Genomic Analysis of γ-Hexachlorocyclohexane-Degrading Sphingopyxis lindanitolerans WS5A3p Strain in the Context of the Pangenome of Sphingopyxis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequencing and Assembly
2.2. Average Nucleotide Identity Based on BLAST (ANIb) and GGDC Analyses
2.3. Pangenomic Analyses
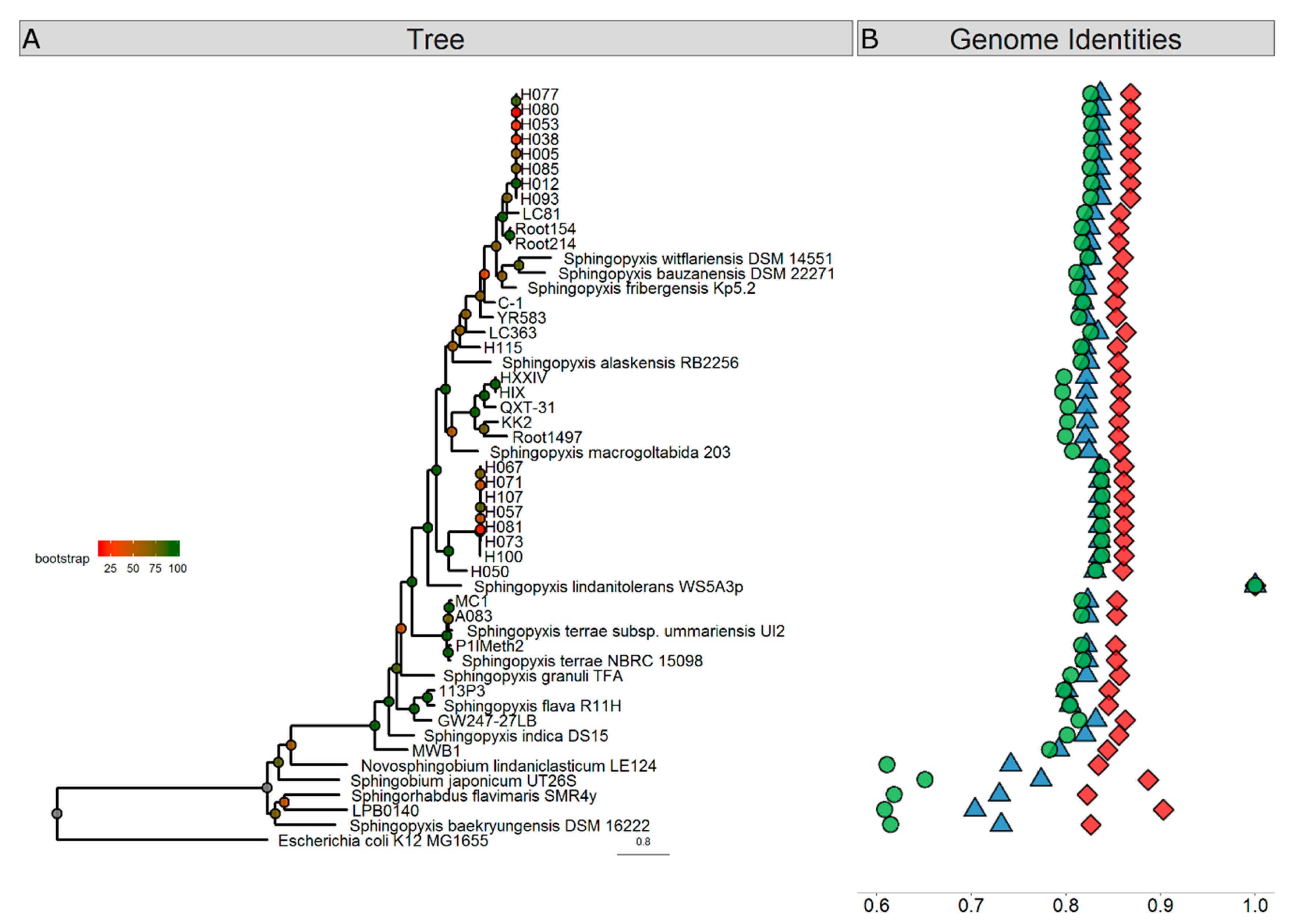
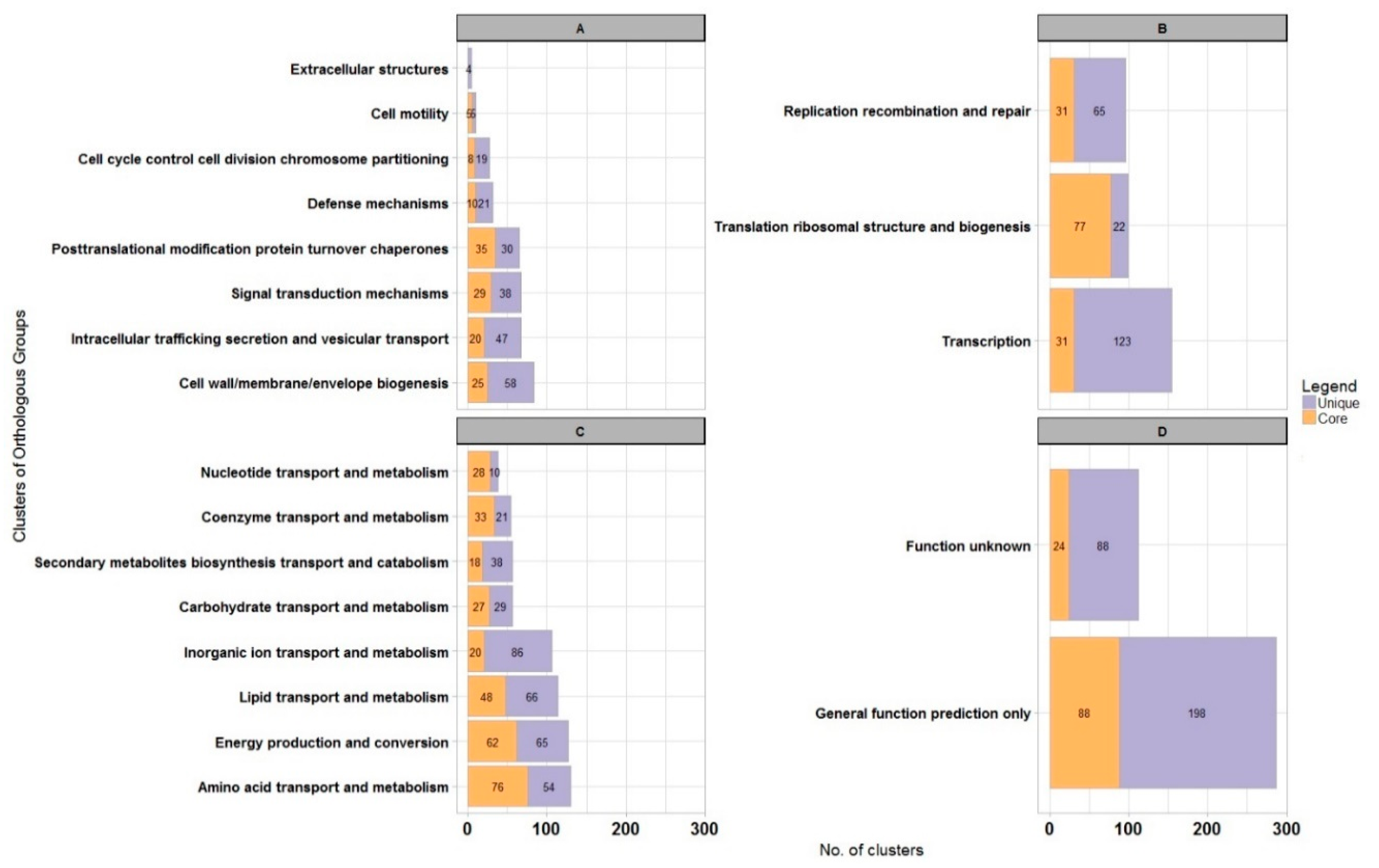
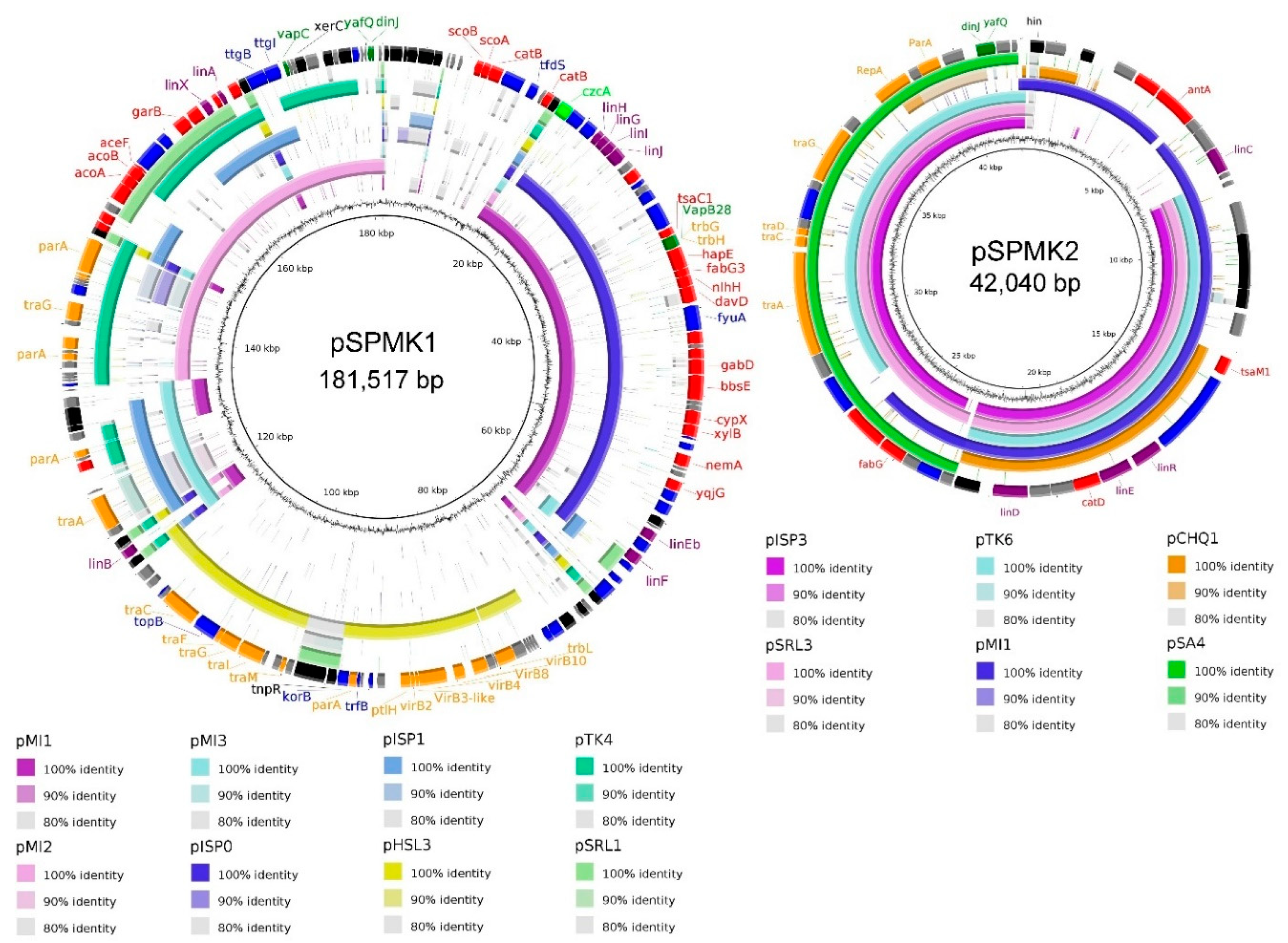
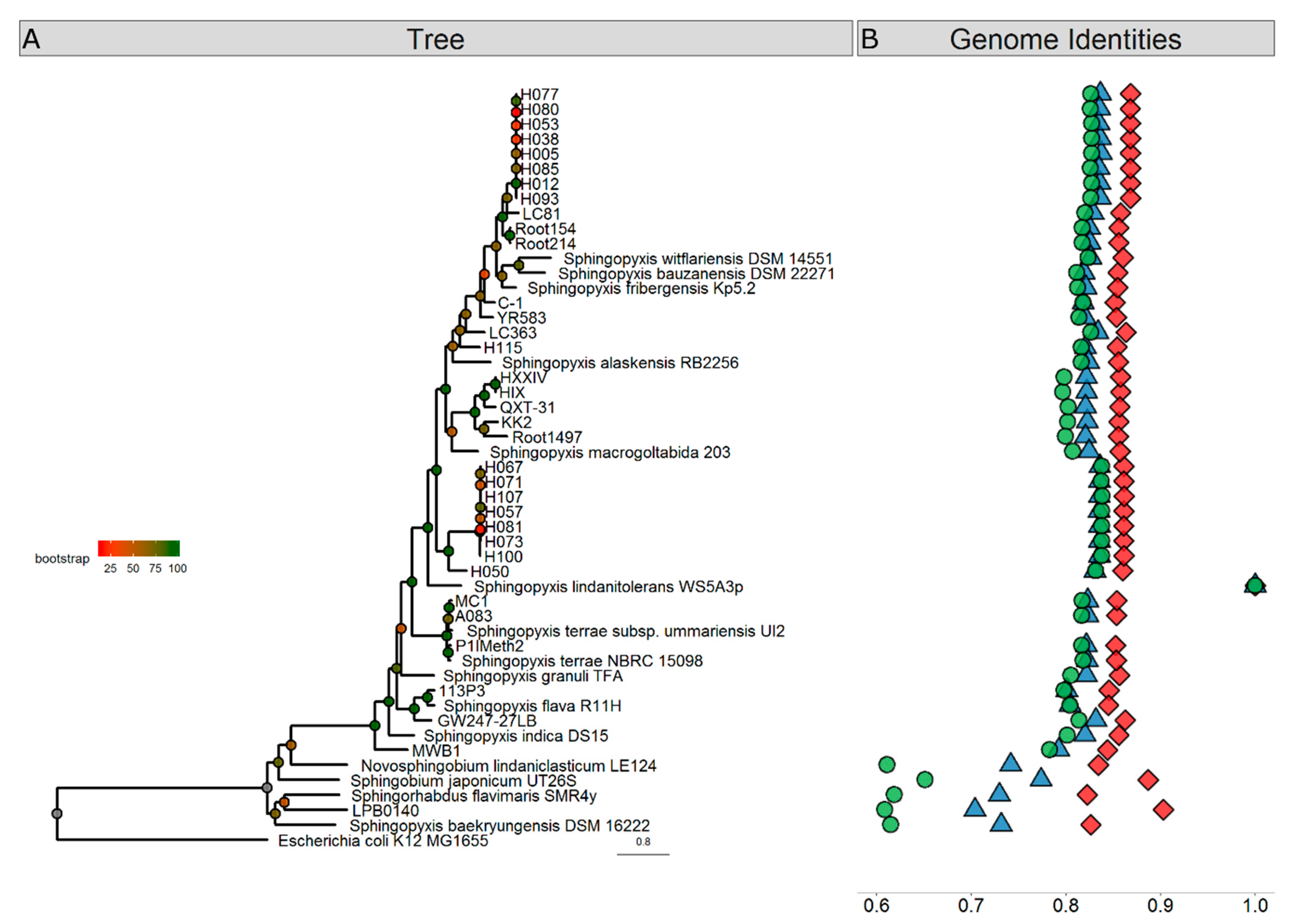
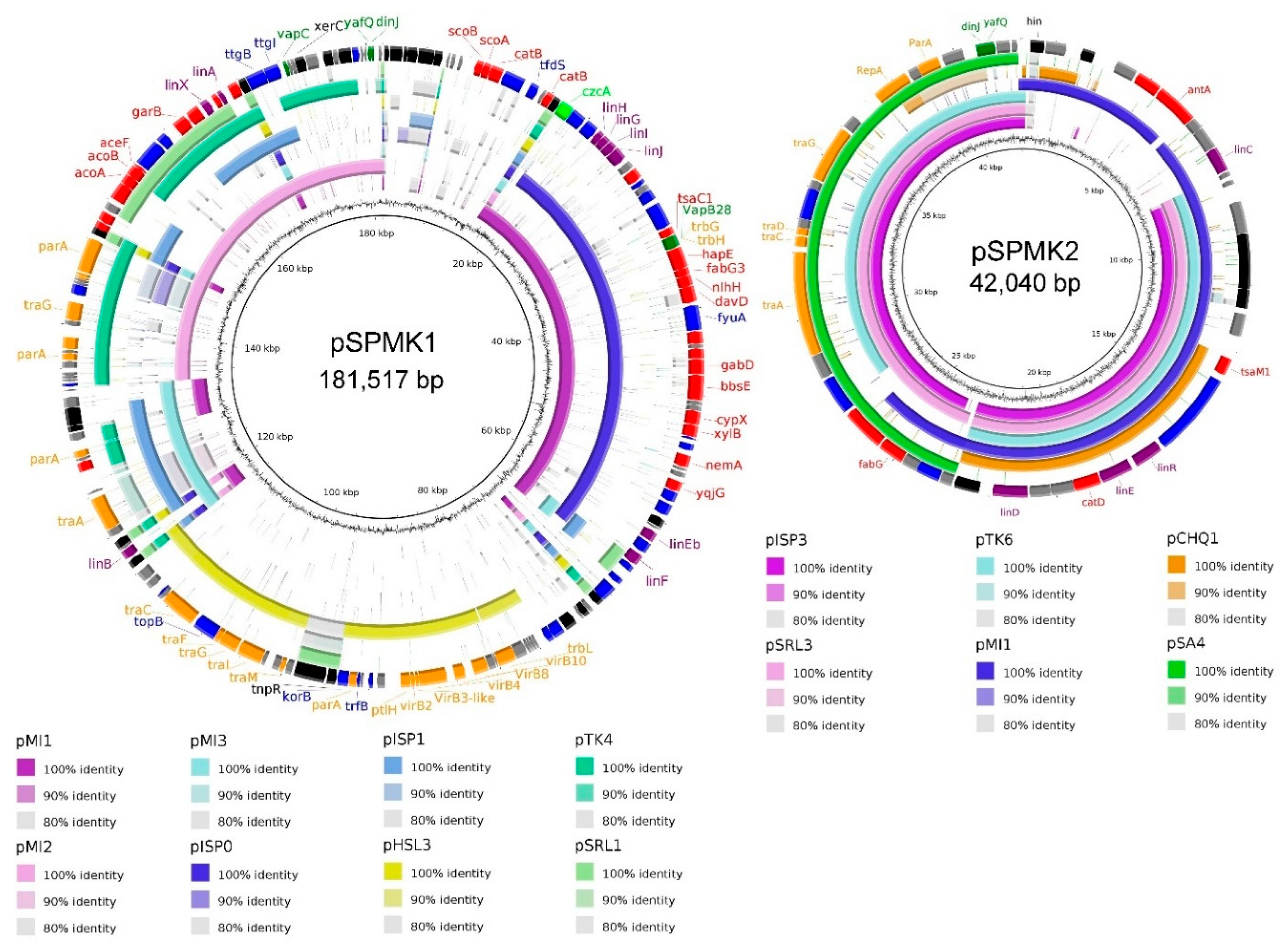
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nolan, K.; Kamrath, J.; Levitt, J. Lindane toxicity: A comprehensive review of the medical literature. Pediatr. Dermatol. 2012, 29, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, J.; Abhilash, P.C.; Li, Y.F.; Lal, R.; Forter, M.; Torres, J.; Singh, N.; Yunus, M.; Tian, C.; Schaffer, A.; et al. Hexachlorocyclohexane (HCH) as new Stockholm Convention POPs—A global perspective on the management of Lindane and its waste isomers. Environ. Sci. Pollut. Res. 2011, 18, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Manickam, N.; Pathak, A.; Saini, H.S.; Mayilraj, S.; Shanker, R. Metabolic profiles and phylogenetic diversity of microbial communities from chlorinated pesticides contaminated sites of different geographical habitats of India. J. Appl. Microbiol. 2010, 109, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Böltner, D.; Moreno-morillas, S.; Ramos, J. 16S rDNA phylogeny and distribution of lin genes in novel hexachlorocyclohexane-degrading Sphingomonas strains. Environ. Microbiol. 2005, 7, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.; Rani, P.; Singh, A.K.; Kumar, R.; Dwivedi, V.; Negi, V.; Lal, R. Sphingopyxis flava sp. nov., isolated from a hexachlorocyclohexane (HCH)-contaminated soil. Int. J. Syst. Evolut. Microbiol. 2015, 65, 3720–3726. [Google Scholar] [CrossRef] [PubMed]
- Jindal, S.; Dua, A.; Lal, R. Sphingopyxis indica sp. nov., isolated from a high dose point hexachlorocyclohexane (HCH)- contaminated dumpsite. Int. J. Syst. Evolut. Microbiol. 2013, 63, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Verma, M.; Bala, K.; Nigam, A.; Lal, R. Sphingopyxis ummariensis sp. nov., isolated from a hexachlorocyclohexane dump site. Int. J. Syst. Evolut. Microbiol. 2010, 60, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.D.; Wang, D.D.; Yang, S.Z.; Li, H.P.; Zhu, H.H. Genome-based reclassification of Sphingopyxis ummariensis as a later heterotypic synonym of Sphingopyxis terrae, with the descriptions of Sphingopyxis terrae subsp terrae subsp. nov. and Sphingopyxis terrae subsp. ummariensis subsp. nov. Int. J. Syst. Evolut. Microbiol. 2017, 67, 5279–5283. [Google Scholar]
