Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries



Abstract
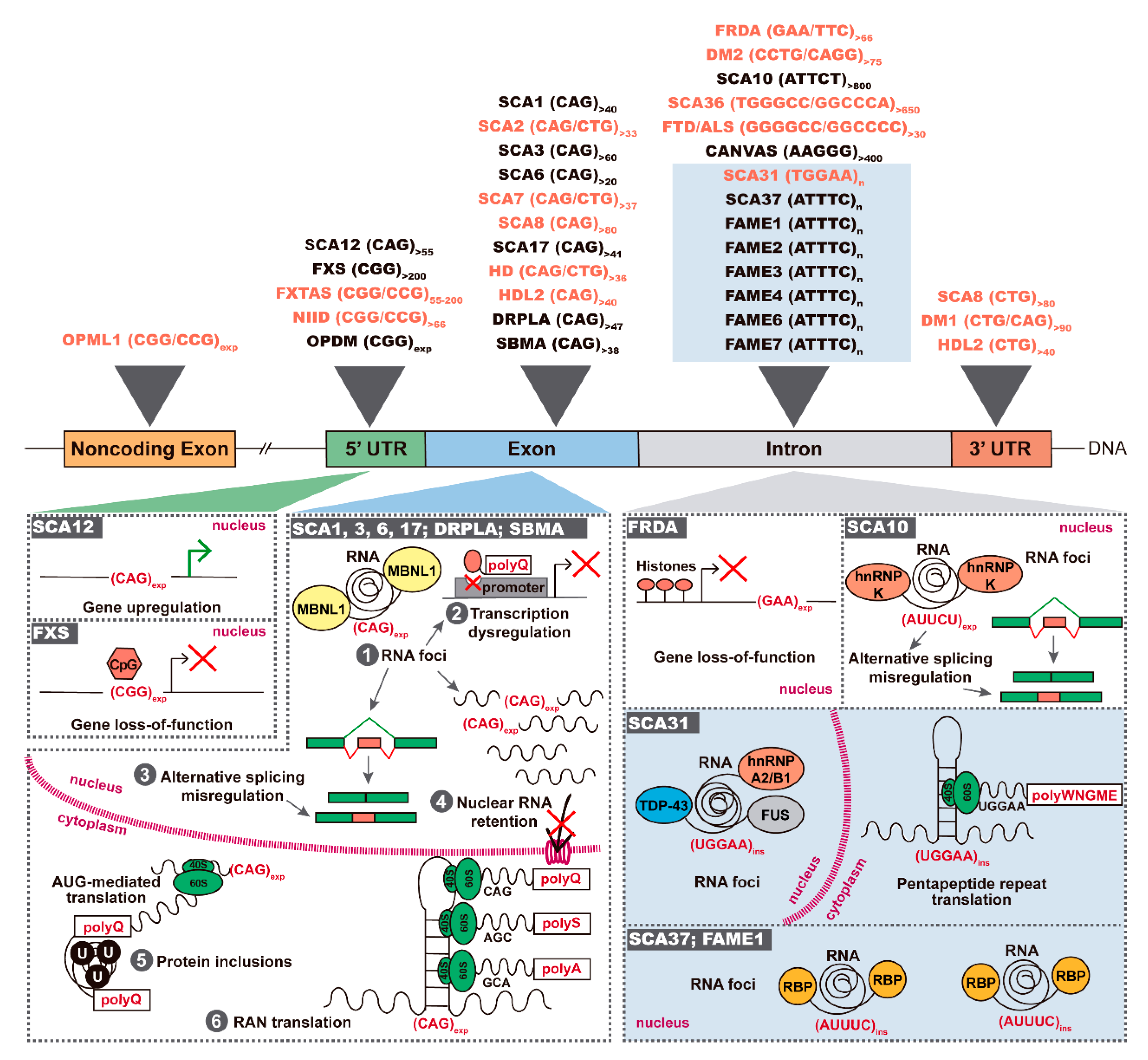
1. Introduction
2. Antisense Expression of Trinucleotide and Pentanucleotide Repeats in Spinocerebellar Ataxias
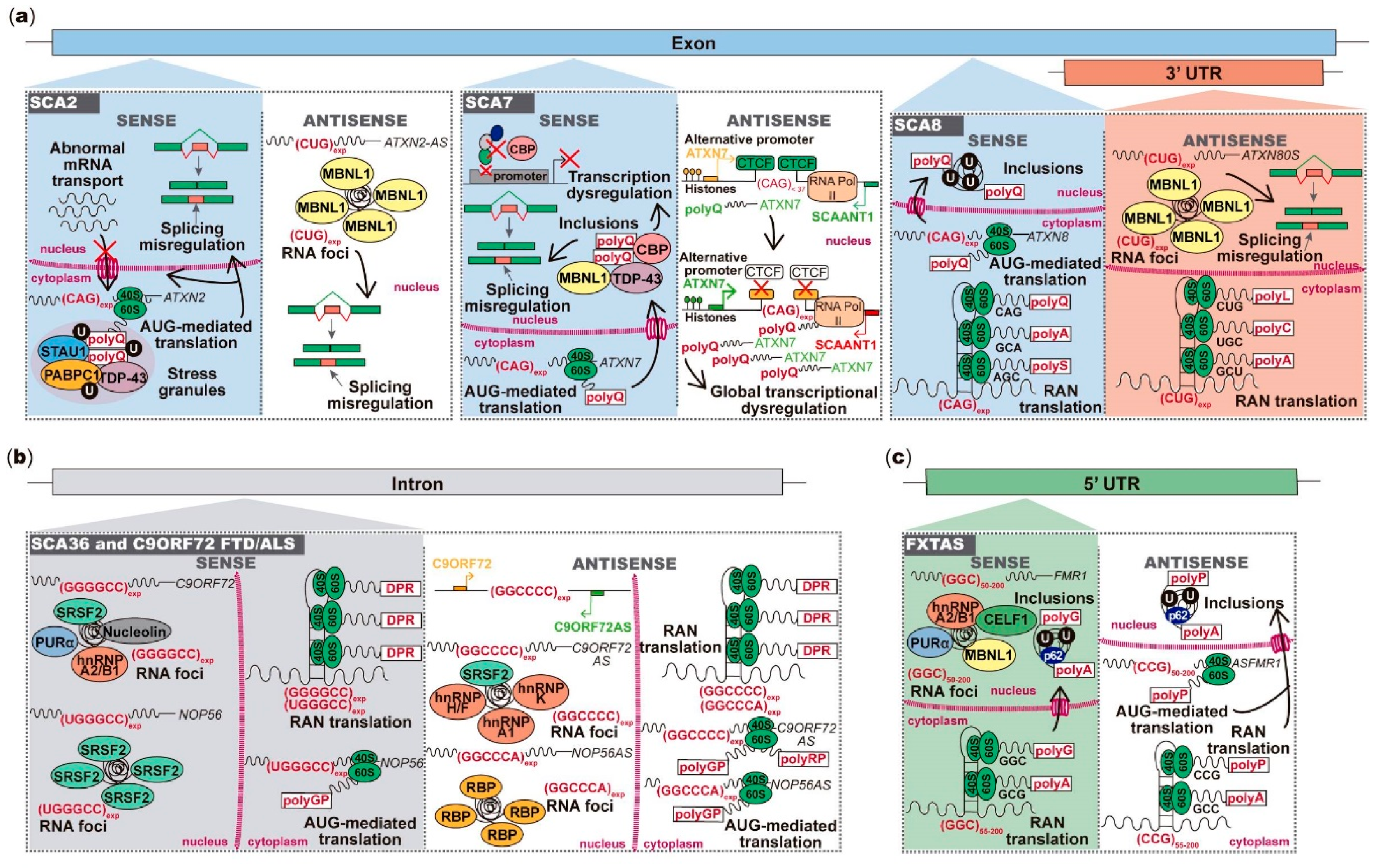
2.1. ATXN2 Sense and Antisense Trinucleotide Repeats in SCA2
2.2. ATXN7 Sense and Antisense Repeat Expression in SCA7
2.3. Bidirectional Transcription across the Repeat Region in SCA8
2.4. Pentanucleotide Repeats in Introns of Genes Transcribed from Opposite Strands in SCA31
3. FXN Antisense Transcript in Friedreich Ataxia
4. Antisense Expression in Hexanucleotide Repeat Expansion Diseases C9ORF72 FTD/ALS and SCA36
5. Antisense Expression in Neuronal CGG Repeat Diseases
5.1. Antisense Transcript Spanning FMR1 Repeat Region in FXTAS
5.2. NOTCH2NLC Repeat Expansions in NIID and Expanded Noncoding RNA LOC642361 in OPML1
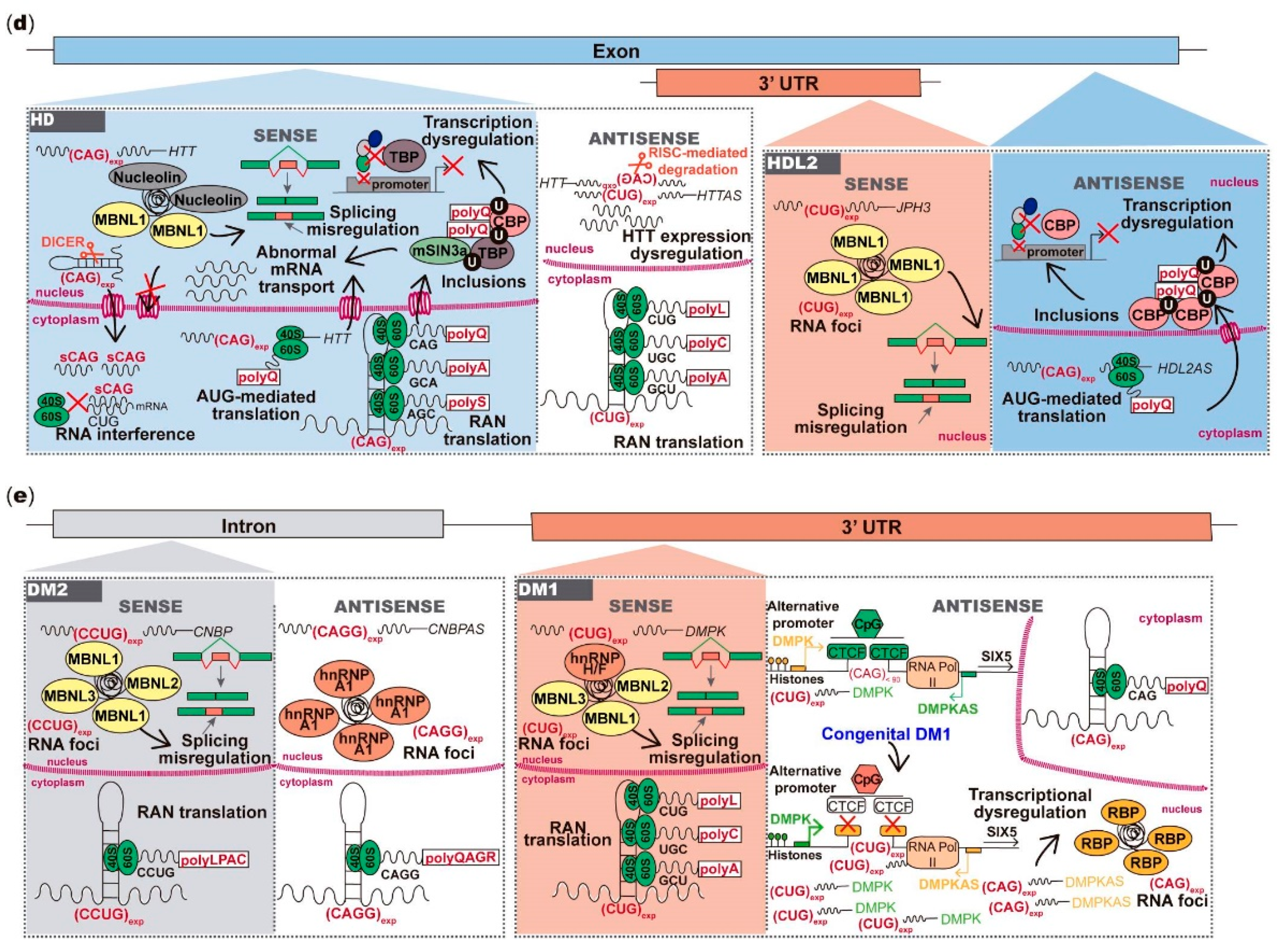
6. Antisense Repeat Expression in HD and HDL2
7. Antisense Repeat Expression in DM1 and DM2
8. Therapeutic Strategies Targeting Toxic RNAs
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Loureiro, J.R.; Oliveira, C.L.; Silveira, I. Unstable repeat expansions in neurodegenerative diseases: Nucleocytoplasmic transport emerges on the scene. Neurobiol. Aging 2016, 39, 174–183. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Robberecht, W.; Bosch, L.V.D. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2020, 39, e101112. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG–initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2010, 108, 260–265. [Google Scholar] [CrossRef]
- Zu, T.; Cleary, J.D.; Liu, Y.; Bañez-Coronel, M.; Bubenik, J.L.; Ayhan, F.; Ashizawa, T.; Xia, G.; Clark, H.B.; Yachnis, A.T.; et al. RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron 2017, 95, 1292–1305.e5. [Google Scholar] [CrossRef]
- Nguyen, L.; Cleary, J.D.; Ranum, L.P. Repeat-Associated Non-ATG Translation: Molecular Mechanisms and Contribution to Neurological Disease. Annu. Rev. Neurosci. 2019, 42, 227–247. [Google Scholar] [CrossRef]
- Cho, D.H.; Thienes, C.P.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense Transcription and Heterochromatin at the DM1 CTG Repeats Are Constrained by CTCF. Mol. Cell 2005, 20, 483–489. [Google Scholar] [CrossRef]
- Moseley, M.L.; Zu, T.; Ikeda, Y.; Gao, W.; Mosemiller, A.K.; Daughters, R.S.; Chen, G.; Weatherspoon, M.R.; Clark, H.B.; Ebner, T.J.; et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 2006, 38, 758–769. [Google Scholar] [CrossRef]
- Margolis, R.L.; Sun, X.; Xia, G.; Arbez, N.; Paul, S.; Zhu, S.; Peng, H.B.; Ross, C.A.; Koeppen, A.H.; Margolis, R.L.; et al. ATXN2-AS, a gene antisense toATXN2, is associated with spinocerebellar ataxia type 2 and amyotrophic lateral sclerosis. Ann. Neurol. 2016, 80, 600–615. [Google Scholar] [CrossRef]
- Sopher, B.L.; Ladd, P.D.; Pineda, V.V.; Libby, R.T.; Sunkin, S.M.; Hurley, J.B.; Thienes, C.P.; Gaasterland, T.; Filippova, G.N.; La Spada, A.R. CTCF Regulates Ataxin-7 Expression through Promotion of a Convergently Transcribed, Antisense Noncoding RNA. Neuron 2011, 70, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Amino, T.; Kobayashi, K.; Asakawa, S.; Ishiguro, T.; Tsunemi, T.; Takahashi, M.; Matsuura, T.; Flanigan, K.M.; Iwasaki, S.; et al. Spinocerebellar Ataxia Type 31 Is Associated with “Inserted” Penta-Nucleotide Repeats Containing (TGGAA)n. Am. J. Hum. Genet. 2009, 85, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Todd, T.W.; McEachin, Z.T.; Chew, J.; Burch, A.R.; Jansen-West, K.; Tong, J.; Yue, M.; Song, Y.; Castanedes-Casey, M.; Kurti, A.; et al. Hexanucleotide Repeat Expansions in c9FTD/ALS and SCA36 Confer Selective Patterns of Neurodegeneration In Vivo. Cell Rep. 2020, 31, 107616. [Google Scholar] [CrossRef] [PubMed]
- McEachin, Z.T.; Gendron, T.F.; Raj, N.; García-Murias, M.; Banerjee, A.; Purcell, R.H.; Ward, P.J.; Todd, T.W.; Merritt-Garza, M.E.; Jansen-West, K.; et al. Chimeric Peptide Species Contribute to Divergent Dipeptide Repeat Pathology in c9ALS/FTD and SCA36. Neuron 2020, 107, 292–305.e6. [Google Scholar] [CrossRef]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.-M.; Schludi, M.H.; Van Der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef]
- Chung, D.W.; Rudnicki, D.D.; Yu, L.; Margolis, R.L. A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum. Mol. Genet. 2011, 20, 3467–3477. [Google Scholar] [CrossRef]
- Seixas, A.I.; Holmes, S.E.; Takeshima, H.; Pavlovich, A.; Sachs, N.; Pruitt, J.L.; Silveira, I.; Ross, C.A.; Margolis, R.L.; Rudnicki, D.D. Loss of junctophilin-3 contributes to huntington disease-like 2 pathogenesis. Ann. Neurol. 2012, 71, 245–257. [Google Scholar] [CrossRef]
- Mikaeili, H.; Sandi, M.; Bayot, A.; Al-Mahdawi, S.; Pook, M.A. FAST-1 antisense RNA epigenetically alters FXN expression. Sci. Rep. 2018, 8, 17217. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, J.-L.; Huang, W.; Zeng, S.; Jiao, B.; Liu, Z.; Chen, Z.; Li, Y.; Wang, Y.; Min, H.-X.; et al. Expansion of Human-Specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am. J. Hum. Genet. 2019, 105, 166–176. [Google Scholar] [CrossRef]
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019, 51, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.; Ruano, L.; Loureiro, J.L.; Cruz, V.T.; Barros, J.; Tuna, A.; Barbot, C.; Guimarães, J.; Alonso, I.; Silveira, I.; et al. Hereditary Ataxia and Spastic Paraplegia in Portugal: A population-based prevalence study. JAMA Neurol. 2013, 70, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Sequeiros, J.; Martins, S.; Silveira, I. Epidemiology and population genetics of degenerative ataxias. Stroke 2012, 103, 227–251. [Google Scholar] [CrossRef]
- Seixas, A.I.; Loureiro, J.R.; Costa, C.; Ordóñez-Ugalde, A.; Marcelino, H.; Oliveira, C.L.; Loureiro, J.L.; Dhingra, A.; Brandão, E.; Cruz, V.T.; et al. A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet. 2017, 101, 87–103. [Google Scholar] [CrossRef]
- Geschwind, D.H.; Perlman, S.; Figueroa, C.P.; Treiman, L.J.; Pulst, S.M. The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am. J. Hum. Genet. 1997, 60, 842–850. [Google Scholar] [PubMed]
- Cancel, G.; Dürr, A.; Didierjean, O.; Imbert, G.; Bürk, K.; Lezin, A.; Belal, S.; Benomar, A.; Abada-Bendib, M.; Vial, C.; et al. Molecular and Clinical Correlations in Spinocerebellar Ataxia 2: A Study of 32 Families. Hum. Mol. Genet. 1997, 6, 709–715. [Google Scholar] [CrossRef]
