Population Genomics of an Anadromous Hilsa Shad Tenualosa ilisha Species across Its Diverse Migratory Habitats: Discrimination by Fine-Scale Local Adaptation

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Habitats of T. ilisha in the Complex Ecosystem of Bangladesh
2.2. Sample Collection of T. ilisha
2.3. NextRAD Genotyping
2.4. Sequence Assembly, Filtering and Discovery of SNPs
2.5. Clustering Analyses
2.6. Data Accessibility
3. Results
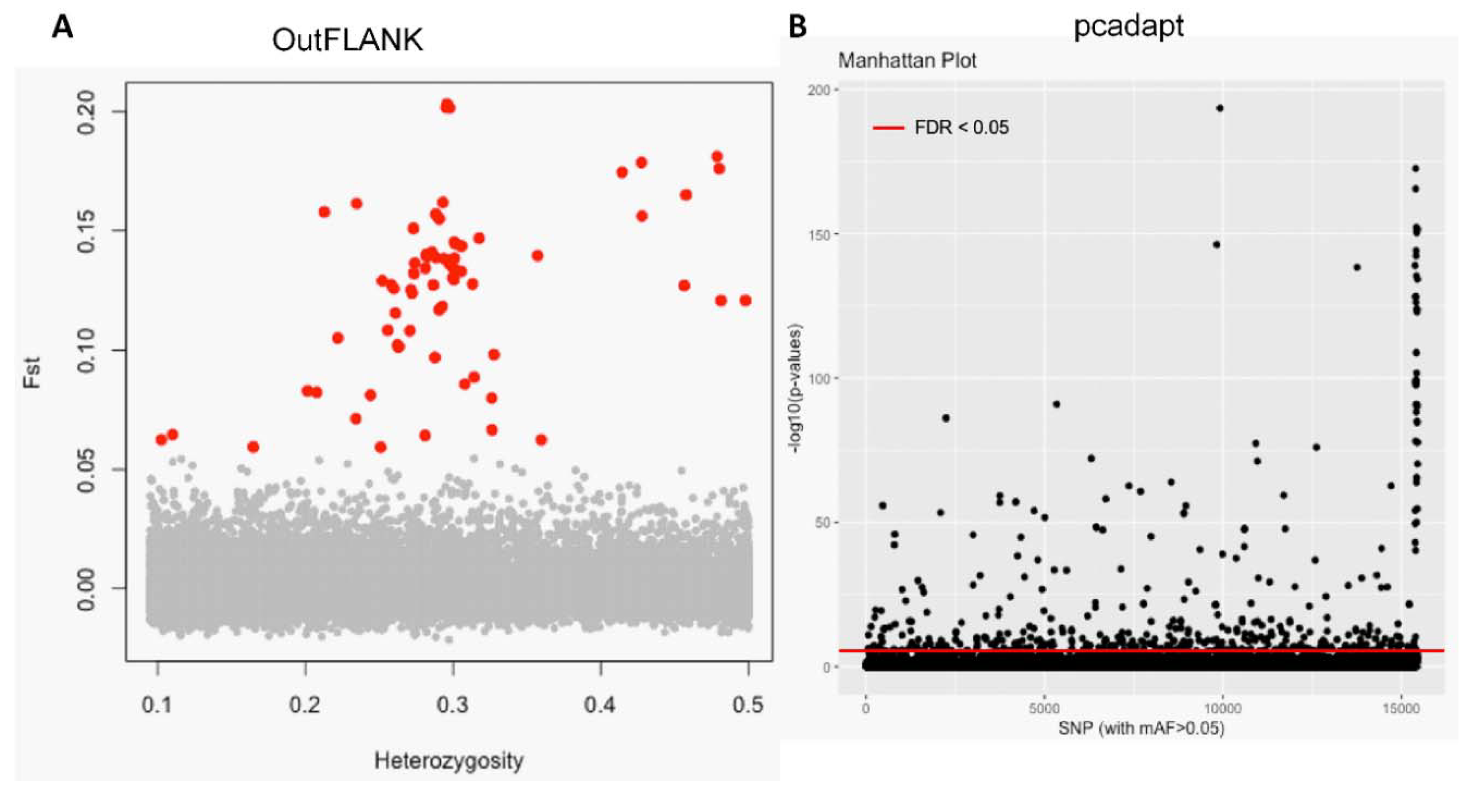
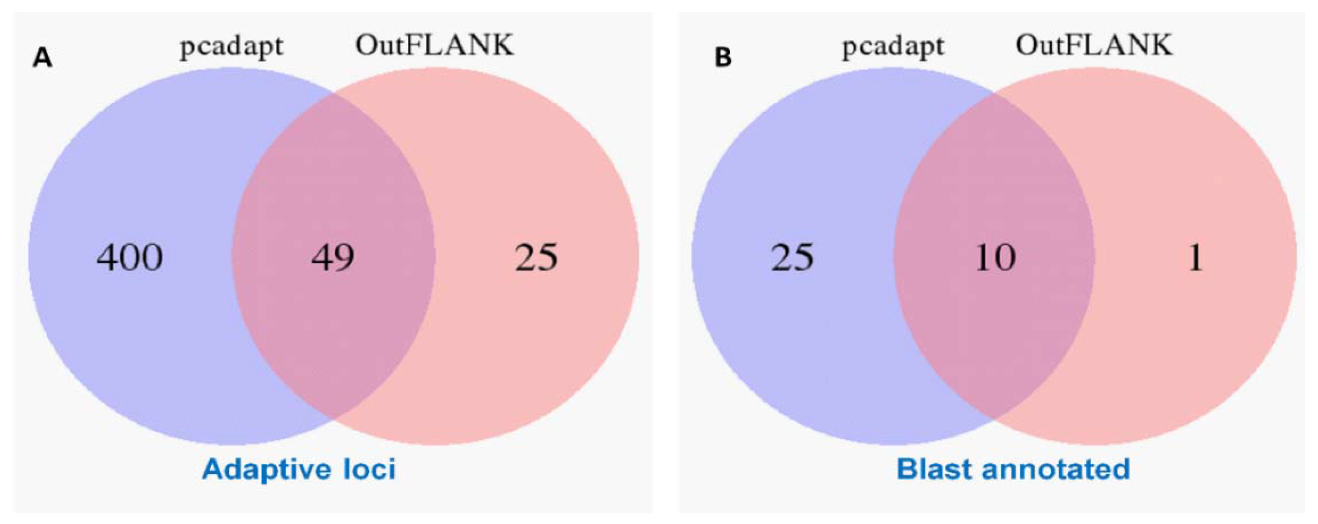
3.1. NextRAD Sequencing and Outlier Analysis
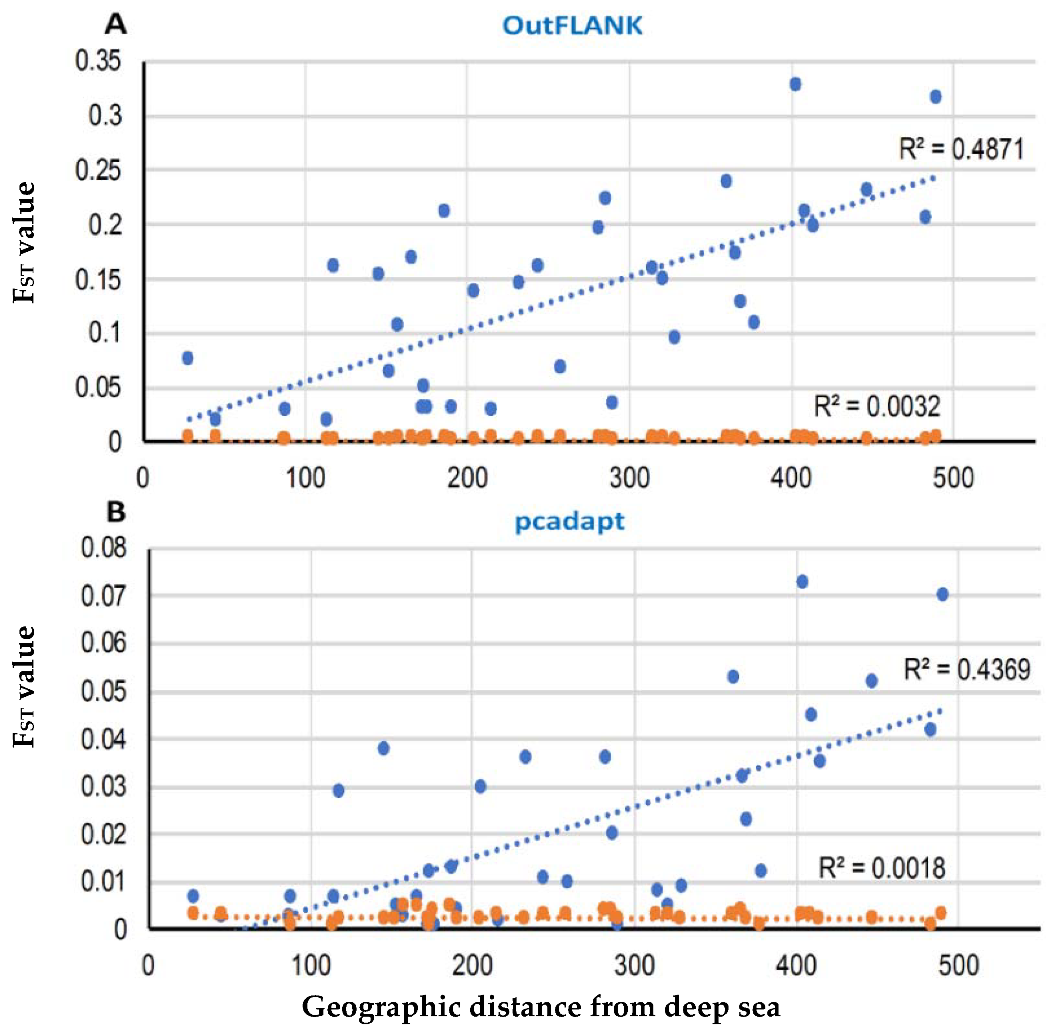
3.2. Demographic Inferences from FST Statistics and AMOVA Analysis
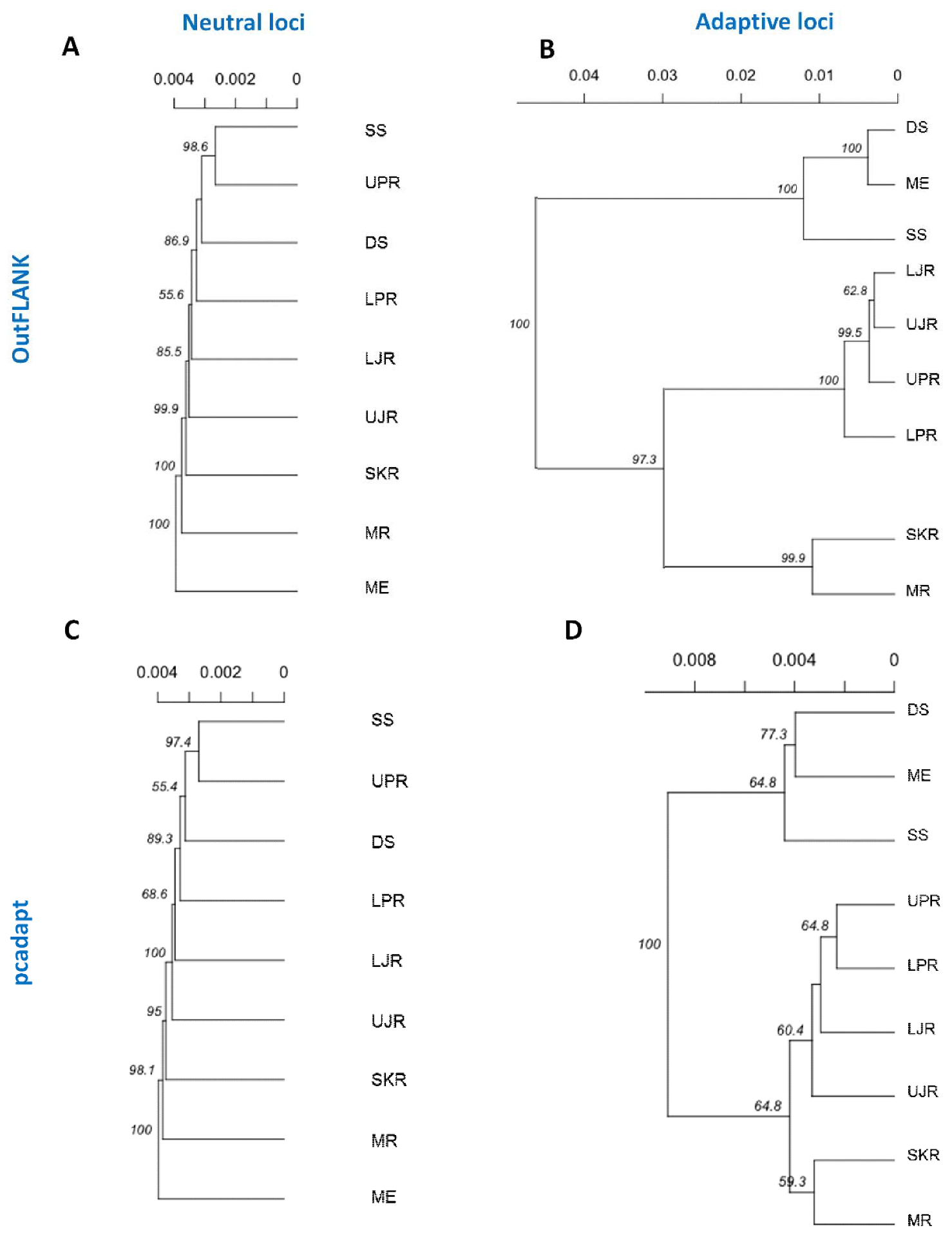
