Gene Expression Profiles and microRNA Regulation Networks in Tiller Primordia, Stem Tips, and Young Spikes of Wheat Guomai 301

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Morphology Observation
2.3. Sample Preparation and RNA Extraction
2.4. Transcriptome Sequencing and Data Analysis
2.5. miRNA Sequencing and Data Analyses
2.6. Prediction of the miRNA Target Genes
2.7. qRT-PCR
3. Results
3.1. Tillering and Spike Differentiating Traits of Guomai 301
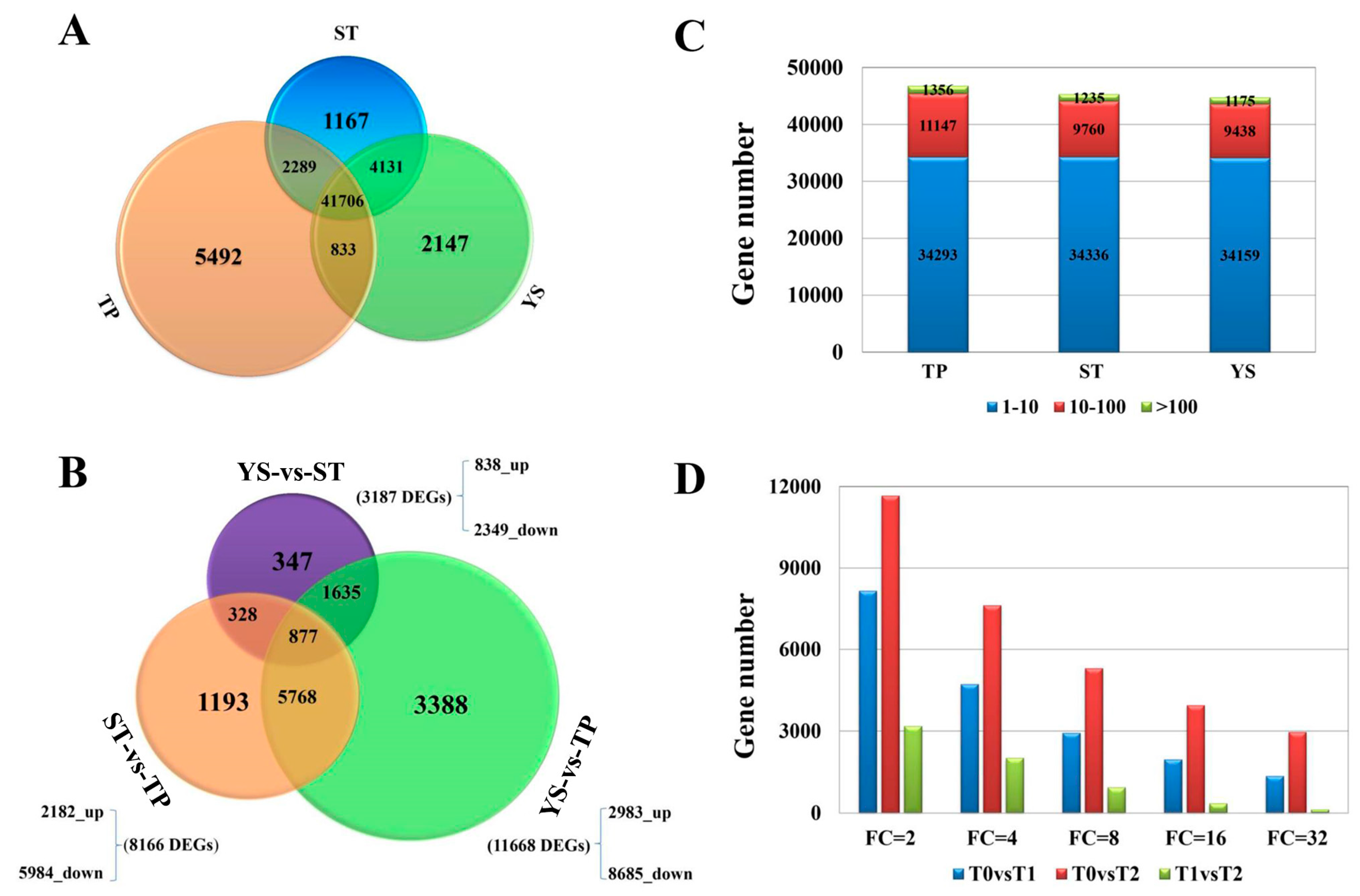
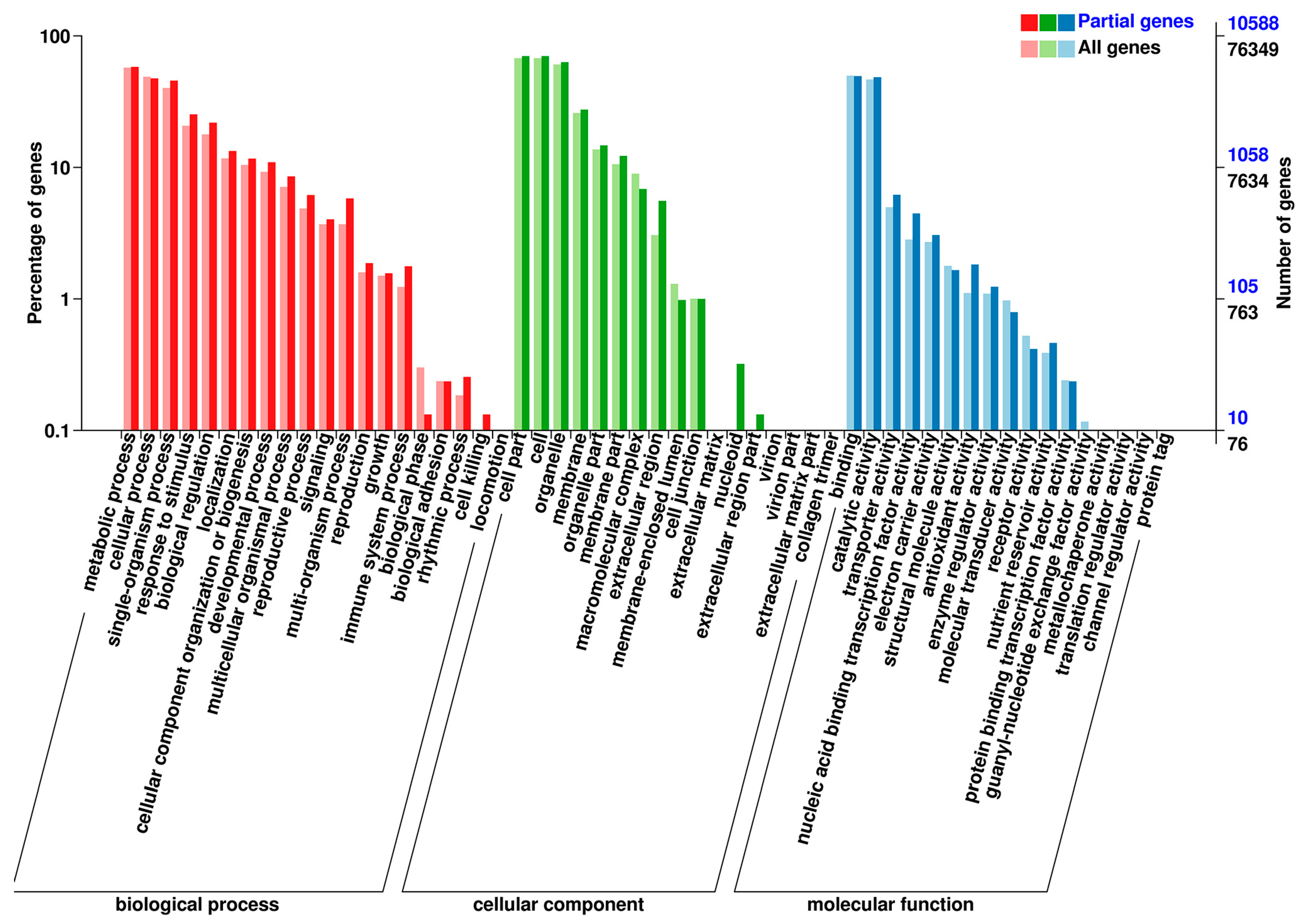
3.2. Overview of Transcriptome Sequencing Data
3.3. Overview of miRNA Sequencing Data
3.4. DEGs between TP and YS
3.5. Tissue-Specific Genes in Tiller Primordia
3.6. Tissue-Specific Genes in Young Spikes
3.7. DEG Co-Expression Clusters
3.8. DE miRNAs Between Tiller Primordia and Young Spikes
3.9. Tissue-Specific miRNAs in TPs
3.10. Tissue-Specific miRNAs in YSs
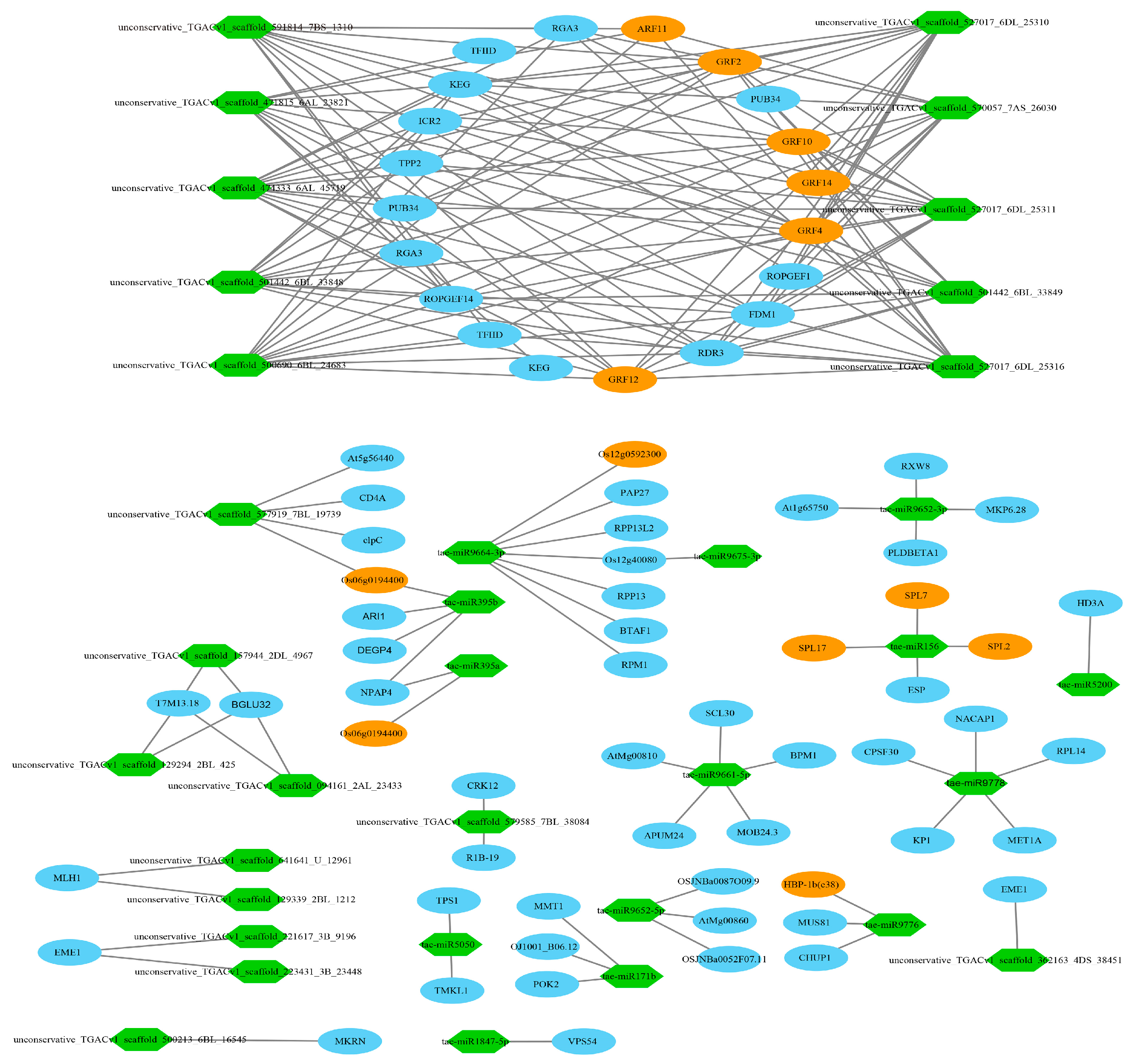
3.11. Target Relationship Between DE miRNAs and DE mRNAs in Tiller Primordia and Young Spikes
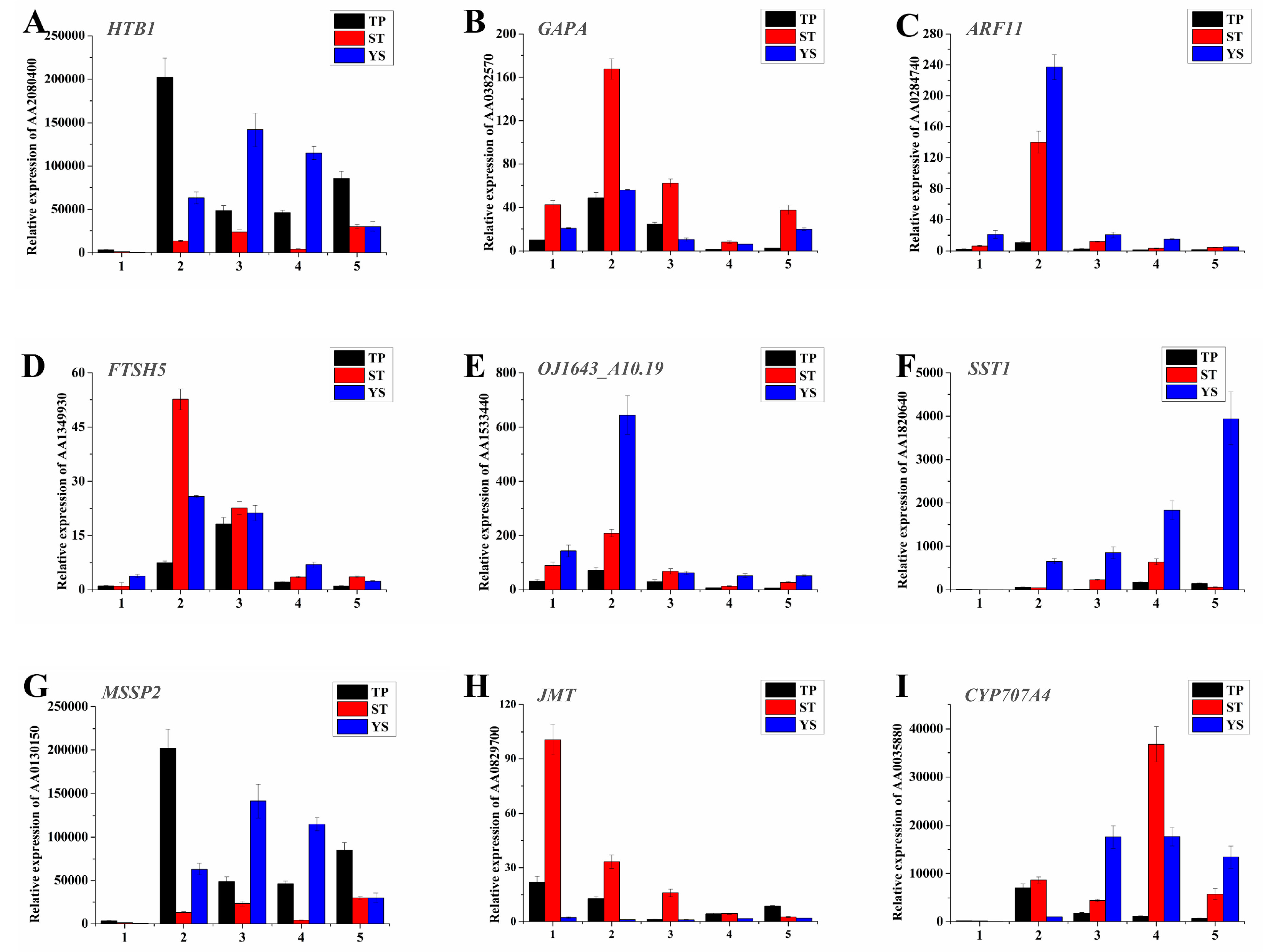
3.12. Expression Profiles of Nine Significant DEGs in Wheat Tiller Primordia, Stem Tips and Young Spikes
4. Discussion
4.1. Typical Expressed Genes During Wheat Tillering and Spike Differentiating
4.2. Typical Expressed miRNAs During Wheat Tillering and Spike Differentiating
4.3. Phytohormones and TFs Played Key Roles During Wheat Tillering and Spike Differentiating
4.4. Post-Transcriptional Regulation of Some Key miRNAs
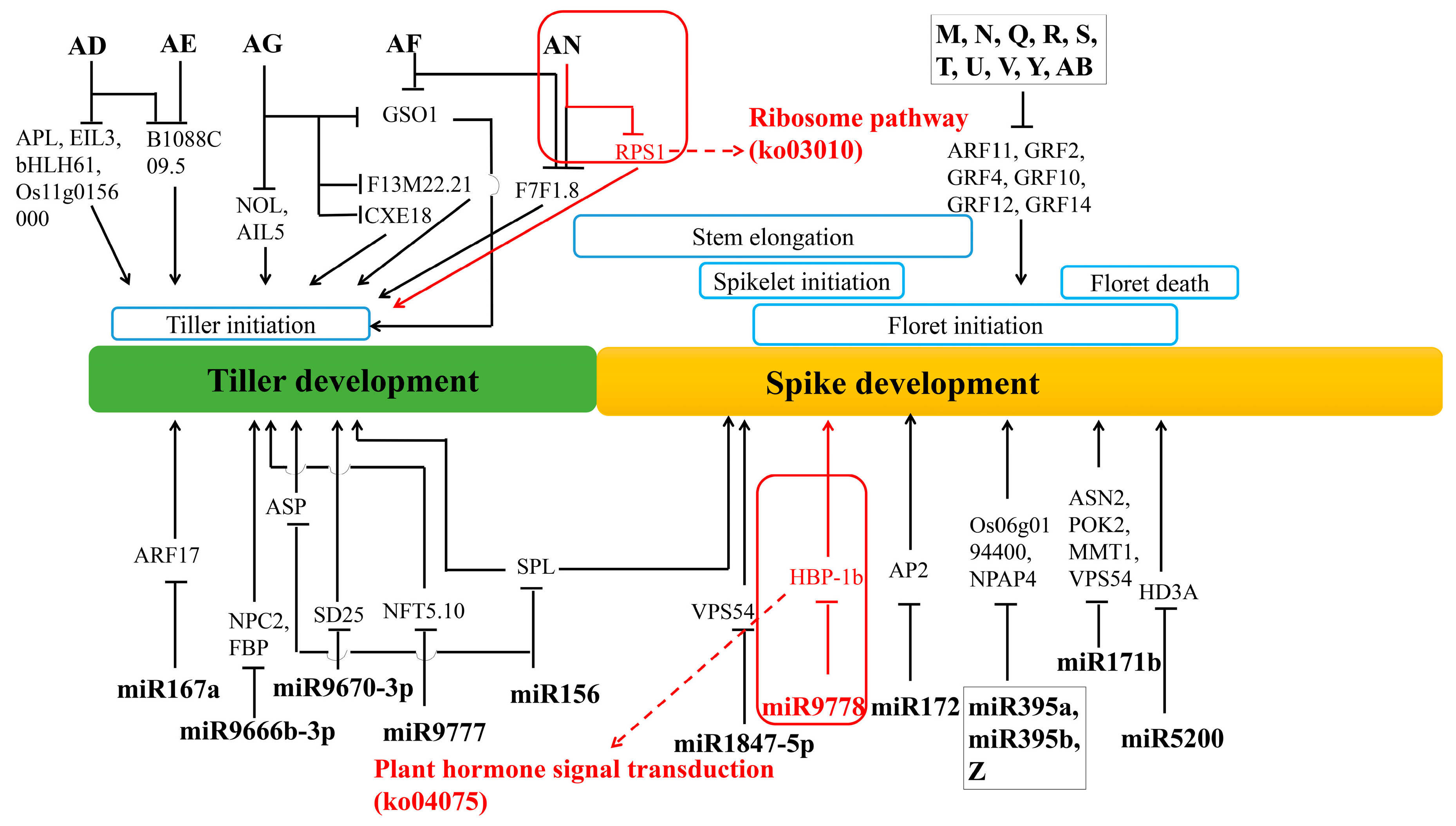
4.5. Regulation Networks of DE mRNA and DE miRNA in Tiller Primordia and Young Spikes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Innes, P.; Blackwell, R.D.; Austin, R.B.; Ford, M.A. The effects of selection for number of ears on the yield and water economy of winter wheat. J. Agric. Sci. 1981, 97, 523–532. [Google Scholar] [CrossRef]
- Kirby, E.J.M. Analysis of leaf, stem and ear growth in wheat from terminal spikelet stage to anthesis. Field Crops Res. 1988, 18, 127–140. [Google Scholar] [CrossRef]
- Feng, N.; Song, G.; Guan, J.; Chen, K.; Jia, M.; Huang, D.; Wu, J.; Zhang, L.; Kong, X.; Geng, S.; et al. Transcriptome profiling of wheat inflorescence development from spikelet initiation to floral patterning identified stage-specific regulatory genes. Plant Physiol. 2017, 174, 1779–1794. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, N.; Schnurbusch, T. A genetic playground for enhancing grain number in cereals. Trends Plant Sci. 2012, 17, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Vahamidis, P.; Karamanos, A.; Economou, G.; Fasseas, C. A new scale for the assessment of wheat spike morphogenesis. Ann. Appl. Biol. 2014, 64, 220–231. [Google Scholar] [CrossRef]
- He, R.; Ni, Y.; Li, J.; Jiao, Z.; Zhu, X.; Jiang, Y.; Li, Q.; Niu, J. Quantitative changes in the transcription of phytohormone-related genes: Some transcription factors are major causes of the wheat mutant dmc not tillering. Int. J. Mol. Sci. 2018, 19, 1324. [Google Scholar] [CrossRef] [PubMed]