- Lopes-Cendesi, I.; Teive, H.G.; Calcagnotto, M.E.; Da Costa, J.C.; Cardoso, F.; Viana, E.; Maciel, J.A.; Radvany, J.; Arruda, W.O.; Trevisol-Bittencourt, P.C.; et al. Frequency of the different mutations causing spinocerebellar ataxia (SCA1, SCA2, MJD/SCA3 and DRPLA) in a large group of Brazilian patients. Arq. Neuro Psiquiatr. 1997, 55, 519–529. [Google Scholar] [CrossRef]
- Silveira, I.; Miranda, C.; Guimarães, L.; Moreira, M.-C.; Alonso, I.; Mendonca, P.; Ferro, A.; Pinto-Basto, J.; Coelho, J.; Ferreirinha, F.; et al. Trinucleotide Repeats in 202 Families With Ataxia: A small expanded (CAG)n allele at the SCA17 locus. Arch. Neurol. 2002, 59, 623–629. [Google Scholar] [CrossRef]
- Pulst, S.M. Spinocerebellar Ataxia Type 2. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2020. [Google Scholar]
- Pulst, S.M.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.-N.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef]
- Ramos, E.M.; Martins, S.; Alonso, I.; Emmel, V.E.; Saraiva-Pereira, M.L.; Jardim, L.B.; Coutinho, P.; Sequeiros, J.; Silveira, I. Common origin of pure and interrupted repeat expansions in spinocerebellar ataxia type 2 (SCA2). Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2009, 9999, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Nechiporuk, T.; Huynh, D.P.; Figueroa, K.; Sahba, S.; Nechiporuk, A.; Pulst, S.M. The mouse SCA2 gene: cDNA sequence, alternative splicing and protein expression. Hum. Mol. Genet. 1998, 7, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Huynh, D.P.; Del Bigio, M.R.; Ho, D.H.; Pulst, S.-M. Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer’s disease and spinocerebellar ataxia 2. Ann. Neurol. 1999, 45, 232–241. [Google Scholar] [CrossRef]
- Kiehl, T.-R.; Nechiporuk, A.; Figueroa, K.P.; Keating, M.T.; Huynh, D.P.; Pulst, S.-M. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem. Biophys. Res. Commun. 2006, 339, 17–24. [Google Scholar] [CrossRef]
- Huynh, D.P.; Figueroa, K.P.; Hoang, N.; Pulst, S.-M. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 2000, 26, 44–50. [Google Scholar] [CrossRef]
- Hansen, S.T.; Meera, P.; Otis, T.S.; Pulst, S.M. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 2013, 22, 271–283. [Google Scholar] [CrossRef]
- Van De Loo, S.; Eich, F.; Nonis, D.; Auburger, G.; Nowock, J. Ataxin-2 associates with rough endoplasmic reticulum. Exp. Neurol. 2009, 215, 110–118. [Google Scholar] [CrossRef]
- Huynh, D.P.; Yang, H.-T.; Vakharia, H.; Nguyen, D.; Pulst, S.M. Expansion of the polyQ repeat in ataxin-2 alters its Golgi localization, disrupts the Golgi complex and causes cell death. Hum. Mol. Genet. 2003, 12, 1485–1496. [Google Scholar] [CrossRef]
- Damrath, E.; Heck, M.V.; Gispert, S.; Azizov, M.; Nowock, J.; Seifried, C.; Rüb, U.; Walter, M.; Auburger, G. ATXN2-CAG42 Sequesters PABPC1 into Insolubility and Induces FBXW8 in Cerebellum of Old Ataxic Knock-In Mice. PLoS Genet. 2012, 8, e1002920. [Google Scholar] [CrossRef]
- Yokoshi, M.; Li, Q.; Yamamoto, M.; Okada, H.; Suzuki, Y.; Kawahara, Y. Direct Binding of Ataxin-2 to Distinct Elements in 3′ UTRs Promotes mRNA Stability and Protein Expression. Mol. Cell 2014, 55, 186–198. [Google Scholar] [CrossRef]
- Dansithong, W.; Paul, S.; Figueroa, K.P.; Rinehart, M.D.; Wiest, S.; Pflieger, L.T.; Scoles, D.R.; Pulst, S.M. Ataxin-2 Regulates RGS8 Translation in a New BAC-SCA2 Transgenic Mouse Model. PLoS Genet. 2015, 11, e1005182. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Dansithong, W.; Figueroa, K.P.; Scoles, D.R.; Pulst, S.M. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Huynh, D.P.; Pulst, S.-M. A novel protein with RNA-binding motifs interacts with ataxin-2. Hum. Mol. Genet. 2000, 9, 1303–1313. [Google Scholar] [CrossRef]
- Nonhoff, U.; Ralser, M.; Welzel, F.; Piccini, I.; Balzereit, D.; Yaspo, M.-L.; Lehrach, H.; Krobitsch, S. Ataxin-2 Interacts with the DEAD/H-Box RNA Helicase DDX6 and Interferes with P-Bodies and Stress Granules. Mol. Biol. Cell 2007, 18, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nat. Cell Biol. 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, K.; Savontaus, M.L.; Stevanin, G.; Holmberg, M.; Digre, K.; Zander, C.; Ehrsson, H.; David, G.; Benomar, A.; Nikoskelainen, E.; et al. An expanded CAG repeat sequence in spinocerebellar ataxia type 7. Genome Res. 1996, 6, 965–971. [Google Scholar] [CrossRef][Green Version]
- David, G.; Abbas, N.; Stevanin, G.; Dürr, A.; Yvert, G.; Cancel, G.; Weber, C.; Imbert, G.; Saudou, F.; Antoniou, E.; et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997, 17, 65–70. [Google Scholar] [CrossRef]
- Martin, J.-J.; Krols, L.; Ceuterick, C.; Van Broeckhoven, C.; Van Regemorter, N.; Hayer-Delatte, F.; Brucher, J.-M.; De Barsy, T.; Szliwowski, H.; Evrard, P.; et al. On an autosomal dominant form of retinal-cerebellar degeneration: An autopsy study of five patients in one family. Acta Neuropathol. 1994, 88, 277–286. [Google Scholar] [CrossRef]
- Yvert, G.; Lindenberg, K.S.; Picaud, S.; Landwehrmeyer, G.B.; Sahel, J.-A.; Mandel, J.-L. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum. Mol. Genet. 2000, 9, 2491–2506. [Google Scholar] [CrossRef]
- La Spada, A.R.; Fu, Y.-H.; Sopher, B.L.; Libby, R.T.; Wang, X.; Li, L.Y.; Einum, D.D.; Huang, J.; Possin, D.E.; Smith, A.C.; et al. Polyglutamine-Expanded Ataxin-7 Antagonizes CRX Function and Induces Cone-Rod Dystrophy in a Mouse Model of SCA7. Neuron 2001, 31, 913–927. [Google Scholar] [CrossRef]
- Garden, G.A.; Libby, R.T.; Fu, Y.-H.; Kinoshita, Y.; Huang, J.; Possin, D.E.; Smith, A.C.; Martinez, R.A.; Fine, G.C.; Grote, S.K.; et al. Polyglutamine-Expanded Ataxin-7 Promotes Non-Cell-Autonomous Purkinje Cell Degeneration and Displays Proteolytic Cleavage in Ataxic Transgenic Mice. J. Neurosci. 2002, 22, 4897–4905. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, D.; Hardy, S.; Abou-Sleymane, G.; Eberlin, A.; Bowman, A.B.; Gansmuller, A.; Picaud, S.; Zoghbi, H.Y.; Trottier, Y.; Tora, L.; et al. Glutamine-Expanded Ataxin-7 Alters TFTC/STAGA Recruitment and Chromatin Structure Leading to Photoreceptor Dysfunction. PLoS Biol. 2006, 4, e67. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-Y.; Pennesi, M.E.; Weeber, E.J.; Xu, B.; Atkinson, R.; Chen, S.; Armstrong, D.L.; Wu, S.M.; Sweatt, J.; Zoghbi, H.Y. SCA7 Knockin Mice Model Human SCA7 and Reveal Gradual Accumulation of Mutant Ataxin-7 in Neurons and Abnormalities in Short-Term Plasticity. Neuron 2003, 37, 383–401. [Google Scholar] [CrossRef]