- Lal, R.; Dadhwal, M.; Kumari, K.; Sharma, P.; Singh, A.; Kumari, H.; Jit, S.; Gupta, S.K.; Nigam, A.; Lal, D.; et al. Pseudomonas sp. to Sphingobium indicum: A journey of microbial degradation and bioremediation of Hexachlorocyclohexane. Indian J. Microbiol. 2008, 48, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Endo, R.; Kamakura, M.; Miyauchi, K.; Ohtsubo, Y.; Tsuda, M.; Fukuda, M.; Nagata, Y.I. Identification and characterization of genes involved in the downstream degradation pathway of γ -hexachlorocyclohexane in Sphingomonas paucimobilis UT26. J. Bacteriol. 2005, 187, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Endo, R.; Ito, M.; Ohtsubo, Y.; Tsuda, M. Aerobic degradation of lindane (γ-hexachlorocyclohexane) in bacteria and its biochemical and molecular basis. Appl. Microbiol. Biotechnol. 2007, 76, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Imai, R.; Nagata, Y.; Fukuda, M.; Takagi, M.; Yano, K. Molecular cloning of a Pseudomonas paucimobilis gene encoding a 17-kilodalton polypeptide that eliminates HCl molecules from γ-hexachlorocyclohexane. J. Bacteriol. 1991, 173, 6811–6819. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Futamura, A.; Miyauchi, K.; Takagi, M. Two different types of dehalogenases, LinA and LinB, involved in γ- hexachlorocyclohexane degradation in Sphingomonas paucimobilis UT26 are localized in the periplasmic space without molecular processing. J. Bacteriol. 1999, 181, 5409–5413. [Google Scholar] [PubMed]
- Nagata, Y.; Hatta, T.; Imai, R.; Kimbara, K.; Fukuda, M.; Yano, K.; Takagi, M. Purification and characterization of γ -hexachlorocyclohexane (γ -HCH) dehydrochlorinase (LinA) from Pseudomonas paucimobilis. Biosci. Biotechnol. Biochem. 1993, 57, 1582–1583. [Google Scholar] [CrossRef]
- Nagata, Y.; Mori, K.; Takagi, M.; Murzin, A.G.; Damborsk, J. Identification of protein fold and catalytic residues of γ-hexachlorocyclohexane dehydrochlorinase LinA. Proteins Struct. Funct. Genet. 2001, 45, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Macwan, A.S.; Kukshal, V.; Srivastava, N.; Javed, S.; Kumar, A.; Ramacheandran, R. Crystal structure of the hexachlorocyclohexane dehydrochlorinase (LinA-Type2): Mutational analysis, thermostability and enantioselectivity. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Okai, M.; Kubota, K.; Fukuda, M.; Nagata, Y.; Nagata, K.; Tanokura, M. Crystallization and preliminary X-ray analysis of- hexachlorocyclohexane dehydrochlorinase LinA from Sphingobium japonicum UT26. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 822–824. [Google Scholar] [CrossRef]
