinase pathway involved in the maintenance of cell integrity |
| Ifh1p | 0.1 | 0 | 44 | 4.53 | 20.7 | Coactivator, regulates transcription of ribosomal protein (RP) genes |
| Met4p | 0.1 | 0 | 63 | 3.04 | 18.7 | Leucine-zipper transcriptional activator; responsible for regulation of sulfur amino acid pathway |
| Mig1p | 0.1 | 0 | 57 | 3.26 | 18.2 | Transcription factor involved in glucose repression; regulates filamentous growth along with Mig2p in response to glucose depletion |
| Mga2p | 0.08 | 0 | 90 | 2.41 | 17.6 | ER membrane protein involved in regulation of OLE1 transcription; inactive ER form dimerizes and one subunit is then activated by ubiquitin/proteasome-dependent processing followed by nuclear targeting |
| Stp2p | 0.09 | 0 | 52 | 3.33 | 16.1 | Transcription factor; activated by proteolytic processing in response to signals from the SPS sensor system for external amino acids; activates transcription of amino acid permease genes |
| Stp1p | 0.08 | 0 | 70 | 2.62 | 14.8 | Transcription factor; undergoes proteolytic processing by SPS sensor component Ssy5p in response to extracellular amino acids; activates transcription of amino acid permease genes and may have a role in tRNA processing |
| Nrg1p | 0.09 | 0 | 46 | 3.29 | 14 | Transcriptional repressor; mediates glucose repression and negatively regulates a variety of processes including filamentous growth and alkaline pH response |
| Gzf3p | 0.1 | 0 | 46 | 3.06 | 13.8 | GATA zinc finger protein; negatively regulates nitrogen catabolic gene expression by competing with Gat1p for GATA site binding; function requires a repressive carbon source; dimerizes with Dal80p and binds to Tor1p |
| Sko1p | 0.08 | 0 | 77 | 2.12 | 13.6 | Basic leucine zipper transcription factor of the ATF/CREB family; cytosolic and nuclear protein involved in osmotic and oxidative stress responses |
| Gat4p | 0.1 | 0 | 42 | 3.06 | 13.3 | Protein containing GATA family zinc finger motifs; involved in spore wall assembly |
| Nrg2p | 0.16 | 0 | 18 | 4.63 | 13 | Transcriptional repressor; mediates glucose repression and negatively regulates filamentous growth |
| Hap1p | 0.09 | 0 | 56 | 2.52 | 13 | Zinc finger transcription factor; involved in the complex regulation of gene expression in response to levels of heme and oxygen |
| Rgm1p | 0.08 | 0 | 49 | 2.67 | 10.9 | Putative zinc finger transcription factor; overproduction impairs cell growth and induces expression of genes involved in monosaccharide catabolism and aldehyde metabolism; regulates expression of subtelomeric genes |
| Sfl1p | 0.08 | 0 | 51 | 2.53 | 10.5 | Transcriptional repressor and activator; involved in repression of flocculation-related genes, and activation of stress responsive genes; has direct role in INO1 transcriptional memory |
| Tbs1p | 0.13 | 0 | 13 | 3.77 | 6.2 | Putative transcription factor of unknown function |
| Rof1p | 0.09 | 0 | 24 | 2.74 | 6.1 | Putative transcription factor containing a WOPR domain |
| Tog1p | 0.1 | 0 | 21 | 2.93 | 5.9 | Transcriptional activator of oleate genes; regulates genes involved in fatty acid utilization |
| Wtm2p | 0.1 | 0 | 17 | 2.98 | 5.1 | Transcriptional modulator; involved in regulation of meiosis, silencing, and expression of RNR genes; involved in response to replication stress |
| Rpi1p | 0.1 | 0 | 17 | 2.89 | 4.8 | Transcription factor, allelic differences between S288C and Sigma1278b; mediates fermentation stress tolerance by modulating cell wall integrity |
| Com2p | 0.09 | 0.0001 | 16 | 2.94 | 4.3 | Transcription factor; COM2 transcription is regulated by Haa1p, Sok2p and Zap1p transcriptional activators; C. albicans homolog (MNL1) plays a role in adaptation to stress |
| Sdd4p | 0.1 | 0.0002 | 12 | 2.93 | 3.5 | Putative transcription factor, induced in response to the DNA-damaging agent MMS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klim, J.; Zielenkiewicz, U.; Kurlandzka, A.; Kaczanowski, S.; Skoneczny, M. Slow Adaptive Response of Budding Yeast Cells to Stable Conditions of Continuous Culture Can Occur without Genome Modifications. Genes 2020, 11, 1419. https://doi.org/10.3390/genes11121419
Klim J, Zielenkiewicz U, Kurlandzka A, Kaczanowski S, Skoneczny M. Slow Adaptive Response of Budding Yeast Cells to Stable Conditions of Continuous Culture Can Occur without Genome Modifications. Genes. 2020; 11(12):1419. https://doi.org/10.3390/genes11121419
Chicago/Turabian StyleKlim, Joanna, Urszula Zielenkiewicz, Anna Kurlandzka, Szymon Kaczanowski, and Marek Skoneczny. 2020. "Slow Adaptive Response of Budding Yeast Cells to Stable Conditions of Continuous Culture Can Occur without Genome Modifications" Genes 11, no. 12: 1419. https://doi.org/10.3390/genes11121419
APA StyleKlim, J., Zielenkiewicz, U., Kurlandzka, A., Kaczanowski, S., & Skoneczny, M. (2020). Slow Adaptive Response of Budding Yeast Cells to Stable Conditions of Continuous Culture Can Occur without Genome Modifications. Genes, 11(12), 1419. https://doi.org/10.3390/genes11121419