- Helmlinger, D.; Hardy, S.; Sasorith, S.; Klein, F.; Robert, F.; Weber, C.; Miguet, L.; Potier, N.; Van-Dorsselaer, A.; Wurtz, J.M.; et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum. Mol. Genet. 2004, 13, 1257–1265. [Google Scholar] [CrossRef]
- Martinez, E.; Palhan, V.B.; Tjernberg, A.; Lymar, E.S.; Gamper, A.M.; Kundu, T.K.; Chait, B.T.; Roeder, R.G. Human STAGA Complex Is a Chromatin-Acetylating Transcription Coactivator That Interacts with Pre-mRNA Splicing and DNA Damage-Binding Factors In Vivo. Mol. Cell. Biol. 2001, 21, 6782–6795. [Google Scholar] [CrossRef]
- Palhan, V.B.; Chen, S.; Peng, G.-H.; Tjernberg, A.; Gamper, A.M.; Fan, Y.; Chait, B.T.; La Spada, A.R.; Roeder, R.G. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 8472–8477. [Google Scholar] [CrossRef]
- Chen, S.; Peng, G.-H.; Wang, X.; Smith, A.C.; Grote, S.K.; Sopher, B.L.; La Spada, A.R. Interference of Crx-dependent transcription by ataxin-7 involves interaction between the glutamine regions and requires the ataxin-7 carboxy-terminal region for nuclear localization. Hum. Mol. Genet. 2003, 13, 53–67. [Google Scholar] [CrossRef][Green Version]
- Alves, S.; Marais, T.; Biferi, M.-G.; Furling, D.; Marinello, M.; El Hachimi, K.; Cartier, N.; Ruberg, M.; Stevanin, G.; Brice, A.; et al. Lentiviral vector-mediated overexpression of mutant ataxin-7 recapitulates SCA7 pathology and promotes accumulation of the FUS/TLS and MBNL1 RNA-binding proteins. Mol. Neurodegener. 2016, 11, 58. [Google Scholar] [CrossRef]
- McCullough, S.D.; Xu, X.; Dent, S.Y.R.; Bekiranov, S.; Roeder, R.G.; Grant, P.A. Reelin is a target of polyglutamine expanded ataxin-7 in human spinocerebellar ataxia type 7 (SCA7) astrocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 21319–21324. [Google Scholar] [CrossRef]
- Yanicostas, C.; Barbieri, E.; Hibi, M.; Brice, A.; Stevanin, G.; Soussi-Yanicostas, N. Requirement for Zebrafish Ataxin-7 in Differentiation of Photoreceptors and Cerebellar Neurons. PLoS ONE 2012, 7, e50705. [Google Scholar] [CrossRef]
- Libby, R.T.; Hagerman, K.A.; Pineda, V.V.; Lau, R.; Cho, D.H.; Baccam, S.L.; Axford, M.M.; Cleary, J.D.; Moore, J.M.; Sopher, B.L.; et al. CTCF cis-Regulates Trinucleotide Repeat Instability in an Epigenetic Manner: A Novel Basis for Mutational Hot Spot Determination. PLoS Genet. 2008, 4, e1000257. [Google Scholar] [CrossRef] [PubMed]
- Koob, M.D.; Moseley, M.L.; Schut, L.J.; Benzow, K.A.; Bird, T.D.; Day, J.W.; Ranum, L.P. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat. Genet. 1999, 21, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Shizuka, M.; Watanabe, M.; Okamoto, K.; Shoji, M. Molecular and clinical analyses of spinocerebellar ataxia type 8 in Japan. Neurology 2000, 54, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Moseley, M.L.; Schut, L.J.; Bird, T.D.; Koob, M.D.; Day, J.W.; Ranum, L.P.W. SCA8 CTG repeat: En masse contractions in sperm and intergenerational sequence changes may play a role in reduced penetrance. Hum. Mol. Genet. 2000, 9, 2125–2130. [Google Scholar] [CrossRef]
- Day, J.W.; Schut, L.J.; Moseley, M.L.; Durand, A.C.; Ranum, L.P.W. Spinocerebellar ataxia type 8: Clinical features in a large family. Neurology 2000, 55, 649–657. [Google Scholar] [CrossRef]
- Mutsuddi, M.; Marshall, C.M.; Benzow, K.A.; Koob, M.D.; Rebay, I. The Spinocerebellar Ataxia 8 Noncoding RNA Causes Neurodegeneration and Associates with Staufen in Drosophila. Curr. Biol. 2004, 14, 302–308. [Google Scholar] [CrossRef]
- Daughters, R.S.; Tuttle, D.L.; Gao, W.; Ikeda, Y.; Moseley, M.L.; Ebner, T.J.; Swanson, M.S.; Ranum, L.P.W. RNA Gain-of-Function in Spinocerebellar Ataxia Type 8. PLoS Genet. 2009, 5, e1000600. [Google Scholar] [CrossRef]
- Ayhan, F.; Perez, B.A.; Shorrock, H.K.; Zu, T.; Banez-Coronel, M.; Reid, T.; Furuya, H.; Clark, H.B.; Troncoso, J.C.; Ross, C.A.; et al. SCA 8 RAN polySer protein preferentially accumulates in white matter regions and is regulated by eIF 3F. EMBO J. 2018, 37, e99023. [Google Scholar] [CrossRef]
- Nemes, J.P.; Benzow, K.A.; Koob, M.D. The SCA8 transcript is an antisense RNA to a brain-specific transcript encoding a novel actin-binding protein (KLHL1). Hum. Mol. Genet. 2000, 9, 1543–1551. [Google Scholar] [CrossRef]
- Benzow, K.A.; Koob, M.D. The transcript () is evolutionarily conserved. Mamm. Genome 2002, 13, 134–141. [Google Scholar] [CrossRef]
- He, Y.; Zu, T.; Benzow, K.A.; Orr, H.T.; Clark, H.B.; Koob, M.D. Targeted Deletion of a Single Sca8 Ataxia Locus Allele in Mice Causes Abnormal Gait, Progressive Loss of Motor Coordination, and Purkinje Cell Dendritic Deficits. J. Neurosci. 2006, 26, 9975–9982. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Daughters, R.S.; Ranum, L.P.W. Bidirectional expression of the SCA8 expansion mutation: One mutation, two genes. Cerebellum 2008, 7, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Niimi, Y.; Takahashi, M.; Sugawara, E.; Umeda, S.; Obayashi, M.; Sato, N.; Ishiguro, T.; Higashi, M.; Eishi, Y.; Mizusawa, H.; et al. Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)nin the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology 2013, 33, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Sato, N.; Ueyama, M.; Fujikake, N.; Sellier, C.; Kanegami, A.; Tokuda, E.; Zamiri, B.; Gall-Duncan, T.; Mirceta, M.; et al. Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron 2017, 94, 108–124.e7. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Nagai, Y. Molecular Mechanisms and Future Therapeutics for Spinocerebellar Ataxia Type 31 (SCA31). Neurother. 2019, 16, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Bidichandani, S.I.; Delatycki, M.B. Friedreich Ataxia. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2020. [Google Scholar]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Naeije, G.; Rai, M.; Allaerts, N.; Sjogard, M.; De Tiège, X.; Pandolfo, M. Cerebellar cognitive disorder parallels cerebellar motor symptoms in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2020, 7, 1050–1054. [Google Scholar] [CrossRef]
- Saveliev, A.; Everett, C.; Sharpe, T.; Webster, Z.; Festenstein, R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nat. Cell Biol. 2003, 422, 909–913. [Google Scholar] [CrossRef]