3.3. Genetic Structure and Recruitment Assignment Based on Different Spatial Analyses
3.4. Gene Function of the Outlier Loci
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next-generation sequencing |
| NextRAD | NGS-based restriction-site-associated DNA |
| RAD-seq | Restriction site-associated DNA sequencing |
| DS | Deep sea |
| SS | Shallow sea |
| ME | Meghna estuary |
| MR | Meghna river |
| SKR | Surma–Kushiara river |
| LPR | Lower Padma river |
| UPR | Upper Padma river |
| LJR | Lower Jamuna river |
| UJR | Upper Jamuna river |
| AMOVA | Analysis of molecular variance |
| PCA | Principal component analysis |
| DAPCs | Discriminant analysis of principal components |
| NJ | Neighbor-joining |
| QDFA | Quadratic discriminant functional analysis |
| SNP | Single nucleotide polymorphism |
References
- Islam, M.M.; Essam, Y.M.; Ali, L. Economic incentives for sustainable Hilsa fishing in Bangladesh: An analysis of the legal and institutional framework. Mar. Policy 2016, 68, 8–22. [Google Scholar] [CrossRef]
- Rahman, M.J.; Wahab, M.A.; Amin, S.M.N.; Nahiduzzaman, M.; Romano, N. Catch Trend and Stock Assessment of Hilsa Tenualosa ilisha Using Digital Image Measured Length-Frequency Data. Mar. Coast. Fish. 2018, 10, 386–401. [Google Scholar] [CrossRef]
- DOF (Department of Fisheries). Yearbook of Fisheries Statistics of Bangladesh 2017–18; Ministry of Fisheries and Livestock and Department of Fisheries: Dhaka, Bangladesh, 2018.