- Spielmeyer, W.; Richards, R.A. Comparative mapping of wheat chromosome 1AS which contains the tiller inhibition gene (tin) with rice chromosome 5S. Theor. Appl. Genet. 2004, 109, 1303–1310. [Google Scholar] [CrossRef]
- Peng, Z.S.; Yen, C.; Yang, J.L. Genetic control of oligo-culms character in common wheat. Wheat Inf. Serv. 1998, 86, 19–24. [Google Scholar]
- Kuraparthy, V.; Sood, S.; Dhaliwal, H.S.; Chhuneja, P.; Gill, B.S. Identification and mapping of a tiller inhibition gene (tin3) in wheat. Theor. Appl. Genet. 2007, 114, 285–294. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, J.; Liu, W.; Lu, X.; Yang, X.; Gao, A.; Li, X.; Lu, Y.; Li, L. Genetic mapping of a fertile tiller inhibition gene, ftin, in wheat. Mol. Breed. 2013, 31, 441–449. [Google Scholar] [CrossRef]
- Xu, T.; Bian, N.; Wen, M.; Xiao, J.; Yuan, C.; Cao, A.; Zhang, S.; Wang, X.; Wang, H. Characterization of a common wheat (Triticum aestivum L.) high-tillering dwarf mutant. Theor. Appl. Genet. 2016, 130, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, Q.; Fu, Z.; Wang, Y.; Xiong, G.; Zeng, D.; Wang, X.; Liu, X.; Teng, S.; Hiroshi, F.; et al. Control of tillering in rice. Nature 2003, 42, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Pysh, L.D.; Wysocka-Diller, J.W.; Camilleri, C.; Bouchez, D.; Benfey, P.N. The GRAS gene family in Arabidopsis: Sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J. 1999, 18, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Shang, F.; Lin, Q.; Lou, C.; Zhang, J. Tillering and panicle branching genes in rice. Gene 2014, 537, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Kyozuka, J. Two-step regulation of LAX PANICLE1 protein accumulation in axillary meristem formation in rice. Plant Cell. 2009, 21, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Challis, R.J.; Hepworth, J.; Mouchel, C.; Waites, R.; Leyser, O. A role for MORE AXILLARY GROWTH 1 (MAX1) in evolutionary diversity in strigolactone signaling upstream of MAX2. Plant Physiol. 2013, 161, 1885–1902. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Maekawa, M.; Arite, T.; Onishi, K.; Takamure, I.; Kyozuka, J. Suppression of tiller bud activity in tillering dwarf mutants of rice. Plant Cell Physiol. 2005, 46, 79–86. [Google Scholar] [CrossRef]
- Zou, J.; Chen, Z.; Zhang, S.; Zhang, W.; Jiang, G.; Zhao, X.; Zhai, W.; Pan, X.; Zhu, L. Characterizations and fine mapping of a mutant gene for high tillering and dwarf in rice (Oryza sativa L.). Planta 2005, 222, 604–612. [Google Scholar] [CrossRef]
- Booker, J.; Auldridge, M.; Wills, S. MAX3/CCD7 is a carotenoid cleavage dioxygenase required for the synthesis of a novel plant signaling molecule. Curr. Biol. 2004, 14, 1232–1238. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, J.; Li, Z.; Gu, F.; Zhang, H.; Tang, S.; Gu, M.; Liang, G. Fine mapping and cloning of MT1, a novel allele of D10. Prog. Nat. Sci. 2009, 19, 1683–1689. [Google Scholar] [CrossRef]
- Arite, T.; Umehara, M.; Ishikawa, S.; Hanada, A.; Maekawa, M.; Yamaguchi, S.; Kyozuka, J. d14, a strigolactone-insensitive mutant of rice, shows an accelerated outgrowth of tillers. Plant Cell Physiol. 2009, 50, 1416–1424. [Google Scholar] [CrossRef]