- Greene, E.; Mahishi, L.; Entezam, A.; Kumari, D.; Usdin, K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 2007, 35, 3383–3390. [Google Scholar] [CrossRef]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef]
- Al-Mahdawi, S.; Pinto, R.M.; Ismail, O.; Varshney, D.; Lymperi, S.; Sandi, C.; Trabzuni, D.; Pook, M.A. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 2007, 17, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Chutake, Y.K.; Costello, W.N.; Lam, C.; Bidichandani, S.I. Altered nucleosome positioning at the transcription start site and deficient transcriptional initiation in Friedreich ataxia. J. Biol. Chem. 2014, 289, 15194–15202. [Google Scholar] [CrossRef] [PubMed]
- Rötig, A.; De Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Singh, A.; Crooks, D.R.; Dai, X.; Cong, Z.; Pan, L.; Ha, D.; Rouault, T.A. Expression of Human Frataxin Is Regulated by Transcription Factors SRF and TFAP2. PLoS ONE 2010, 5, e12286. [Google Scholar] [CrossRef] [PubMed]
- De Biase, I.; Chutake, Y.K.; Rindler, P.M.; Bidichandani, S.I. Epigenetic Silencing in Friedreich Ataxia Is Associated with Depletion of CTCF (CCCTC-Binding Factor) and Antisense Transcription. PLoS ONE 2009, 4, e7914. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; Van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; MacKenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Kobayashi, H.; Abe, K.; Matsuura, T.; Ikeda, Y.; Hitomi, T.; Akechi, Y.; Habu, T.; Liu, W.; Okuda, H.; Koizumi, A. Expansion of Intronic GGCCTG Hexanucleotide Repeat in NOP56 Causes SCA36, a Type of Spinocerebellar Ataxia Accompanied by Motor Neuron Involvement. Am. J. Hum. Genet. 2011, 89, 121–130. [Google Scholar] [CrossRef]
- García-Murias, M.; Quintáns, B.; Arias, M.; Seixas, A.I.; Cacheiro, P.; Tarrío, R.; Pardo, J.; Millán, M.J.; Arias-Rivas, S.; Blanco-Arias, P.; et al. ‘Costa da Morte’ ataxia is spinocerebellar ataxia 36: Clinical and genetic characterization. Brain 2012, 135, 1423–1435. [Google Scholar] [CrossRef]
- Liu, W.; Ikeda, Y.; Hishikawa, N.; Yamashita, T.; Deguchi, K.; Abe, K. Characteristic RNA foci of the abnormal hexanucleotide GGCCUG repeat expansion in spinocerebellar ataxia type 36 (Asidan). Eur. J. Neurol. 2014, 21, 1377–1386. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Highley, J.R.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. Deletion of C9ORF72 Results in Motor Neuron Degeneration and Stress Sensitivity in C. elegans. PLoS ONE 2013, 8, e83450. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Rizzu, P.; Blauwendraat, C.; Heetveld, S.; Lynes, E.M.; Castillo-Lizardo, M.; Dhingra, A.; Pyz, E.; Hobert, M.; Synofzik, M.; Simón-Sánchez, J.; et al. C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol. Commun. 2016, 4, 37. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.-J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef]
- Matsuzono, K.; Imamura, K.; Murakami, N.; Tsukita, K.; Yamamoto, T.; Izumi, Y.; Kaji, R.; Ohta, Y.; Yamashita, T.; Abe, K.; et al. Antisense Oligonucleotides Reduce RNA Foci in Spinocerebellar Ataxia 36 Patient iPSCs. Mol. Ther. Nucleic Acids 2017, 8, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Moens, T.G.; Mizielinska, S.; Niccoli, T.; Mitchell, J.S.; Thoeng, A.; Ridler, C.E.; Grönke, S.; Esser, J.; Heslegrave, A.; Zetterberg, H.; et al. Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathol. 2018, 135, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Bento-Abreu, A.; Gendron, T.F.; Boeynaems, S.; Bogaert, E.; Nuyts, R.; Timmers, M.; Scheveneels, W.; Hersmus, N.; Wang, J.; et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018, 135, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Gartler, S.M.; Scott, C.; Chen, S.-H.; Xlaird, C.M. Methylation analysis of CGG sites in the CpG island of the human FMR1 gene. Hum. Mol. Genet. 1992, 1, 571–578. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.; Fu, Y.-H.; Warren, S.T.; Oostra, B.A.; Caskey, C.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Coffee, B.; Zhang, F.; Warren, S.T.; Reines, D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999, 22, 98–101. [Google Scholar] [CrossRef]
- Coffee, B.; Zhang, F.; Ceman, S.; Warren, S.T.; Reines, D. Histone Modifications Depict an Aberrantly Heterochromatinized FMR1 Gene in Fragile X Syndrome. Am. J. Hum. Genet. 2002, 71, 923–932. [Google Scholar] [CrossRef]
- Tassone, F.; Beilina, A.; Carosi, C.; Albertosi, S.; Bagni, C.; Li, L.; Glover, K.; Bentley, D.; Hagerman, P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA 2007, 13, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated Levels of FMR1 mRNA in Carrier Males: A New Mechanism of Involvement in the Fragile-X Syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Zhang, F.; Hagedorn, C.H.; Warren, S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 2001, 10, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.M.; Hagerman, R.J.; Tassone, F.; Chudley, A.E.; Del Bigio, M.R.; Jacquemont, S.; Leehey, M. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, 125, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Iwahashi, C.; Hagerman, P.J. FMR1 RNA within the Intranuclear Inclusions of Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). RNA Biol. 2004, 1, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, C.K.; Yasui, D.H.; An, H.-J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, P. Protein composition of the intranuclear inclusions of FXTAS. Brain 2005, 129, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Bontekoe, C.J.M.; Bakker, C.E.; Nieuwenhuizen, I.M.; Van Der Linde, H.; Lans, H.; De Lange, D.; Hirst, M.C.; Oostra, B.A. Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum. Mol. Genet. 2001, 10, 1693–1699. [Google Scholar] [CrossRef]
- Willemsen, R.; Hoogeveen-Westerveld, M.; Reis, S.; Holstege, J.; Severijnen, L.-A.W.; Nieuwenhuizen, I.M.; Schrier, M.; Van Unen, L.; Tassone, F.; Hoogeveen, A.T.; et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum. Mol. Genet. 2003, 12, 949–959. [Google Scholar] [CrossRef]
- Hashem, V.; Galloway, J.N.; Mori, M.; Willemsen, R.; Oostra, B.A.; Paylor, R.; Nelson, D.L. Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum. Mol. Genet. 2009, 18, 2443–2451. [Google Scholar] [CrossRef]