- Bernatchez, L. On the maintenance of genetic variation and adaptation to environmental change: Considerations from population genomics in fishes. J. Fish Biol. 2016, 89, 2519–2556. [Google Scholar] [CrossRef]
- Blaber, S.J.M.; Milton, D.A.; Chenery, S.R.; Fry, G.C. New insights into the life history of Tenualosa ilisha and fishery implications. Am. Fish. Soc. Symp. 2003, 35, 223–240. [Google Scholar]
- Milton, D.A.; Chenery, S.R. Can Otolith chemistry detect the population structure of the shad hilsa Tenualosa ilisha? Comparison with genetic and morphological studies. Mar. Ecol. Prog. Ser. 2001, 222, 239–251. [Google Scholar] [CrossRef]
- Hossain, M.S.; Sarker, S.; Sharifuzzaman, S.M.; Chowdhury, S.R. Discovering spawning ground of Hilsa Shad (Tenualosa ilisha) in the coastal waters of Bangladesh. Ecol. Model. 2014, 282, 59–68. [Google Scholar] [CrossRef]
- Hess, J.E.; Campbell, N.R.; Close, D.A.; Docker, M.F.; Narum, S.R. Population genomics of Pacific lamprey: Adaptive variation in a highly dispersive species. Mol. Ecol. 2012, 22, 2898–2916. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef]
- Carreras, C.; Ordóñez, V.; Zane, L.; Kruschel, C.; Nasto, I.; Macpherson, E.; Pascual, M. Population genomics of an endemic Mediterranean fish: Differentiation by fine scale dispersal and adaptation. Sci. Rep. 2016, 7, 43417. [Google Scholar] [CrossRef]
- Drinan, D.P.; Gruenthal, K.M.; Canino, M.F.; Lowry, D.; Fisher, M.C.; Hauser, L. Population assignment and local adaptation along an isolation-by-distance gradient in Pacific cod (Gadus macrocephalus). Evol. Appl. 2018, 11, 1448–1464. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.; Bishop, J.M.; Matthee, C.A.; von der Heyden, S. Genetic isolation by distance reveals restricted dispersal across a range of life histories: Implications for biodiversity conservation planning across highly variable marine environments. Divers. Distrib. 2015, 21, 698–710. [Google Scholar] [CrossRef]
- Gamboa, M.; Watanabe, K. Genome-wide signatures of local adaptation among seven stoneflies species along a nationwide latitudinal gradient in Japan. BMC Genom. 2019, 20, 84. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Naevdal, G. Population genetics studies of Hilsa shad, Tenualosa ilisha (Hamilton), in Bangladesh waters: Evidence for the existence of separate gene pools. Fish. Manag. Ecol. 2000, 7, 401–412. [Google Scholar] [CrossRef]
- Salini, J.P.; Milton, D.A.; Rahman, M.J.; Hussain, M.G. Allozyme and morphological variation throughout the geographic range of the tropical shad, Hilsa Tenualosa ilisha. Fish. Res. 2004, 66, 53–69. [Google Scholar] [CrossRef]
- Ahmed, A.S.I.; Islam, M.S.; Azam, M.S.; Khan, M.M.R.; Alam, M.S. RFLP analysis of the mtDNA D-loop region in Hilsa shad (Tenualosa ilisha) population from Bangladesh. Indian J. Fish 2004, 51, 25–31. [Google Scholar]
- Mazumder, S.K.; Alam, M.S. High levels of genetic variability and differentiation in Hilsa shad, Tenualosa ilisha (Clupeidae, Clupeiformes) populations revealed by PCR-RFLP analysis of the mitochondrial DNA D-loop region. Genet. Mol. Biol. 2009, 32, 190–196. [Google Scholar] [CrossRef]
- Brahmane, M.P.; Kundu, S.N.; Das, M.K.; Sharma, A.P. Low genetic diversity and absence of population differentiation of Hilsa (Tenualosa ilisha) revealed by mitochondrial DNA cytochrome b region in Ganga and Hooghly rivers. Afr. J. Biotechnol. 2013, 12, 3383–3389. [Google Scholar]
- Behera, B.K.; Singh, N.S.; Paria, P.; Sahoo, A.K.; Panda, D.; Meena, D.K.; Das, P.; Pakrashi, S.; Biswas, D.K.; Sharma, A.P. Population genetic structure of Indian shad, Tenualosa ilisha Inferred from variation in mitochondrial DNA sequences. J. Environ. Biol. 2015, 36, 1193–1197. [Google Scholar]
- Hemmer-Hansen, J.; Nielsen, E.E.; Frydenberg, J.; Loeschcke, V. Adaptive divergence in a high gene flow environment: Hsc70 variation in the European flounder (Platichthys flesus L.). Heredity 2007, 99, 592–600. [Google Scholar] [CrossRef]
- Schunter, C.; Garza, J.C.; Macpherson, E.; Pascual, M. SNP development from RNA-seq data in a nonmodel fish: How many individuals are needed for accurate allele frequency prediction? Mol. Ecol. Resour. 2014, 14, 157–165. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef]
- Star, B.; Nederbragt, A.J.; Jentoft, S.; Grimholt, U.; Malmstrom, M.; Gregers, T.F.; Rounge, T.B.; Paulsen, J.; Solbakken, M.H.; Sharma, A.; et al. The genome sequence of Atlantic cod reveals a unique immune system. Nature 2011, 477, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Barson, N.J.; Aykanat, T.; Hindar, K.; Baranski, M.; Bolstad, G.H.; Fiske, P.; Jacq, C.; Jensen, A.J.; Johnston, S.E.; Karlsson, S.; et al. Sex-dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature 2015, 528, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Hohenlohe, P.A.; Bassham, S.; Etter, P.D.; Stiffler, N.; Johnson, E.A.; Cresko, W.A. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010, 6, e1000862. [Google Scholar] [CrossRef] [PubMed]