- Lin, H.; Wang, R.; Qian, Q.; Yan, M.; Meng, X.; Fu, Z.; Yan, C.; Jiang, B.; Su, Z.; Li, J.; et al. DWARF27, an iron-containing protein required for the biosynthesis of strigolactones, regulates rice tiller bud outgrowth. Plant Cell. 2009, 21, 1512–1525. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, W.; Eggert, K.; Charnikhova, T.; Bouwmeester, H.; Schweizer, P.; Hajirezaei, M.R.; Seiler, C.; Sreenivasulu, N.; Wirén, N.; et al. Abscisic acid influences tillering by modulation of strigolactones in barley. J. Exp. Bot. 2018, 69, 3883–3898. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, W.; Pautler, M.; Jackson, D.; Hirano, H. Grass meristems II: Inflorescence architecture, flower development and meristem fate. Plant Cell Physiol. 2013, 54, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Guo, Y.; Xu, Y.; Li, R.; Li, S. Identification of a distal spikelet sterility mutant in wheat and spikelet morphological development. J. Triticeae Crop. 2008, 28, 249–253. [Google Scholar] [CrossRef]
- Peng, Z.; Yang, Z.; Ouyang, Z.; Yang, H. Characterization of a novel pistillody mutant in common wheat. Aust. J. Crop. Sci. 2013, 7, 159–164. [Google Scholar]
- Derbyshire, P.; Byrne, M.E. MORE SPIKELETS1 is required for spikelet fate in the inflorescence of Brachypodium. Plant Physiol. 2013, 161, 1291–1302. [Google Scholar] [CrossRef]
- Duan, Z.; Shen, C.; Li, Q.; Lv, G.; Ni, Y.; Yu, D.; Niu, J. Identification of a novel male sterile wheat mutant dms conferring dwarf status and multi-pistils. J. Integr. Agric. 2015, 14, 1706–1714. [Google Scholar] [CrossRef]
- Sormacheva, I.; Golovnina, K.; Vavilova, V.; Kosuge, K.; Watanabe, N.; Blinov, A.; Goncharov, N.P. Q gene variability in wheat species with different spike morphology. Genet. Resour. Crop Evol. 2015, 62, 1–16. [Google Scholar] [CrossRef]
- Faris, J.D.; Fellers, J.P.; Brooks, S.A.; Gill, B.S. A bacterial artificial chromosome contig spanning the major domestication locus Q in wheat and identification of a candidate gene. Genetics 2003, 164, 311–321. [Google Scholar]
- Guo, Z.; Chen, D.; Schnurbusch, T. Plant and floret growth at distinct developmental stages during the stem elongation phase in wheat. Front. Plant Sci. 2018, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhao, Y.; Röder, M.S.; Reif, J.C.; Ganal, M.W.; Chen, D.; Schnurbusch, T. Manipulation and prediction of spike morphology traits for the improvement of grain yield in wheat. Sci. Rep. 2018, 8, 14435. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, S.; Golan, G.; Guo, Z.; Ogawa, T.; Tagiri, A.; Sugimoto k Bernhardt, N.; Brassac, J.; Mascher, M.; Hensel, G.; Ohnishi, S.; et al. Unleashing floret fertility in wheat through the mutation of a homeobox gene. Proc. Natl. Acad. Sci. USA 2019, 116, 5182–5187. [Google Scholar] [CrossRef] [PubMed]