- Jin, P.; Zarnescu, D.C.; Zhang, F.; Pearson, C.E.; Lucchesi, J.C.; Moses, K.; Warren, S.T. RNA-Mediated Neurodegeneration Caused by the Fragile X Premutation rCGG Repeats in Drosophila. Neuron 2003, 39, 739–747. [Google Scholar] [CrossRef]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; De Haro, M.; Nelson, D.L.; Botas, J. RNA-Binding Proteins hnRNP A2/B1 and CUGBP1 Suppress Fragile X CGG Premutation Repeat-Induced Neurodegeneration in a Drosophila Model of FXTAS. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Duan, R.; Qurashi, A.; Qin, Y.; Tian, D.; Rosser, T.C.; Liu, H.; Feng, Y.; Warren, S.T. Pur α Binds to rCGG Repeats and Modulates Repeat-Mediated Neurodegeneration in a Drosophila Model of Fragile X Tremor/Ataxia Syndrome. Neuron 2007, 55, 556–564. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Krzyzosiak, W.J. Cellular toxicity of expanded RNA repeats: Focus on RNA foci. Hum. Mol. Genet. 2011, 20, 3811–3821. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.-C.; Scaglione, K.M.; Basrur, V.; et al. CGG Repeat-Associated Translation Mediates Neurodegeneration in Fragile X Tremor Ataxia Syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Krans, A.; Skariah, G.; Zhang, Y.; Bayly, B.; Todd, P.K. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2019, 7, 152. [Google Scholar] [CrossRef]
- Buijsen, R.A.M.; Sellier, C.; Severijnen, L.-A.W.F.M.; Oulad-Abdelghani, M.; Verhagen, R.F.M.; Berman, R.F.; Charlet-Berguerand, N.; Willemsen, R.; Hukema, R.K. FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2014, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sofola, O.A.; Jin, P.; Botas, J.; Nelson, D.L. Argonaute-2-dependent rescue of a Drosophila model of FXTAS by FRAXE premutation repeat. Hum. Mol. Genet. 2007, 16, 2326–2332. [Google Scholar] [CrossRef]
- Krans, A.; Kearse, M.G.; Todd, P.K. Repeat-associated non-AUG translation from antisense CCG repeats in fragile X tremor/ataxia syndrome. Ann. Neurol. 2016, 80, 871–881. [Google Scholar] [CrossRef]
- Sone, J.; Mori, K.; Inagaki, T.; Katsumata, R.; Takagi, S.; Yokoi, S.; Araki, K.; Kato, T.; Nakamura, T.; Koike, H.; et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain 2016, 139, 3170–3186. [Google Scholar] [CrossRef]
- Fiddes, I.T.; Lodewijk, G.A.; Mooring, M.; Bosworth, C.M.; Ewing, A.D.; Mantalas, G.L.; Novak, A.M.; Bout, A.V.D.; Bishara, A.; Rosenkrantz, J.L.; et al. Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell 2018, 173, 1356–1369.e22. [Google Scholar] [CrossRef]
- Suzuki, I.K.; Gacquer, D.; Van Heurck, R.; Kumar, D.; Wojno, M.; Bilheu, A.; Herpoel, A.; Lambert, N.; Cheron, J.; Polleux, F.; et al. Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell 2018, 173, 1370–1384.e16. [Google Scholar] [CrossRef] [PubMed]
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019, 51, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, D.D.; Pletnikova, O.; Vonsattel, J.-P.G.; Ross, C.A.; Margolis, R.L. A Comparison of Huntington Disease and Huntington Disease-Like 2 Neuropathology. J. Neuropathol. Exp. Neurol. 2008, 67, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Margolis, R.L.; O’Hearn, E.; Rosenblatt, A.; Willour, V.; Holmes, S.E.; Franz, M.L.; Callahan, C.; Hwang, H.S.; Troncoso, J.C.; Ross, C.A. A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann. Neurol. 2001, 50, 373–380. [Google Scholar] [CrossRef]
- Holmes, S.E.; O’Hearn, E.; Rosenblatt, A.; Callahan, C.; Hwang, H.S.; Ingersoll-Ashworth, R.G.; Fleisher, A.; Stevanin, G.; Brice, A.; Potter, N.T.; et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease–like 2. Nat. Genet. 2001, 29, 377–378. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD Gene with an Expanded CAG Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of Neuronal Intranuclear Inclusions Underlies the Neurological Dysfunction in Mice Transgenic for the HD Mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Schilling, G.; Becher, M.W.; Sharp, A.H.; Jinnah, H.A.; Duan, K.; Kotzuk, J.A.; Slunt, H.H.; Ratovitski, T.; Cooper, J.K.; Jenkins, Z.W.N.G.C.N.A.; et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin [published erratum appears in Hum Mol Genet 1999 May;8(5):943]. Hum. Mol. Genet. 1999, 8, 397–407. [Google Scholar] [CrossRef]
- Cooper, J.K.; Schilling, G.; Peters, M.F.; Herring, W.J.; Sharp, A.H.; Kaminsky, Z.; Masone, J.; Khan, F.A.; Delanoy, M.; Borchelt, D.R.; et al. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum. Mol. Genet. 1998, 7, 783–790. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Becher, M.W.; Kotzuk, J.A.; Sharp, A.H.; Davies, S.W.; Bates, G.P.; Priceaef, D.L.; Rossbfg, C.A. Intranuclear Neuronal Inclusions in Huntington’s Disease and Dentatorubral and Pallidoluysian Atrophy: Correlation between the Density of Inclusions andIT15CAG Triplet Repeat Length. Neurobiol. Dis. 1998, 4, 387–397. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.-Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, N.C., Jr.; Sasaki, M.; Peters, M.F.; Huang, H.; Cooper, J.K.; Yamada, M.; Takahashi, H.; Tsuji, S.; Troncoso, J.; Dawson, V.L.; et al. Interference by Huntingtin and Atrophin-1 with CBP-Mediated Transcription Leading to Cellular Toxicity. Science 2001, 291, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Nucifora, F.C.; Ross, C.A.; DeFranco, D.B. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet. 2003, 12, 1–12. [Google Scholar] [CrossRef]
- Boutell, J.M.; Thomas, P.; Neal, J.W.; Weston, V.J.; Duce, J.A.; Harper, P.S.; Jones, A.L. Aberrant interactions of transcriptional repressor proteins with the Huntington’s disease gene product, huntingtin. Hum. Mol. Genet. 1999, 8, 1647–1655. [Google Scholar] [CrossRef]
- Huang, C.C.; Faber, P.W.; Persichetti, F.; Mittal, V.; Vonsattel, J.-P.; Macdonald, M.E.; Gusella, J.F. Amyloid formation by mutant huntingtin: Threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 1998, 24, 217–233. [Google Scholar] [CrossRef]
- De Mezer, M.; Wojciechowska, M.; Napierala, M.; Sobczak, K.; Krzyzosiak, W.J. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011, 39, 3852–3863. [Google Scholar] [CrossRef]
- Sun, X.; Li, P.P.; Zhu, S.; Cohen, R.E.; Marque, L.O.; Ross, C.A.; Pulst, S.M.; Chan, H.Y.E.; Margolis, R.L.; Rudnicki, D.D. Nuclear retention of full-length HTT RNA is mediated by splicing factors MBNL1 and U2AF65. Sci. Rep. 2015, 5, 12521. [Google Scholar] [CrossRef]