- Nagata, Y.; Nariya, T.; Ohtomo, R.; Fukuda, M.; Yano, K.; Takagi, M. Cloning and sequencing of a dehalogenase gene encoding an enzyme with hydrolase γ-hexachlorocyclohexane involved in the degradation of y-hexachlorocyclohexane. J. Bacteriol. 1993, 175, 6403–6410. [Google Scholar] [CrossRef]
- Kmunícek, J.; Hynková, K.; Jedlicka, T.; Nagata, Y.; Negri, A.; Gago, F.; Wade, R.C.; Damborsky, J. Quantitative analysis of substrate specificity of haloalkane dehalogenase LinB from Sphingomonas paucimobilis UT26. Biochemistry 2005, 44, 3390–3401. [Google Scholar] [CrossRef]
- Nagata, Y.; Ohtomo, R.; Miyauchi, K.; Fukuda, M.; Yano, K.; Takagi, M. Cloning and sequencing of a 2,5-dichloro-2,5-cyclohexadiene-1,4-diol dehydrogenase gene involved in the degradation of γ-hexachlorocyclohexane in Pseudomonas paucimobilis. J. Bacteriol. 1994, 176, 3117–3125. [Google Scholar] [CrossRef]
- Miyauchi, K.; Suh, S.K.; Nagata, Y.; Takagi, M. Cloning and sequencing of a 2,5-dichlorohydroquinone reductive dehalogenase gene whose product is involved in degradation of γ-hexachlorocyclohexane by Sphingomonas paucimobilis. J. Bacteriol. 1998, 180, 1354–1359. [Google Scholar] [PubMed]
- Miyauchi, K.; Adachi, Y.; Nagata, Y. Cloning and sequencing of a novel meta-cleavage dioxygenase gene whose product is involved in degradation of γ -Hexachlorocyclohexane in Sphingomonas paucimobilis. J. Bacteriol. 1999, 181, 6712–6719. [Google Scholar] [PubMed]
- Kaminski, M.A.; Sobczak, A.; Spolnik, G.; Danikiewicz, W.; Dziembowski, A.; Lipinski, L. Sphingopyxis lindanitolerans sp. nov. strain WS5A3pT enriched from a pesticide disposal site. Int. J. Syst. Evol. Microbiol. 2018, 68, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. Journal 2011, 17, 10–12. [Google Scholar]