- Wolde, G.M.; Mascher, M.; Schnurbusch, T. Genetic modification of spikelet arrangement in wheat increases grain number without significantly affecting grain weight. Mol. Genet. Genom. 2019, 294, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Boden, S.A.; Cavanagh, C.; Cullis, B.R.; Ramm, K.; Greenwood, J.; Finnegan, E.J.; Trevaskis, B.; Swain, S.M. Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat. Plants 2015, 1, 14016. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaya, O.; Pont, C.; Sibout, R.; Martinek, P.; Badaeva, E.; Murat, F.; Chosson, A.; Watanabe, N.; Prat, E.; Gautier, N.; et al. Frizzy panicle drives supernumerary spikelets in bread wheat. Plant Physiol. 2015, 167, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Pautler, M.; Tanaka, W.; Hirano, H.; Jackson, D. Grass meristems I: Shoot apical meristem maintenance, axillary meristem determinacy and the floral transition. Plant Cell Physiol. 2013, 54, 302–312. [Google Scholar] [CrossRef]
- Chuck, G.; Meeley, R.; Irish, E.; Sakai, H.; Hake, S. The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1. Nat. Genet. 2007, 39, 1517–1521. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Helliwell, C.A. Regulation of flowering time and floral patterning by miR172. J. Exp. Bot. 2011, 62, 487–495. [Google Scholar] [CrossRef]
- Wang, L.; Sun, S.; Jin, J.; Fu, D.; Yang, X.; Weng, X.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. Coordinated regulation of vegetative and reproductive branching in rice. Proc. Natl. Acad. Sci. USA 2015, 112, 15504–15509. [Google Scholar] [CrossRef]
- Yu, N.; Niu, Q.W.; Ng, K.H. The role of miR156/SPLs modules in Arabidopsis lateral root development. Plant J. 2015, 83, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cheng, X.; Liu, P.; Sun, J. miR156-Targeted SBP-Box transcription factors interact with DWARF53 to regulate TEOSINTE BRANCHED1 and BARREN STALK1 expression in bread wheat. Plant Physiol. 2017, 174, 1931. [Google Scholar] [CrossRef]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Salinas, M.; Höhmann, S.; Berndtgen, R.; Huijser, P. miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell. 2010, 22, 3935–3950. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Grande, A.V.; Bujdoso, N.; Saedler, H.; Huijser, P. The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol. Biol. 2008, 67, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Gou, J.; Felippes, F.; Liu, C.; Weigel, D.; Wang, J. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell. 2011, 23, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Gandikota, M.; Birkenbihl, R.P.; Höhmann, S.; Cardon, G.H.; Saedle, H.; Huijser, P. The miRNA156/157 recognition element in the 3’ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2006, 49, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Cardon, G.; Hohmann, S.; Klein, J.; Nettesheim, K.; Saedler, H.; Huijser, P. Molecular characterisation of the Arabidopsis SBP-box genes. Gene 1999, 237, 91–104. [Google Scholar] [CrossRef]
- Wang, J.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef]
- Alonso-Peral, M.M.; Li, J.; Li, Y.; Allen, R.S.; Schnippenkoetter, W.; Ohms, S.; White, R.G.; Millar, A.A. The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol. 2010, 154, 757–771. [Google Scholar] [CrossRef]