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.C.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 2017, 94, 93–107.e6. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Chan, H.Y.E. Expression of Expanded CAG Transcripts Triggers Nucleolar Stress in Huntington’s Disease. Cerebellum 2013, 12, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Schut, M.H.; Pepers, B.A.; Atalar, M.; Van Belzen, M.J.; Faull, R.L.; Roos, R.A.; Van Roon-Mom, W.M.C. Making (anti-) sense out of huntingtin levels in Huntington disease. Mol. Neurodegener. 2015, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bañez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN Translation in Huntington Disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Báñez-Coronel, M.; Porta, S.; Kagerbauer, B.; Mateu-Huertas, E.; Pantano, L.; Ferrer, I.; Guzmán, M.; Estivill, X.; Martí, E. A Pathogenic Mechanism in Huntington’s Disease Involves Small CAG-Repeated RNAs with Neurotoxic Activity. PLoS Genet. 2012, 8, e1002481. [Google Scholar] [CrossRef]
- Rudnicki, D.D.; Margolis, R.L.; Pearson, C.E.; Krzyzosiak, W.J. Diced triplets expose neurons to RISC. PLoS Genet. 2012, 8, e1002545. [Google Scholar] [CrossRef]
- Lawlor, K.T.; O’Keefe, L.V.; Samaraweera, S.E.; McLeod, C.J.; Dang, T.H.; Suter, C.M.; Van Eyk, C.L.; Maloney, C.A.; Richards, R.I. Double-stranded RNA is pathogenic in Drosophila models of expanded repeat neurodegenerative diseases. Hum. Mol. Genet. 2011, 20, 3757–3768. [Google Scholar] [CrossRef]
- Nishi, M.; Mizushima, A.; Nakagawara, K.-I.; Takeshima, H. Characterization of Human Junctophilin Subtype Genes. Biochem. Biophys. Res. Commun. 2000, 273, 920–927. [Google Scholar] [CrossRef]
- Takeshima, H. JunctophilinsA Novel Family of Junctional Membrane Complex Proteins. Mol. Cell 2000, 6, 11–22. [Google Scholar] [CrossRef]
- Nishia, M.; Hashimotobc, K.; Kuriyamad, K.; Komazakid, S.; Kanobc, M.; Shibatad, S.; Takeshima, H. Motor Discoordination in Mutant Mice Lacking Junctophilin Type 3. Biochem. Biophys. Res. Commun. 2002, 292, 318–324. [Google Scholar] [CrossRef]
- Rudnicki, B.D.; Holmes, S.E.; Lin, M.W.; Thornton, C.A.; Ross, C.A.; Margolis, R.L. Huntington’s disease-like 2 is associated with CUG repeat-containing RNA foci. Ann. Neurol. 2007, 61, 272–282. [Google Scholar] [CrossRef]
- Wilburn, B.; Rudnicki, D.D.; Zhao, J.; Weitz, T.M.; Cheng, Y.; Gu, X.; Greiner, E.; Park, C.S.; Wang, N.; Sopher, B.L.; et al. An Antisense CAG Repeat Transcript at JPH3 Locus Mediates Expanded Polyglutamine Protein Toxicity in Huntington’s Disease-like 2 Mice. Neuron 2011, 70, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.A. Myotonic Dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.-P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, G.B.; Cleary, J.D.; Pearson, C.E. In Vitro(CTG)·(CAG) Expansions and Deletions by Human Cell Extracts. J. Biol. Chem. 2002, 277, 13926–13934. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P.W. Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, P.; Gläser, D.; Vogel, W.; Wolf, M.; Schwemmle, S. The DMPK Gene of Severely Affected Myotonic Dystrophy Patients Is Hypermethylated Proximal to the Largely Expanded CTG Repeat. Am. J. Hum. Genet. 1998, 62, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.E.; Singer, R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef]
- Davis, B.M.; McCurrach, M.E.; Taneja, K.L.; Singer, R.H.; Housman, D.E. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. USA 1997, 94, 7388–7393. [Google Scholar] [CrossRef]
- Fu, Y.; Friedman, D.; Richards, S.; Pearlman, J.; Gibbs, R.; Pizzuti, A.; Ashizawa, T.; Perryman, M.; Scarlato, G.; Fenwick, R.; et al. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science 1993, 260, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Smith, D.B.J.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Berul, C.I.; Maguire, C.T.; Aronovitz, M.J.; Greenwood, J.; Miller, C.; Gehrmann, J.; Housman, D.; Mendelsohn, M.E.; Reddy, S. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Investig. 1999, 103, R1–R7. [Google Scholar] [CrossRef]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic Dystrophy in Transgenic Mice Expressing an Expanded CUG Repeat. Sci. 2000, 289, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, Y.; Abe, Y.; Cheney, L.; Udd, B.; Liu, X.X. Department of Infectious DiseasesThe Fifth Affiliated Hospital of Sun Yat-sen University 519000 Zhuhai China Haploinsuffciency for Znf9 in Znf9+/− Mice Is Associated with Multiorgan Abnormalities Resembling Myotonic Dystrophy. J. Mol. Biol. 2007, 368, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Stock, L.; Schneider-Gold, C.; Sommer, C.; Timchenko, N.A.; Timchenko, L. Reduction of Cellular Nucleic Acid Binding Protein Encoded by a Myotonic Dystrophy Type 2 Gene Causes Muscle Atrophy. Mol. Cell. Biol. 2018, 38, MCB.00649-17. [Google Scholar] [CrossRef]
- Huichalaf, C.; Schoser, B.; Schneider-Gold, C.; Jin, B.; Sarkar, P.; Timchenko, L.T. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J. Neurosci. 2009, 29, 9042–9049. [Google Scholar] [CrossRef]
- Raheem, O.; Olufemi, S.-E.; Bachinski, L.L.; Vihola, A.; Sirito, M.; Holmlund-Hampf, J.; Haapasalo, H.; Li, Y.-P.; Udd, B.; Krahe, R. Mutant (CCTG)n Expansion Causes Abnormal Expression of Zinc Finger Protein 9 (ZNF9) in Myotonic Dystrophy Type 2. Am. J. Pathol. 2010, 177, 3025–3036. [Google Scholar] [CrossRef]
- Mankodi, A.; Urbinati, C.R.; Yuan, Q.-P.; Moxley, R.T.; Sansone, V.; Krym, M.; Henderson, D.; Schalling, M.; Swanson, M.S.; Thornton, C.A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001, 10, 2165–2170. [Google Scholar] [CrossRef]
- Timchenko, L.T.; Timchenko, N.A.; Caskey, C.T.; Roberts, R. Novel Proteins with Binding Specificity for DNA CTG Repeats And RNA Cug Repeats: Implications for Myotonic Dystrophy. Hum. Mol. Genet. 1996, 5, 115–121. [Google Scholar] [CrossRef][Green Version]
- Timchenko, L.T.; Miller, J.W.; Timchenko, N.A.; Devore, D.R.; Datar, K.V.; Lin, L.; Roberts, R.; Caskey, C.T.; Swanson, M.S. Identification of a (CUG)n Triplet Repeat RNA-Binding Protein and Its Expression in Myotonic Dystrophy. Nucleic Acids Res. 1996, 24, 4407–4414. [Google Scholar] [CrossRef] [PubMed]