- Joshi, N.; Fass, J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files [Internet]. 2011. Available online: https://github.com/najoshi/sickle (accessed on 1 December 2017).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibrlski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; OSTELL, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acid. Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; Heijne, G.V.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Method 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Bose, T.; Haque, M.M.; Reddy, C.; Mande, S.S. COGNIZER: A framework for functional annotation of metagenomic datasets. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef]
- Calteau, A.; Gachet, M.; Gautreau, G.; Josso, A.; Langlois, J.; Médigue, C.; Cruveiller, S.; Lajus, A.; Pereira, H.; Planel, P.; et al. MicroScope-an integrated resource for community expertise of gene functions and comparative analysis of microbial genomic and metabolic data. Brief Bioinform. 2017, 1–14. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.J.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Lauro, F.M.; Dougald, D.M.; Thomas, T.; Williams, T.J.; Egan, S.; Rice, S.; DeMaere, M.Z.; Ting, L.; Ertan, H.; Johnson, J.; et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci.USA 2009, 106, 15527–15533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsubo, Y.; Nonoyama, S.; Nagata, Y.; Numata, M.; Tsuchikane, K.; Hosoyama, A.; Yamazoe, A.; Tsuda, M.; Fujita, N.; Kawai, F. Complete genome sequence of Sphingopyxis terrae strain 203-1 (NBRC 111660), a polyethylene glycol degrader. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, Y.; Nagata, Y.; Numata, M.; Tsuchikane, K.; Hosoyama, A.; Yamazoe, A.; Tsuda, M.; Fujita, N.; Kawai, F. Complete genome sequence of polyvinyl alcohol-degrading strain Sphingopyxis sp. 113P3 (NBRC 111507). Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.Y.; Gan, H.M.; Tarasco, A.M.; Busairi, N.I.; Barton, H.A.; Hudson, A.O.; Savka, M.A. Whole-genome sequences of five oligotrophic bacteria isolated from deep within Lechuguilla Cave, New Mexico. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Shimizu, K.; Maseda, H.; Kawauchi, Y.; Utsumi, M.; Itayama, T.; Zhang, Z.Y.; Sugiura, N. Whole-genome sequence of the microcystin-degrading bacterium Sphingopyxis sp. strain C-1. Genome Announc. 2015, 3. [Google Scholar] [CrossRef]
- Kaminski, M.A.; Furmanczyk, E.M.; Dziembowski, A.; Sobczak, A.; Lipinski, L. Draft genome sequence of the type strain Sphingopyxis witflariensis DSM 14551. Genome Announc. 2017, 5. [Google Scholar] [CrossRef]
- Kaminski, M.A.; Furmanczyk, E.M.; Dziembowski, A.; Sobczak, A.; Lipinski, L. Draft genome sequence of the type strain Sphingopyxis bauzanensis DSM 22271. Genome Announc. 2017, 5. [Google Scholar] [CrossRef]