- Glazińska, P.; Wojciechowski, W.; Wilmowicz, E.; Zienkiewicz, A.; Frankowski, K.; Kopcewicz, J. The involvement of InMIR167 in the regulation of expression of its target gene InARF8, and their participation in the vegetative and generative development of Ipomoea nil plants. J. Plant Physiol. 2014, 171, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Schommer, C.; Debernardi, J.M.; Bresso, E.G.; Rodriguez, R.E.; Palatnik, J.F. Repression of cell proliferation by miR319-regulated TCP4. Mol. Plant. 2014, 7, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, H.; Tian, C.; Sajjad, M.; Gao, C.; Tong, Y.; Wang, X.; Jiao, Y. Transcriptome association identifies regulators of wheat spike architecture. Plant Physiol. 2017, 175, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Duncan, D.; Morrow, D.J.; Fernandes, J.; Walbot, V. Transcriptome profiling of maize anthers using genetic ablation to analyze pre-meiotic and tapetal cell types. Plant J. 2010, 50, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Niu, J.; Yang, X.; He, D.; Wang, C. Impacts of powdery mildew on wheat grain sugar metabolism and sstarch accumulation in de veloping grains. Starke 2015, 66, 947–958. [Google Scholar] [CrossRef]
- Li, Q.; Qin, Z.; Jiang, Y.; Shen, C.; Duan, Z.; Niu, J. Screening for resistance to black point in wheat and the effects of diseased kernels on seed germination. J. Plant Dis. Prot. 2014, 121, 79–88. [Google Scholar] [CrossRef]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enricheded pathways and diseases. Nucleic Acids Res. 2011, 39 (Suppl. 2), W316–W322. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res 2013, 2, 188. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Curr. Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Pearson, E. The Test of Significance for the Correlation Coefficient. J. Am. Stat. Assoc. 1931, 26, 128–134. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T.; Tibshirani, R. Sparse Principal Component Analysis. J. Comput. Graph. Stat. 2010, 15, 265–286. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A K-means clustering algorithm. Appl. Stat. 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, L.; Wang, J.; Gu, P.Z.; Chen, M. MTide: An integrated tool for the identification of miRNA-target interaction in plants. Bioinformatics 2015, 31, 290–291. [Google Scholar] [CrossRef]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan1, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; et al. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef]
- Bo, X.; Wang, S. TargetFinder: A software for antisense oligonucleotide target site selection based on MAST and secondary structures of target mRNA. Bioinformatics 2005, 21, 1401–1402. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wu, S.; Houten, V.J.; Wang, Y.; Ding, B.; Fei, Z.; Clarke, T.H.; Reed, J.W.; Knaap, E.V.D. Down-regulation of AUXIN RESPONSE FACTORS 6 and 8 by microRNA 167 leads to floral development defects and female sterility in tomato. J. Exp. Bot. 2014, 65, 2507–2520. [Google Scholar] [CrossRef] [PubMed]