- Bélanger-Deschênes, S.; Couture, P.; Campbell, P.G.C.; Bernatchez, L. Evolutionary change driven by metal exposure as revealed by coding SNP genome scan in wild yellow perch (Perca flavescens). Ecotoxicology 2013, 22, 938–957. [Google Scholar] [CrossRef] [PubMed]
- Laporte, M.; Rogers, S.M.; Dion-Cote, A.M.; Normandeau, E.; Gagnaire, P.A.; Dalziel, A.C.; Chebib, J.; Bernatchez, L. RAD-QTL Mapping reveals both genome-level parallelism and different genetic architecture underlying the evolution of body shape in lake whitefish (Coregonus clupeaformis) species pairs. G3: Genes Genom. Genet. 2015, 5, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Hauser, L.; Carvalho, G.R. Paradigm shifts in marine fisheries genetics: Ugly hypotheses slain by beautiful facts. Fish Fish. 2008, 9, 333–362. [Google Scholar] [CrossRef]
- Bradbury, I.R.; Hubert, S.; Higgins, B.; Borza, T.; Bowman, S.; Paterson, I.G.; Snelgrove, P.V.R.; Morris, C.J.; Gregory, R.S.; Hardie, D.C.; et al. Parallel adaptive evolution of Atlantic cod on both sides of the Atlantic Ocean in response to temperature. Proc. R. Soc. B 2010, 1701, 3725–3734. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Barrio, A.M.; Rafati, N.; Sundström, G.; Rubin, C.J.; Gilbert, E.R.; Berglund, J.; Wetterbom, A.; Laikre, L.; Grabherr, M.; et al. Population-scale sequencing reveals genetic differentiation due to local adaptation in Atlantic herring. Proc. Natl. Acad. Sci. USA 2012, 109, 19345–19350. [Google Scholar] [CrossRef]
- Ogden, R.; Gharbi, K.; Mugue, N.; Martinsohn, J.; Senn, H.; Davey, J.W.; Pourkazemi, M.; McEwing, R.; Eland, C.; Vidotto, M.; et al. Sturgeon conservation genomics: SNP discovery and validation using RAD sequencing. Mol. Ecol. 2013, 22, 3112–3123. [Google Scholar] [CrossRef]
- Benestan, L.; Gosselin, T.; Perrier, C.; Sainte-Marie, B.; Rochette, R.; Bernatchez, L. RAD genotyping reveals fine-scale genetic structuring and provides powerful population assignment in a widely distributed marine species, the American lobster (Homarus americanus). Mol. Ecol. 2015, 24, 3299–3315. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.L.; Blaxter, M.W. RADseq: Next-generation population genetics. Brief. Funct. Genom. 2010, 9, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Allison, M.A. Geologic Framework and Environmental Status of the Ganges-Brahmaputra Delta. J. Coast. Res. 1998, 14, 826–836. [Google Scholar]
- Barua, D.K. Suspended sediment movement in the estuary of the Ganges-Brahmaputra-Meghna River system. Mar. Geol. 1990, 91, 243–253. [Google Scholar] [CrossRef]
- Hasan, K.M.M.; Wahab, A.; Ahmed, Z.F.; Mohammed, E.Y. The Biophysical Assessments of the Hilsa Fish (Tenualosa Ilisha) Habitat in the Lower Meghna, Bangladesh; IIED Working Paper; IIED: London, UK, 2015. [Google Scholar]
- Gilbert, S.F. Mechanisms for the environmental regulation of gene expression: Ecological aspects of animal development. J. Biosci. 2005, 30, 65–74. [Google Scholar] [CrossRef]
- Emerson, K.J.; Conn, J.E.; Bergo, E.S.; Randel, M.A.; Sallum, M.A. Brazilian Anopheles darlingi root (Diptera: Culicidae) clusters by major biogeographical region. PLoS ONE 2015, 10, e0130773. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: An R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Whitlock, M.C.; Lotterhos, K.E. Reliable Detection of Loci Responsible for Local Adaptation: Inference of a Null Model through Trimming the Distribution of FST. Am. Nat. 2015, 186, S24–S36. [Google Scholar] [CrossRef]
- Luu, K.; Bazin, E.; Blum, M.G.B. pcadapt: An R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 2017, 17, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Duforet-Frebourg, N.; Luu, K.; Laval, G.; Bazin, E.; Blum, M.G. Detecting genomic signatures of natural selection with principal component analysis: Application to the 1000 Genomes data. Mol. Biol. Evol. 2015, 334, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G.; van Tienderen, P.H. GENOTYPE and GENODIVE: Two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 2004, 4, 792–794. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Funk, W.C.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 2012, 27, 489–496. [Google Scholar] [CrossRef]
- Milano, I.; Babbucci, M.; Cariani, A.; Atanassova, M.; Bekkevold, D.; Carvalho, G.R.; Espiñeira, M.; Fiorentino, F.; Garofalo, G.; Geffen, A.J.; et al. Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius). Mol. Ecol. 2014, 23, 118–135. [Google Scholar] [CrossRef]