- Oelschlägel, M.; Rückert, C.; Kalinowski, J.; Schmidt, G.; Schlömann, M.; Tischler, D. Sphingopyxis fribergensis sp. nov., a soil bacterium with the ability to degrade styrene and phenylacetic acid. Int. J. Syst. Evolut. Microbiol. 2015, 65, 3008–3015. [Google Scholar] [CrossRef] [PubMed]
- García-Romero, I.; Pérez-Pulido, A.J.; González-Flores, Y.E.; Reyes-Ramírez, F.; Santero, E.; Floriano, B. Genomic analysis of the nitrate-respiring Sphingopyxis granuli (formerly Sphingomonas macrogoltabida) strain TFA. BMC Genom. 2016, 17, 93. [Google Scholar] [CrossRef]
- Ohtsubo, Y.; Nonoyama, S.; Nagata, Y.; Numata, M.; Tsuchikane, K.; Hosoyama, A.; Yamazoe, A.; Tsuda, M.; Fujita, N.; Kawai, F. Complete genome sequence of Sphingopyxis macrogoltabida strain 203N (NBRC 111659), a polyethylene glycol degrader. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Segata, N.; Börnigen, D.; Morgan, X.C.; Huttenhower, C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 2013, 4, 2304. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evolut. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Kato, H.; Mori, H.; Maruyama, F.; Toyoda, A.; Oshima, K.; Endo, R.; Fuchu, G.; Miyakoshi, M.; Dozono, A.; Ohtsubo, Y. Time-series metagenomic analysis reveals robustness of soil microbiome against chemical disturbance. DNA Res. 2015, 22, 413–424. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr Opin Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Lefébure, T.; Stanhope, M.J. Evolution of the core and pan-genome of Streptococcus: Positive selection, recombination, and genome composition. Genome Biol. 2007, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Bottacini, F.; Medini, D.; Pavesi, A.; Turroni, F.; Foroni, E.; Riley, D.; Giubellini, V.; Tettelin, H.; Sinderen, D.V.; Ventura, M. Comparative genomics of the genus Bifidobacterium. Microbiology 2010, 156, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Azam, S.; Lakshman Sagar, A.; Narasimha Rao, V.; Gudla, R.; Parapatla, H.; Yakkala, H.; Vemuri, S.G.; Siddavattam, D. Genome-guided insights reveal organophosphate-degrading Brevundimonas diminuta as Sphingopyxis wildii and define its versatile metabolic capabilities and environmental adaptations. Genome Biol. Evolut. 2017, 9, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.M.; Gocayne, J.D.; White, O.; Adams, M.D.; Clayton, R.A.; Fleischmann, R.D.; Bult, C.J.; Kerlavage, A.R.; Sutton, G.; Kelley, J.M.; et al. The minimal gene complement of Mycoplasma genitalium. Science 1995, 270, 397–403. [Google Scholar] [CrossRef]