- Fardaei, M.; Rogers, M.T.; Thorpe, H.M.; Larkin, K.; Hamshere, M.G.; Harper, P.S.; Brook, J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002, 11, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.W.; Day, J.W. Myotonic Dystrophy: RNA Pathogenesis Comes into Focus. Am. J. Hum. Genet. 2004, 74, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Cooper, T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009, 37, 1281–1286. [Google Scholar] [CrossRef]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef]
- Mankodi, A.; Lin, X.; Blaxall, B.C.; Swanson, M.S.; Thornton, C.A. Nuclear RNA Foci in the Heart in Myotonic Dystrophy. Circ. Res. 2005, 97, 1152–1155. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A Muscleblind Knockout Model for Myotonic Dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef]
- De Haro, M.; Al-Ramahi, I.; De Gouyon, B.; Ukani, L.; Rosa, A.; Faustino, N.A.; Ashizawa, T.; Cooper, T.A.; Botas, J. MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum. Mol. Genet. 2006, 15, 2138–2145. [Google Scholar] [CrossRef]
- Todd, P.K.; Ackall, F.Y.; Hur, J.; Sharma, K.; Paulson, H.L.; Dowling, J.J. Transcriptional changes and developmental abnormalities in a zebrafish model of myotonic dystrophy type 1. Dis. Model. Mech. 2013, 7, 143–155. [Google Scholar] [CrossRef]
- Sicot, G.; Gomes-Pereira, M. RNA toxicity in human disease and animal models: From the uncovering of a new mechanism to the development of promising therapies. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1390–1409. [Google Scholar] [CrossRef]
- Otten, A.D.; Tapscott, S.J. Triplet repeat expansion in myotonic dystrophy alters the adjacent chromatin structure. Proc. Natl. Acad. Sci. USA 1995, 92, 5465–5469. [Google Scholar] [CrossRef]
- Thornton, C.A.; Wymer, J.P.; Simmons, Z.; McClain, C.; Moxley, R.T. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet. 1997, 16, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Klesert, T.R.; Otten, A.D.; Bird, T.D.; Tapscott, S.J. Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat. Genet. 1997, 16, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Boucher, C.; King, S.; Carey, N.; Krahe, R.; Winchester, C.L.; Creavin, T.; Mehji, P.; Bailey, M.E.S.; Chartier, F.; Brown, S.; et al. A novel homeodomain-encoding gene is associated with a large CpG island interrupted by the myotonic dystrophy unstable (CTG) n repeat. Hum. Mol. Genet. 1995, 4, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Filippova, G.N.; Thienes, C.P.; Penn, B.H.; Cho, D.H.; Hu, Y.J.; Moore, J.M.; Klesert, T.R.; Lobanenkov, V.V.; Tapscott, S.J. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 2001, 28, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, M.; Hamanaka, K.; Thomas, J.D.; Wang, E.T.; Hayashi, Y.K.; Takahashi, M.P.; Swanson, M.S.; Nishino, I.; Mochizuki, H. Aberrant Myokine Signaling in Congenital Myotonic Dystrophy. Cell Rep. 2017, 21, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.-Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, Physiological, and Motor Performance Defects in DMSXL Mice Carrying >1000 CTG Repeats from the Human DM1 Locus. PLoS Genet. 2012, 8, e1003043. [Google Scholar] [CrossRef]
- Scoles, D.R.; Pulst, S.M. Oligonucleotide therapeutics in neurodegenerative diseases. RNA Biol. 2018, 15, 1–8. [Google Scholar] [CrossRef]
- Ramachandran, P.S.; Boudreau, R.L.; Schaefer, K.A.; La Spada, A.R.; Davidson, B.L. Nonallele Specific Silencing of Ataxin-7 Improves Disease Phenotypes in a Mouse Model of SCA7. Mol. Ther. 2014, 22, 1635–1642. [Google Scholar] [CrossRef]
- Scholefield, J.; Greenberg, L.J.; Weinberg, M.S.; Arbuthnot, P.B.; Abdelgany, A.; Wood, M.J.A. Design of RNAi Hairpins for Mutation-Specific Silencing of Ataxin-7 and Correction of a SCA7 Phenotype. PLoS ONE 2009, 4, e7232. [Google Scholar] [CrossRef]
- Fiszer, A.; Wroblewska, J.P.; Nowak, B.M.; Krzyzosiak, W.J. Mutant CAG Repeats Effectively Targeted by RNA Interference in SCA7 Cells. Genes 2016, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- Miniarikova, J.; Zimmer, V.; Martier, R.; Brouwers, C.C.; Pythoud, C.; Richetin, K.; Rey, M.; Lubelski, J.; Evers, M.M.; Van Deventer, S.J.; et al. AAV5-miHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington’s disease. Gene Ther. 2017, 24, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Miniarikova, J.; Juhas, S.; Vallès, A.; Bohuslavova, B.; Juhasova, J.; Skalnikova, H.K.; Vodicka, P.; Valekova, I.; Brouwers, C.; et al. AAV5-miHTT Gene Therapy Demonstrates Broad Distribution and Strong Human Mutant Huntingtin Lowering in a Huntington’s Disease Minipig Model. Mol. Ther. 2018, 26, 2163–2177. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Pfister, E.L.; Liu, W.; Andre, R.; Träger, U.; Kennington, L.A.; Lo, K.; Dijkstra, S.; Macdonald, U.; Ostroff, G.; et al. Allele-Selective Suppression of Mutant Huntingtin in Primary Human Blood Cells. Sci. Rep. 2017, 7, 46740. [Google Scholar] [CrossRef]
- Pfister, E.L.; Chase, K.O.; Sun, H.; Kennington, L.A.; Conroy, F.; Johnson, E.; Miller, R.; Borel, F.; Aronin, N.; Mueller, C. Safe and Efficient Silencing with a Pol II, but Not a Pol lII, Promoter Expressing an Artificial miRNA Targeting Human Huntingtin. Mol. Ther. Nucleic Acids 2017, 7, 324–334. [Google Scholar] [CrossRef]
- Sobczak, K.; Wheeler, T.M.; Wang, W.; Thornton, C.A. RNA Interference Targeting CUG Repeats in a Mouse Model of Myotonic Dystrophy. Mol. Ther. 2012, 21, 380–387. [Google Scholar] [CrossRef]
- Langlois, M.-A.; Boniface, C.; Wang, G.; Alluin, J.; Salvaterra, P.M.; Puymirat, J.; Rossi, J.J.; Lee, N.S. Cytoplasmic and Nuclear Retained DMPK mRNAs Are Targets for RNA Interference in Myotonic Dystrophy Cells. J. Biol. Chem. 2005, 280, 16949–16954. [Google Scholar] [CrossRef]
- Bisset, D.R.; Stepniak-Konieczna, E.A.; Zavaljevski, M.; Wei, J.; Carter, G.T.; Weiss, M.D.; Gabellini, D. Therapeutic impact of systemic AAV-mediated RNA interference in a mouse model of myotonic dystrophy. Hum. Mol. Genet. 2015, 24, 4971–4983. [Google Scholar] [CrossRef]
- Spronck, E.A.; Brouwers, C.C.; Vallès, A.; De Haan, M.; Petry, H.; Van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT Gene Therapy Demonstrates Sustained Huntingtin Lowering and Functional Improvement in Huntington Disease Mouse Models. Mol. Ther. Methods Clin. Dev. 2019, 13, 334–343. [Google Scholar] [CrossRef]