- Hollenbeck, C.M.; Portnoy, D.S.; Gold, J.R. Evolution of population structure in an estuarine-dependent marine fish. Ecol. Evol. 2019, 9, 3141–3152. [Google Scholar] [CrossRef]
- Salas, E.M.; Bernardi, G.; Berumen, M.L.; Gaither, M.R.; Rocha, L.A. RADseq analyses reveal concordant Indian Ocean biogeographic and phylogeographic boundaries in the reef fish Dascyllus trimaculatus. R. Soc. Open Sci. 2019, 6, 172413. [Google Scholar] [CrossRef] [PubMed]
- Dahle, G.; Rahman, M.; Eriksen, A.G. RAPD finger printing used for discriminating among three populations of Hilsa shad (Tenualosa ilisha). Fish. Res. 1997, 32, 263–269. [Google Scholar] [CrossRef]
- Shifat, R.; Begum, A.; Khan, H. Use of RAPD fingerprinting for discriminating two populations of Hilsa shad (Tenualosa ilisha Ham.) from inland rivers of Bangladesh. J. Biochem. Mol. Biol. 2003, 36, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Brahmane, M.P.; Das, M.K.; Sinha, M.R.; Sugunan, V.V.; Mukherjee, A.; Singh, S.N.; Prakash, S.; Maurye, P.; Hajra, A. Use of RAPD fingerprinting for delineating populations of hilsa shad Tenualosa ilisha (Hamilton, 1822). Genet. Mol. Res. 2006, 5, 643–652. [Google Scholar] [PubMed]
- Jorfi, E.; Farhad, A.; Ghorezshi, S.A.; Mortezaei, S.R.S. A study on population genetic of hilsa shad, Tenualosa iliisha in Khoozestan, Iran using molecular method (RAPD). Iran. Sci. Fish. J. 2008, 17, 31–44. [Google Scholar]
- Gralka, M.; Stiewe, F.; Farrell, F.; Möbius, W.; Waclaw, B.; Hallatschek, O. Allele surfing promotes microbial adaptation from standing variation. Ecol. Lett. 2016, 19, 889–898. [Google Scholar] [CrossRef]
- Asaduzzaman, M.; Wahab, M.A.; Rahman, M.J.; Nahiduzzaman, M.; Dickson, M.W.; Igarashi, Y.; Asakawa, S.; Wong, L.L. Fine-scale population structure and ecotypes of anadromous Hilsa shad (Tenualosa ilisha) across complex aquatic ecosystems revealed by NextRAD genotyping. Sci. Rep. 2019, 9, 16050. [Google Scholar] [CrossRef]
- Hossain, M.S.; Sharifuzzaman, S.M.; Chowdhury, S.R. Habitats across the life cycle of Hilsa shad (Tenualosa ilisha) in aquatic ecosystem of Bangladesh. Fish. Manag. Ecol. 2016, 23, 450–462. [Google Scholar] [CrossRef]
- Pecoraro, C.; Babbucci, M.; Franch, R.; Rico, C.; Papetti, C.; Chassot, E.; Bodin, N.; Cariani, A.; Bargelloni, L.; Tinti, F. The population genomics of yellowfin tuna (Thunnus albacares) at global geographic scale challenges current stock delineation. Sci. Rep. 2018, 8, 13890. [Google Scholar] [CrossRef]
- Mojumdar, C.H. Culture of Hilsa. Mod. Rev. 1939, 66, 293–295. [Google Scholar]
- Quddus, M.M.A.; Shimizu, M.; Nose, Y. Meristic and morphometric differences in two types of Hilsa ilisha in Bangladesh waters. Bull. Jpn. Soc. Sci. Fish. 1984, 50, 43–49. [Google Scholar] [CrossRef]
- Ghosh, A.N.; Bhattacharya, R.K.; Rao, K.V. On the identification of the sub-populations of Hilsa ilisha (Ham.) in the Gangetic system with a note on their distribution. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 1968, 34, 44–57. [Google Scholar]
- Shafi, M.; Quddus, M.M.A.; Hossain, H. A morphometric study of the population of Hilsa ilisha (Hamilton-Buchanan) from the river Meghna. Proc. Second Bangladesh Sci. Conf. 1977, 2, 22–24. [Google Scholar]
- Pillay, S.R.; Rao, K.V.; Mathur, P.K. Preliminary report on the tagging of the Hilsa, Hilsa ilisha (Hamilton). Proc. Indo Pac. Fish. Counc. 1962, 10, 28–36. [Google Scholar]
- Lotterhos, K.E.; Whitlock, M.C. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol. Ecol. 2014, 23, 2178–2192. [Google Scholar] [CrossRef]
- Meirmans, P.G. The trouble with isolation by distance. Mol. Ecol. 2012, 21, 2839–2846. [Google Scholar] [CrossRef]
- Russello, M.A.; Kirk, S.L.; Frazer, K.K.; Askey, P.J. Detection of outlier loci and their utility for fisheries management. Evol. Appl. 2012, 5, 39–52. [Google Scholar] [CrossRef]
- Picq, S.; McMillan, W.O.; Puebla, O. Population genomics of local adaptation versus speciation in coral reef fishes (Hypoplectrus spp., Serranidae). Ecol. Evol. 2016, 6, 2109–2124. [Google Scholar] [CrossRef]
- Barth, J.M.; Damerau, M.; Matschiner, M.; Jentoft, S.; Hanel, R. Genomic differentiation and demographic histories of Atlantic and Indo-Pacific yellowfin tuna (Thunnus albacares) populations. Genome Biol. Evol. 2017, 9, 1084–1098. [Google Scholar] [CrossRef]