- Alshalalfa, M. MicroRNA response elements-mediated mirna-mirna interactions in prostate cancer. Adv. Bioinf. 2014, 2012, 839837. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Martino, M.T.D.; Tagliaferri, P.; Tassone, P.; Cannataro, M. Analysis of miRNA, mRNA, and TF interactions through network-based methods. EURASIP J. Bioinf. Syst. Biol. 2015, 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- King, L.S.; Kozono, D.; Agre, P. From structure to disease: The evolving tale of aquaporin biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 687–698. [Google Scholar] [CrossRef]
- Kamiya, T.; Tanaka, M.; Mitani, N.; Ma, J.F.; Maeshima, M.; Fujiwara, T. NIP1;1, an aquaporin homolog, determines the arsenite sensitivity of Arabidopsis thaliana. J. Biol. Chem. 2009, 284, 2114–2120. [Google Scholar] [CrossRef]
- Xu, W.; Dai, W.; Yan, H.; Li, S.; Shen, H.; Chen, Y.; Xu, H.; Sun, Y.; He, Z.; Ma, M. Arabidopsis NIP3;1 plays an important role in arsenic uptake and root-to-shoot translocation under arsenite stress conditions. Mol. Plant. 2015, 8, 722–733. [Google Scholar] [CrossRef]
- Lee, S.H.; Chung, G.C.; Jang, J.Y.; Ahn, S.J.; Zwiazek, J.J. Overexpression of PIP2;5 aquaporin alleviates effects of low root temperature on cell hydraulic conductivity and growth in Arabidopsis. Plant Physiol. 2012, 159, 479–488. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, W.; Hong, C.; Lur, H.; Chang, M. Comprehensive analysis of differentially expressed rice actin depolymerizing factor gene family and heterologous overexpression of confers drought tolerance. Rice 2012, 5, 33. [Google Scholar] [CrossRef]
- Fukaki, H.; Tameda, S.; Masuda, H.; Tasaka, M. Lateral root formation is blocked by a gain-of-function mutation in the SOLITARY-ROOT/IAA14 gene of Arabidopsis. Plant J. 2002, 29, 153–168. [Google Scholar] [CrossRef]
- Tian, C.; Muto, H.; Higuchi, K.; Matamura, T.; Tatematsu, K.; Koshiba, T.; Yamamoto, K.T. Disruption and overexpression of auxin response factor 8 gene of Arabidopsis affect hypocotyl elongation and root growth habit, indicating its possible involvement in auxin homeostasis in light condition. Plant J. 2004, 40, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Alboresi, A.; Gerotto, C.; Cazzaniga, S.; Bassi, R.; Morosinotto, T. A red-shifted antenna protein associated with photosystem II in Physcomitrella patens. J. Biol. Chem. 2011, 286, 28978. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, L.; Cai, Q.; Hu, Y.; Jin, Z.; Zhao, X.; Fan, W.; Huang, Q.; Luo, Z.; Chen, M.; et al. OsMADS32 interacts with PI-like proteins and regulates rice flower development. J. Integr. Plant Biol. 2015, 57, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Khanday, I.; Yadav, S.R.; Vijayraghavan, U. Rice LHS1/OsMADS1 controls floret meristem specification by coordinated regulation of transcription factors and hormone signaling pathways. Plant Physiol. 2013, 161, 1970–1983. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.H.; Lee, S.; Cho, L.H.; Kim, S.L.; Lee, Y.S.; Choi, S.C.; Jeong, H.J.; Yi, J.; Park, S.J.; Han, C.D.; et al. OsMADS50 and OsMADS56 function antagonistically in regulating long day (LD)-dependent flowering in rice. Plant Cell Environ. 