- Koebnik, R.; Locher, K.P.; Van Gelder, P. Structure and function of bacterial outer membrane proteins: Barrels in a nutshell. Mol. Microbiol. 2000, 37, 239–353. [Google Scholar] [CrossRef]
- Tabata, M.; Ohhata, S.; Nikawadori, Y.; Sato, T.; Kishida, K.; Ohtsubo, Y.; Tsuda, M.; Nagata, Y. Complete genome sequence of a γ-hexachlorocyclohexane-degrading bacterium, Sphingobium sp. strain MI1205. Genome Announc. 2016, 4. [Google Scholar] [CrossRef]
- Kumar, S.; Mukerji, K.G.; Lai, R. Molecular aspects of pesticide degradation by microorganisms. Crit. Rev. Microbiol. 1996, 22, 1–26. [Google Scholar] [CrossRef]
- Ramakrishnan, B.; Venkateswarlu, K.; Sethunathan, N.; Megharaj, M. Local applications but global implications: Can pesticides drive microorganisms to develop antimicrobial resistance? Sci. Total Environ. 2019, 654, 177–189. [Google Scholar] [CrossRef]
- Tabata, M.; Ohhata, S.; Nikawadori, Y.; Kishida, K.; Sato, T.; Kawasumi, T.; Kato, H.; Ohtsubo, Y.; Tsuda, M.; Nagata, Y. Comparison of the complete genome sequences of four γ-hexachlorocyclohexane-degrading bacterial strains: Insights into the evolution of bacteria able to degrade a recalcitrant man-made pesticide. DNA Res. 2016, 23, 581–599. [Google Scholar] [CrossRef]
- Tabata, M.; Ohtsubo, Y.; Ohhata, S.; Tsuda, M.; Nagata, Y. Complete genome sequence of the γ-hexachlorocyclohexane-degrading bacterium Sphingomonas sp. strain MM-1. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Sangwan, N.; Lata, P.; Kaur, J.; Dua, A.; Singh, A.K.; Verma, M.; Kaur, J.; Khurana, J.P.; Khurana, P.; et al. Genome sequence of Sphingobium indicum B90A, a hexachlorocyclohexane-degrading bacterium. J Bacteriol. 2012, 194, 4471–4472. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.; Bajaj, A.; Kumar, R.; Kaur, J.; Anand, S.; Nayyar, N.; Puri, A.; Singh, Y.; Khurana, J.P.; Lal, R. Genome organization of Sphingobium indicum B90A: An archetypal hexachlorocyclohexane (HCH) degrading genotype. Genome Biol. Evolut. 2017, 9, 2191–2197. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, B.; Kos, P.B.; Balint, B.; Maroti, G.; Gan, H.M.; Perei, K.; Rákhely, G. Complete genome sequence of Novosphingobium resinovorum SA1, a versatile xenobiotic-degrading bacterium capable of utilizing sulfanilic acid. J. Biotechnol. 