- Boyer, N.P.; Monkiewicz, C.; Menon, S.; Moy, S.S.; Gupton, S.L. Mammalian TRIM67 functions in brain development and behavior. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Schrick, C.; Fischer, A.; Srivastava, D.P.; Tronson, N.C.; Penzes, P.; Radulovic, J. N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron 2007, 55, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Sakurai, T.; Bozdagi-Gunal, O.; Dorr, N.P.; Moy, J.; Krug, L.; Gama-Sosa, M.; Elder, G.A.; Koch, R.J.; Walker, R.H.; et al. Increased expression of receptor phosphotyrosine phosphatase-β/ζ is associated with molecular, cellular, behavioral and cognitive schizophrenia phenotypes. Transl. Psychiatry 2011, 1, e8. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Sites | Migratory Habitat | Year of Collection | Latitude | Longitude | Sample Size |
|---|---|---|---|---|---|
| DS | Deep sea (Bay of Bengal) | 2018 | 21.2722 | 91.0632 | 30 |
| SS | Shallow sea (Bay of Bengal) | 2017 | 21.7927 | 90.1120 | 30 |
| ME | Meghna estuary | 2017 | 22.0482 | 90.9188 | 30 |
| MR | Meghna river | 2018 | 23.0805 | 90.6413 | 30 |
| SKR | Surma–Kushiara river | 2018 | 24.5794 | 91.2256 | 30 |
| LPR | Lower Padma river | 2018 | 23.3117 | 90.5208 | 30 |
| UPR | Upper Padma river | 2017 | 24.6338 | 88.0582 | 60 |
| LJR | Lower Jamuna river | 2018 | 25.1046 | 89.6869 | 30 |
| UJR | Upper Jamuna river | 2017 | 25.5138 | 89.6827 | 30 |
| Habitats | DS | LJR | MR | LPR | ME | SKR | SS | UJR | UPR |
|---|---|---|---|---|---|---|---|---|---|
| FST OutFLANK | |||||||||
| DS | -- | 0.002 *** | 0.002 *** | 0.002 *** | 0.002 *** | 0.003 *** | 0.001 *** | 0.003 *** | 0.001 *** |
| LJR | 0.230 *** | -- | 0.003 *** | 0.003 *** | 0.003 *** | 0.004 *** | 0.002 *** | 0.003 *** | 0.002 *** |
| MR | 0.138 *** | 0.160 *** | -- | 0.003 *** | 0.001 ** | 0.003 *** | 0.001 *** | 0.004 *** | 0.003 *** |
| LPR | 0.145 *** | 0.028 * | 0.076 ** | -- | 0.002 *** | 0.004 *** | 0.001 *** | 0.003 *** | 0.002 *** |
| ME | 0.010 NS | 0.238 *** | 0.161 *** | 0.152 *** | -- | 0.004 *** | 0.001 ** | 0.003 *** | 0.003 *** |
| SKR | 0.173 *** | 0.169 *** | 0.030 * | 0.106 *** | 0.195 *** | -- | 0.002 *** | 0.004 *** | 0.003 *** |
| SS | 0.020 * | 0.127 *** | 0.064 ** | 0.050 ** | 0.028 * | 0.095 *** | -- | 0.002 *** | 0.001 *** |
| UJR | 0.315 *** | 0.020 * | 0.223 *** | 0.067 *** | 0.327 *** | 0.211 *** | 0.197 *** | -- | 0.002 *** |
| UPR | 0.206 *** | 0.012 NS | 0.159 *** | 0.034 * | 0.212 *** | 0.150 *** | 0.109 *** | 0.031 ** | -- |
| pcadapt | |||||||||
| DS | -- | 0.002 *** | 0.002 *** | 0.002 *** | 0.002 *** | 0.004 *** | 0.001 *** | 0.003 *** | 0.001 *** |
| LJR | 0.052 *** | -- | 0.003 *** | 0.003 *** | 0.003 *** | 0.005 *** | 0.002 *** | 0.003 *** | 0.002 *** |
| MR | 0.030 *** | 0.011 ** | -- | 0.003 *** | 0.002 *** | 0.004 *** | 0.002 *** | 0.004 *** | 0.003 *** |
| LPR | 0.036 *** | 0.002 NS | 0.007 * | -- | 0.002 *** | 0.005 *** | 0.001 *** | 0.003 *** | 0.002 *** |
| ME | 0.003 NS | 0.053 *** | 0.029 *** | 0.038 *** | -- | 0.004 *** | 0.001 *** | 0.003 *** | 0.003 *** |
| SKR | 0.032 *** | 0.007 * | 0.001 NS | 0.003 NS | 0.036 *** | -- | 0.002 *** | 0.005 *** | 0.003 *** |
| SS | 0.007 * | 0.023 ** | 0.005 NS | 0.012 ** | 0.007 * | 0.009 * | -- | 0.002 *** | 0.001 *** |
| UJR | 0.070 *** | 0.003 NS | 0.020 *** | 0.010 *** | 0.073 *** | 0.013 *** | 0.035 *** | -- | 0.002 *** |
| UPR | 0.042 *** | 0.001 NS | 0.008 ** | 0.001 NS | 0.045 *** | 0.005 * | 0.016 *** | 0.004 * | -- |
| Loci Category | Source of Variation | Sum of Square | Variance Components | % of Variation | Statistics | p-Value |
|---|---|---|---|---|---|---|
| Adaptive panel of SNP loci | FST OutFLANK | |||||
| Within individuals | 3468 | 11.56 | 103.1 | F_it = −0.031 | - | |
| Among individuals | 2529.7 | −1.433 | −12.8 | F_is = −0.142 | 1.00 | |
| Among collection sites | 645.337 | 1.091 | 9.7 | F_st = 0.097 | 0.000 | |
| pcadapt | ||||||
| Within individuals | 17,112.5 | 57.042 | 101.7 | F_it = −0.017 | - | |
| Among individuals | 15,559.02 | −1.787 | −3.2 | F_is = −0.032 | 1.00 | |
| Among collection sites | 871.202 | 0.84 | 1.5 | F_st = 0.015 | 0.000 | |
| Neutral panel of SNP loci | FST OutFLANK | |||||
| Within individuals | 599,152.5 | 1997.175 | 100.5 | F_it = −0.005 | - | |
| Among individuals | 573,245.4 | −13.63 | −0.7 | F_is = −0.007 | 1.00 | |
| Among collection sites | 17,503.28 | 3.303 | 0.2 | F_st = 0.002 | 0.000 | |
| pcadapt | ||||||
| Within individuals | 585,923 | 1953.077 | 100.5 | F_it = −0.005 | - | |
| Among individuals | 560,559.6 | −13.377 | −0.7 | F_is = −0.007 | 1.00 | |
| Among collection sites | 17,326.43 | 3.629 | 0.2 | F_st = 0.002 | 0.000 | |
| Locus Name | GenBank Accession Number | Gene Name 1 | Species | Biological Function 2 |
|---|---|---|---|---|
| SCED01086260.1:268 | XM_012815555.1 | nrxn3b * | Clupea harengus | Neuronal cell surface protein |
| SCED01010228.1:165 | XM_012823864.1 | TRIM67 * | C. harengus | Neuron projection development |
| SCED01084234.1:74 | XM_012823924.1 | TTC13 * | C. harengus | Unknown |