2009, 32, 1412–1427. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tian, L.; Latoszek-Green, M.; Brown, D.; Wu, K. Arabidopsis ERF4 is a transcriptional repressor capable of modulating ethylene and abscisic acid responses. Plant Mol. Biol. 2005, 58, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wu, T.; Liu, S.; Liu, X.; Jiang, L.; Wan, J. Disruption of OsARF19 is critical for floral organ development and plant architecture in Rice (Oryza sativa L.). Plant Mol. Biol. Rep. 2015, 34, 748–760. [Google Scholar] [CrossRef]
- Varaud, E.; Brioudes, F.; Szécsi, J.; Leroux, J.; Brown, S.; Perrot-Rechenmann, C.; Bendahmane, M. AUXIN RESPONSE FACTOR8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell 2011, 449–453. [Google Scholar] [CrossRef]
- Mulki, M.A.; Bi, X.; Korff, M. FLOWERING LOCUS T3 control spikelet initiation but not floral development. Plant Physiol. 2018, 178, 1170–1186. [Google Scholar]
- Zhu, C.; Ding, Y.; Liu, H. MiR398 and plant stress responses. Physiol. Plant. 2011, 143, 1–9. [Google Scholar] [CrossRef]
- Rodriguez, R.E.; Ercoli, M.F.; Debernardi, J.M.; Breakfield, N.W.; Mecchia, M.A.; Sabatini, M.; Cools, T.; Veylder, L.D.; Benfey, P.N.; Palatnik, J.F. MicroRNA miR396 regulates the switch between stem cells and transit-amplifying cells in Arabidopsis roots. Plant Cell. 2015, 27, 3354. [Google Scholar] [CrossRef] [PubMed]
- Curaba, J.; Talbot, M.; Li, Z.; Helliwell, C. Over-expression of microRNA171 affects phase transitions and floral meristem determinancy in barley. BMC Plant Biol. 2013, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Xu, Q. Progress in research of microRNA regulation on angiosperm flower development. Plant Physiol. 2012, 48, 929–940. [Google Scholar] [CrossRef]
- Naya, L.; Khan, G.A.; Sorin, C.; Hartmann, C.; Crespi, M.; Lelandais-Brière, C. Cleavage of a non-conserved target by a specific miR156 isoform in root apexes of Medicago truncatula. Plant Signal Behav. 2010, 5, 328. [Google Scholar] [CrossRef] [PubMed]
- Vysotskaya, L.B.; Cherkoz’Yanova, A.V.; Veselov, S.Y.; Kudoyarova, G.R. Role of auxins and cytokinins in the development of lateral roots in wheat plants with several roots removed. J. Plant Physiol. 2007, 54, 402–406. [Google Scholar] [CrossRef]
- Brocard, I.M.; Lynch, T.J.; Finkelstein, R.R. Regulation and role of the Arabidopsis abscisic acid-insensitive 5 gene in abscisic acid, sugar, and stress response. Plant Physiol. 2002, 129, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Ueguchi-tanaka, M.; Matsuoka, M. Brassinosteroids and rice architecture. J. Pestic. Sci. 2004, 29, 184–188. [Google Scholar] [CrossRef]
- Liu, Y.J.; Gao, S.Q.; Tang, Y.M.; Gong, J.; Zhang, X.; Wang, Y.B.; Zhang, L.P.; Sun, R.W.; Zhang, Q.; Chen, Z.B.; et al. Transcriptome analysis of wheat seedling and spike tissues in the hybrid Jingmai 8 uncovered genes involved in heterosis. Planta 2018, 247, 1307–1321. [Google Scholar] [CrossRef]
- Paolo, S.D.; Gaudio, L.; Aceto, S. Analysis of the TCP genes expressed in the inflorescence of the orchid Orchis italica. Sci. Rep. 2015, 5, 16265. [Google Scholar] [CrossRef] [PubMed]