2017, 241, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Tabata, M.; Endo, R.; Ito, M.; Ohtsubo, Y.; Kumar, A.; Tsuda, M.; Nagata, Y. The lin genes for γ-hexachlorocyclohexane degradation in Sphingomonas sp. MM-1 proved to be dispersed across multiple plasmids. Biosci. Biotechnol. Biochem. 2011, 75, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.L.; Oakeshott, J.G.; Pandey, G. Insights into ongoing evolution of the hexachlorocyclohexane catabolic pathway from comparative genomics of ten Sphingomonadaceae strains. G3 (Bethesda). 2015, 5, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Dogra, C.; Raina, V.; Pal, R.; Suar, M.; Lal, S.; Gartemann, K.H.; Holliger, C.; Meer, J.R.V.D.; Lal, R. Organization of lin genes and IS6100 among different strains of hexachlorocyclohexane-degrading Sphingomonas paucimobilis: Evidence for horizontal gene transfer. J. Bacteriol. 2004, 186, 2225–2235. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, 32–36. [Google Scholar] [CrossRef]
- Sharma, P.; Pandey, R.; Kumari, K.; Pandey, G.; Jackson, C.J.; Russell, R.J.; Oakeshott, J.G.; Lal, R. Kinetic and sequence-structure-function analysis of known LinA variants with different hexachlorocyclohexane isomers. PLoS ONE. 2011, 6. [Google Scholar] [CrossRef]
- Kumari, R.; Subudhi, S.; Suar, M.; Dhingra, G.; Raina, V.; Dogra, C.; Lal, S.; Meer, J.R.V.D.; Holliger, C.; Lal, R. Cloning and characterization of lin genes responsible for the degradation of hexachlorocyclohexane isomers by Sphingomonas paucimobilis strain B90. Appl. Environ Microbiol. 2002, 68, 6021–6028. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.; Kumar, R.; Oldach, P.; Sangwan, N.; Khurana, J.P.; Gilbert, J.A.; Lal, R. Comparative genomic analysis of nine Sphingobium strains: Insights into their evolution and hexachlorocyclohexane (HCH) degradation pathways. BMC Genomics 2014, 15, 1014. [Google Scholar] [CrossRef] [PubMed]
- Macwan, A.S.; Javed, S.; Kumar, A. Isolation of a novel thermostable dehydrochlorinase (LinA) from a soil metagenome. 3Biotech 2011, 1, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, N.; Prokop, Z.; Kumar, A. Novel LinA type 3 δ-hexachlorocyclohexane dehydrochlorinase. Appl. Environ. Microbiol. 2015, 81, 7553–7559. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, N.; Macwan, A.S.; Kohler, H.P.E.; Kumar, A. Important amino acid residues of hexachlorocyclohexane dehydrochlorinases (LinA) for enantioselective transformation of hexachlorocyclohexane isomers. Biodegradation 2017, 28, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