| SCED01037310.1:1343 | XM_012823959.1 | MTHFD1 * | C. harengus | Somite and heart development |
| SCED01066042.1:124 | XM_012828951.1 | Grik2 * | C. harengus | Neuronal activity |
| SCED01072826.1:68 | XM_012835569.1 | EXTL3 * | C. harengus | Biosynthesis of polysaccharides |
| SCED01077141.1:127 | XM_012839387.1 | PLCB1 * | C. harengus | Lipid degradation and metabolism |
| SCED01094462.1:170 | XM_017701045.1 | ITPKB * | Pygocentrus nattereri | Metabolic process, ATP binding |
| SCED01094733.1:323 | XM_023923858.1 | EXOC8 * | Cyanistes caeruleus | Exocytosis and protein transport |
| SCED01029258.1:4937 | XM_028970071.1 | NFIB * | Denticeps clupeoides | Differentiation, DNA replication |
| SCED01007742.1:237924 | XM_012823803.1 | FBXO11 a | C. harengus | Cellular protein modification |
| SCED01001444.1:144867 | XM_008399996.2 | PRLHR | Poecilia reticulata | Feeding behavior, metabolic process |
| SCED01000736.1:64807 | XM_012815189.1 | U2SURP | C. harengus | mRNA splicing |
| SCED01012000.1:38243 | XM_012822717.1 | MYO6 | C. harengus | Movement, endocytosis |
| SCED01013770.1:62161 | XM_012824084.1 | Gmps | C. harengus | Guanosine monophosphate biosynthesis |
| SCED01008867.1:80237 | XM_012827367.1 | EPR1 | C. harengus | Cellular membrane biosynthesis |
| SCED01003175.1:15453 | XM_012827500.1 | Elfn2 | C. harengus | Protein phosphatase inhibitor |
| SCED01030760.1:318 | XM_012828774.1 | SLC7A10 | C. harengus | Amino acids transport |
| SCED01009040.1:131462 | XM_012828861.1 | BLMH | C. harengus | Response to toxic substances |
| SCED01011615.1:5090 | XM_012830641.1 | AADAT | C. harengus | Biosynthetic and metabolic process |
| SCED01010819.1:27239 | XM_012830963.1 | SIDT2 | C. harengus | Glucose homeostasis |
| SCED01020677.1:12909 | XM_012832078.1 | MYRF | C. harengus | Differentiation and transcription |
| SCED01010957.1:39086 | XM_012832114.1 | SFI1 | C. harengus | Mitotic cell cycle regulation |
| SCED01005892.1:34793 | XM_012832603.1 | GABBR1 | C. harengus | Neuronal cell signaling |
| SCED01000975.1:25910 | XM_012833200.1 | PDHA1 | C. harengus | Glucose metabolic process |
| SCED01010852.1:64989 | XM_012835295.1 | ACIN1 | C. harengus | Apoptosis and mRNA processing |
| SCED01002831.1:133084 | XM_012837338.1 | TUBGCP6 | C. harengus | Microtubule nucleation |
| SCED01000696.1:30199 | XM_012837568.1 | KDM6A | C. harengus | Embryonic development |
| SCED01003627.1:221050 | XM_012838801.1 | CCNF | C. harengus | Mitotic cell division control |
| SCED01010418.1:19123 | XM_012840278.1 | Herc1 | C. harengus | Neuromuscular process |
| SCED01024554.1:1059 | XM_012841400.1 | ARAP1 | C. harengus | GTPase activity |
| SCED01022525.1:8702 | XM_012841845.1 | PPP1R9B | C. harengus | Cell cycle control and neurogenesis |
| SCED01000852.1:137973 | XM_012842171.1 | PGM1 | C. harengus | Synthesis glucose and glycogen |
| SCED01062845.1:131 | XM_015395566.1 | MPEG1 | Cyprinodon variegatus | Immune response against bacteria |
| SCED01008207.1:36816 | XM_026272763.1 | UBR3 | Carassius auratus | Embryonic development |
| SCED01003543.1:101682 | XR_001162390.1 | SRP68 | C. harengus | Role in targeting secreting protein |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asaduzzaman, M.; Igarashi, Y.; Wahab, M.A.; Nahiduzzaman, M.; Rahman, M.J.; Phillips, M.J.; Huang, S.; Asakawa, S.; Rahman, M.M.; Wong, L.L. Population Genomics of an Anadromous Hilsa Shad Tenualosa ilisha Species across Its Diverse Migratory Habitats: Discrimination by Fine-Scale Local Adaptation. Genes 2020, 11, 46. https://doi.org/10.3390/genes11010046
Asaduzzaman M, Igarashi Y, Wahab MA, Nahiduzzaman M, Rahman MJ, Phillips MJ, Huang S, Asakawa S, Rahman MM, Wong LL. Population Genomics of an Anadromous Hilsa Shad Tenualosa ilisha Species across Its Diverse Migratory Habitats: Discrimination by Fine-Scale Local Adaptation. Genes. 2020; 11(1):46. https://doi.org/10.3390/genes11010046
Chicago/Turabian StyleAsaduzzaman, Md, Yoji Igarashi, Md Abdul Wahab, Md Nahiduzzaman, Md Jalilur Rahman, Michael J. Phillips, Songqian Huang, Shuichi Asakawa, Md Moshiur Rahman, and Li Lian Wong. 2020. "Population Genomics of an Anadromous Hilsa Shad Tenualosa ilisha Species across Its Diverse Migratory Habitats: Discrimination by Fine-Scale Local Adaptation" Genes 11, no. 1: 46. https://doi.org/10.3390/genes11010046
APA StyleAsaduzzaman, M., Igarashi, Y., Wahab, M. A., Nahiduzzaman, M., Rahman, M. J., Phillips, M. J., Huang, S., Asakawa, S., Rahman, M. M., & Wong, L. L. (2020). Population Genomics of an Anadromous Hilsa Shad Tenualosa ilisha Species across Its Diverse Migratory Habitats: Discrimination by Fine-Scale Local Adaptation. Genes, 11(1), 46. https://doi.org/10.3390/genes11010046