- Keskin, S.; Brouwers, C.C.; Sogorb-Gonzalez, M.; Martier, R.; Depla, J.A.; Vallès, A.; Van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT Lowers Huntingtin mRNA and Protein without Off-Target Effects in Patient-Derived Neuronal Cultures and Astrocytes. Mol. Ther. Methods Clin. Dev. 2019, 15, 275–284. [Google Scholar] [CrossRef]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense oligonucleotides: A primer. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Silveira, I.; Veiga, F.J.; Ribeiro, A. Recent advances in characterization of nonviral vectors for delivery of nucleic acids: Impact on their biological performance. Expert Opin. Drug Deliv. 2014, 12, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Toonen, L.J.; Van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, G.H.F.R.C.F.; Otis, P.M.T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nat. Cell Biol. 2017, 544, 362–366. [Google Scholar] [CrossRef]
- Scoles, D.R.; Dansithong, W.; Pflieger, L.T.; Paul, S.; Gandelman, M.; Figueroa, K.P.; Rigo, F.; Bennett, C.F.; Pulst, S.M. ALS-associated genes in SCA2 mouse spinal cord transcriptomes. Hum. Mol. Genet. 2020, 29, 1658–1672. [Google Scholar] [CrossRef]
- Niu, C.; Prakash, T.P.; Kim, A.; Quach, J.L.; Huryn, L.A.; Yang, Y.; Lopez, E.; Jazayeri, A.; Hung, G.; Sopher, B.L.; et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci. Transl. Med. 2018, 10, eaap8677. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Jauvin, D.; Chrétien, J.; Pandey, S.K.; Martineau, L.; Revillod, L.; Bassez, G.; Lachon, A.; MacLeod, A.R.; Gourdon, G.; Wheeler, T.M.; et al. Targeting DMPK with Antisense Oligonucleotide Improves Muscle Strength in Myotonic Dystrophy Type 1 Mice. Mol. Ther. Nucleic Acids 2017, 7, 465–474. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.-W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.-R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Chew, J.; Stankowski, J.N.; Hayes, L.R.; Zhang, Y.-J.; Prudencio, M.; Carlomagno, Y.; Daughrity, L.M.; Jansen-West, K.; Perkerson, E.A.; et al. Poly(GP) proteins are a useful pharmacodynamic marker forC9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaai7866. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Kordasiewicz, H.; Langbehn, D.R.; Skotte, N.H.; Parsons, M.P.; Villanueva, E.B.; Caron, N.S.; Østergaard, M.E.; Anderson, L.M.; Xie, Y.; et al. Huntingtin suppression restores cognitive function in a mouse model of Huntington’s disease. Sci. Transl. Med. 2018, 10, eaar3959. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease | Gene | Function of the Encoded Protein or RNA | Pathogenic Alleles | Proteins Sequestered in RNA foci | Proteins in Intranuclear Inclusions | Proteins in Cytoplasmic Stress Granules | Translated Repeat Polypeptides |
|---|---|---|---|---|---|---|---|
| SCA2 | ATXN2 | Endocytosis, translation, and mitochondrial function | (CAG)>33 | ND | ND | STAU1 1, FOX/A2BP1 1, TDP-43 1, PolyQ 1, ubiquitin 1, DDX6 2, PABPC1 3 | ND * |
| ATXN2-AS | Unknown | (CTG)>33 | MBNL1 2 | ND | NF 2 | ||
| SCA7 | ATXN7 | Assembly and maintenance of TFTC/STAGA complexes | (CAG)>37 | ND | CBP 3, PolyQ 1, MBNL1 1, pTDP-43 1 and FUS/TLS 1, GCN5 2 | ND | ND |
| SCAANT1 | Transcriptional regulation of ATNX7 | (CTG)>37 | ND | ||||
| SCA8 | ATXN8 | Unknown | (CAG)>80 | ND | PolyQ 1, ubiquitin 1 | ND | PolyQ 1, PolyA 1, PolyS 1 |
| ATXN8OS | (CTG)>80 | MBNL1 1 | ND | PolyL 2, PolyC 2, PolyA 2 | |||
| SCA31 | BEAN1 | NEDD4-mediated ubiquitination | (TGGAA)ins | TDP-43 1, FUS 4, hnRNPA2/B1 4 | ND | ND | PolyWNGME 1 |
| SCA31AS | Phosphorylation | (TTCCA)ins | NF 1 | ND | |||
| FRDA | FXN | Iron metabolism | (GAA)>66 | ND | ND | ND | ND |
| FAST-1 | Unknown | - | |||||
| SCA36 | NOP56 | Pre-rRNA processing | (TGGGCC)>650 | SRSF2 1 | ND | ND | PolyGP 1, PolyWA 2, PolyGL 2 |
| NOP56AS | Unknown | (GGCCCA)>650 | ND | PolyPR 1, PolyGP 1, PolyAQ 2, | |||
| C9ORF72 FTD/ALS | C9ORF72 | Regulation of endosomal trafficking | (GGGGCC)>30 | SRSF1 1, SRSF2 1, ALYREF 1, ADARB2 1, nucleolin1, Purα 1, hnRNPA2/B1 1, hnRNPH 1, hnRNPF 1 | ND | pTDP-43 1, polyGR 3 | PolyGA 1, PolyGR 1, PolyGP 1 |
| C9ORF72AS | Unknown | (CCCCGG)>30 | SRSF2 1, ALYREF 1, hnRNPA1 1, hnRNPH/F 1, hnRNPK 1 | ND | PolyPR 1, PolyPA 1, PolyGP 1 | ||
| FXTAS | FMR1 | mRNA trafficking from the nucleus to the cytoplasm | (CGG)55–200 | Purα 1, hnRNPA2/B1 1, Sam68 1, CELF 1, MBNL1 1, Rm62 1, DGCR8 1 | SUMO2 1, MLF2 1, MBP 1, ubiquitin 1, p62 1, hnRNPL 1, hnRNPA1 1, hnRNPA3 1, hnRNPC 1, U2AF 1, SFPQ 1 RAD50 1, RPA1 1, XRCC6 1 | ND | PolyG 1, PolyA 2 |
| ASFMR1 | Unknown | (CCG)55–200 | ND | ND | ND | PolyP 1, PolyA 1, PolyR2 | |
| NIID | NOTCH2NLC | Neuronal proliferation and differentiation | (CGG)>66 | ND | ND | ND | ND |
| NOTCH2NLC-AS | Unknown | (CCG)>66 | |||||
| OPML1 | LOC642361 | Unknown | (CGG)exp | ND | ND | ND | ND |
| NUTM2B-AS2 | Unknown | (CCG)exp | |||||
| HD | HTT | Unknown | (CAG)>36 | MBNL1 1, Nucleolin 3 | CBP 1, mSIN3a 1, TBP 1, PolyQ 1, ubiquitin 1, p53 3 | Caprin-12, G3BP2 | PolyQ 1, PolyA 1, PolyS 1 |
| HTTAS1 | HTT regulation | (CTG)>36 | ND | NA | ND | PolyC 1, PolyL 1, PolyA 1 | |
| HDL2 | JPH3 | Formation of junctional membrane complexes | (CTG)>40 | MBNL1 1 | ND | ND | ND |
| HDL2AS | Unknown | (CAG)>40 | ND | CBP 1, PolyQ 1, ubiquitin 1 | |||
| DM1 | DMPK | Serine-threonine kinase | (CTG)>50 | MBNL1 1,5, MBNL2 1,5, MBNL3 5, hnRNPH 1, hnRNPF 1, Proteosome subunits 1 | ND | ND | PolyL 2, PolyC 2, PolyA 2 |
| DMPKAS | Regulation of DMPK expression | (CAG)>50 | ND | PolyQ 5, PolyS 2, PolyA 2 | |||
| DM2 | CNBP | Nucleic acid-binding protein | (CCTG)>75 | MBNL1 5, MBNL2 5, MBNL3 5, RNA Pol II 5, CStF 5, PML 5 | ND | ND | PolyLPAC 1 |
| CNBPAS | Unknown | (CAGG)>75 | hnRNPA1 1 | PolyQAGR 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, A.F.; Loureiro, J.R.; Bessa, J.; Silveira, I. Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries. Genes 2020, 11, 1418. https://doi.org/10.3390/genes11121418
Castro AF, Loureiro JR, Bessa J, Silveira I. Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries. Genes. 2020; 11(12):1418. https://doi.org/10.3390/genes11121418
Chicago/Turabian StyleCastro, Ana F., Joana R. Loureiro, José Bessa, and Isabel Silveira. 2020. "Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries" Genes 11, no. 12: 1418. https://doi.org/10.3390/genes11121418
APA StyleCastro, A. F., Loureiro, J. R., Bessa, J., & Silveira, I. (2020). Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries. Genes, 11(12), 1418. https://doi.org/10.3390/genes11121418