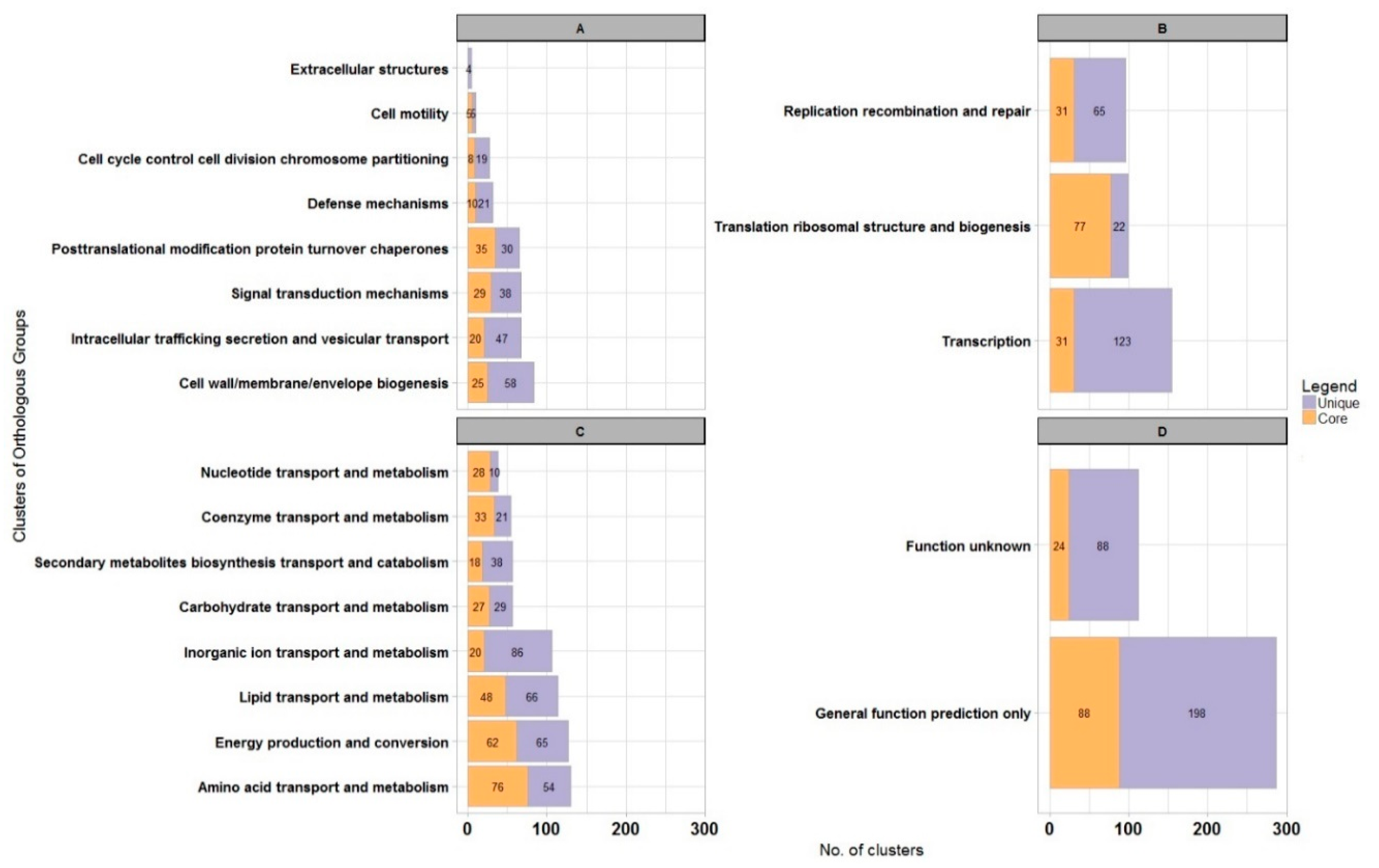
| Code | Description | WS5A3p | |
|---|---|---|---|
| Value | % | ||
| J | Translation, ribosomal structure and biogenesis | 241 | 6.8% |
| K | Transcription | 287 | 8.0% |
| L | Replication, recombination and repair | 319 | 8.9% |
| D | Cell cycle control, cell division, chromosome partitioning | 38 | 1.1% |
| V | Defense mechanisms | 81 | 2.3% |
| T | Signal transduction mechanisms | 179 | 5.0% |
| M | Cell wall/membrane/envelope biogenesis | 232 | 6.5% |
| N | Cell motility | 57 | 1.6% |
| W | Extracellular structures | 8 | 0.2% |
| U | Intracellular trafficking, secretion, and vesicular transport | 209 | 5.9% |
| O | Posttranslational modification, protein turnover, chaperones | 189 | 5.3% |
| C | Energy production and conversion | 381 | 10.7% |
| G | Carbohydrate transport and metabolism | 218 | 6.1% |
| E | Amino acid transport and metabolism | 512 | 14.3% |
| F | Nucleotide transport and metabolism | 91 | 2.5% |
| H | Coenzyme transport and metabolism | 175 | 4.9% |
| I | Lipid transport and metabolism | 481 | 13.5% |
| P | Inorganic ion transport and metabolism | 481 | 13.5% |
| Q | Secondary metabolites biosynthesis, transport and catabolism | 302 | 8.5% |
| R | General function prediction only | 651 | 18.2% |
| S | Function unknown | 277 | 7.8% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaminski, M.A.; Sobczak, A.; Dziembowski, A.; Lipinski, L. Genomic Analysis of γ-Hexachlorocyclohexane-Degrading Sphingopyxis lindanitolerans WS5A3p Strain in the Context of the Pangenome of Sphingopyxis. Genes 2019, 10, 688. https://doi.org/10.3390/genes10090688
Kaminski MA, Sobczak A, Dziembowski A, Lipinski L. Genomic Analysis of γ-Hexachlorocyclohexane-Degrading Sphingopyxis lindanitolerans WS5A3p Strain in the Context of the Pangenome of Sphingopyxis. Genes. 2019; 10(9):688. https://doi.org/10.3390/genes10090688
Chicago/Turabian StyleKaminski, Michal A., Adam Sobczak, Andrzej Dziembowski, and Leszek Lipinski. 2019. "Genomic Analysis of γ-Hexachlorocyclohexane-Degrading Sphingopyxis lindanitolerans WS5A3p Strain in the Context of the Pangenome of Sphingopyxis" Genes 10, no. 9: 688. https://doi.org/10.3390/genes10090688
APA StyleKaminski, M. A., Sobczak, A., Dziembowski, A., & Lipinski, L. (2019). Genomic Analysis of γ-Hexachlorocyclohexane-Degrading Sphingopyxis lindanitolerans WS5A3p Strain in the Context of the Pangenome of Sphingopyxis. Genes, 10(9), 688. https://doi.org/10.3390/genes10090688