- Sorin, C.; Bussell, J.D.; Camus, I.; Ljung, K.; Kowalczyk, M.; Geiss, G.; McKhann, H.; Garcion, C.; Vaucheret, H.; Sandberg, G.; et al. Auxin and light control of adventitious rooting in Arabidopsis require ARGONAUTE1. Plant Cell 2005, 17, 1343–1359. [Google Scholar] [CrossRef]
- Ohashi-Ito, K.; Saegusa, M.; Iwamoto, K.; Oda, Y.; Katayama, H.; Kojima, M.; Sakakibara, H.; Fukuda, H. A bHLH complex activates vascular cell division via cytokinin action in root apical meristem. Curr. Biol. 2014, 24, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Gupta, S.M.; Gupta, A.K.; Gaur, V.S.; Kumar, A. (2014) Fluctuation of Dof1/Dof2 expression ratio under the influence of varying nitrogen and light conditions: Involvement in differential regulation of nitrogen metabolism in two genotypes of finger millet (Eleusine coracana L.). Gene 2014, 546, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.M.; Silva, E.M.; Saldanha, L.L.; Adachi, S.A.; Schley, T.R.; Rodrigues, T.M.; Dokkedal, A.L.; Nogueira, F.T.S.; Almeida, L.F.R. Flavonoids modify root growth and modulate expression of SHORT-ROOT and HD-ZIP III. J. Plant Physiol. 2015, 188, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; An, G. Isolation and characterization of a rice MADS box gene belonging to the AGL2 gene family. Mol. Cells 1997, 7, 45. [Google Scholar] [PubMed]
- Liu, Z.; Makaroff, C.A. Arabidopsis separase AESP is essential for embryo development and the release of cohesin during meiosis. Plant Cell 2006, 18, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Miao, L.; Huo, R.; Song, X.; Johnson, C.; Kong, L.; Sundaresan, V.; Yu, X. ARF2–ARF4 and ARF5 are essential for female and male gametophyte development in Arabidopsis. Plant Cell Physiol. 2018, 59, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Bartel, B.; Bartel, B. microRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Pathway | Ko id | p-Value | Corrected p-Value |
|---|---|---|---|---|
| Tiller primordia | Ribosome | ko03010 | 3.24 × 10−13 | 3.34 × 10−11 |
| Biosynthesis of amino acids | ko01230 | 2.83 × 10−7 | 2.91 × 10−5 | |
| Nitrogen metabolism | ko00910 | 3.28 × 10−7 | 3.38 × 10−5 | |
| Stem tips | Photosynthesis - antenna proteins | ko00196 | 0 | 0 |
| Carbon fixation in photosynthesis | ko00710 | 2.84 × 10−11 | 1.30 × 10−9 | |
| Carbon metabolism | ko01200 | 1.52 × 10−6 | 6.98 × 10−5 | |
| Young spikes | Plant hormone signal transduction | ko04075 | 3.03 × 10−5 | 0 |
| DNA replication | ko03030 | 0.01 | 0.23 |
| miRNA ID 1 | miRNA Sequence | log2FC | Target Genes or Transcription Factor Genes 2 |
|---|---|---|---|
| miR156 | TGACAGAAGAGAGTGAGCACA | −5.9 | ESP, SPL2, SPL7, SPL17 |
| miR167a | TGAAGCTGCCAGCATGATCTA | 2.6 | CER1, FH2, WRKY19, ARF17, MNJ7.15 |
| miR9776 | TTGGACGAGGATGTGCAACTG | −3.61 | MUS81, CHUP1, HBP-1b(c38) |
| M | UUCCACAGCUUUCUUGAACUG | −3.76 | GRF12, ARF11, RDR3, FDM1, KEG, GRF4, ROPGEF14, GRF10, GRF2, RGA3, TFIID |
| N | UUCCACAGCUUUCUUGAACUU | −3.7 | TPP2, ICR2, GRF12, RDR3, FDM1, KEG, GRF4, ROPGEF14, PUB34, GRF10, GRF2, RGA3 |
| P | UGUUCUGAAGAAACUGUCACC | −2.19 | MKRN |
| Q | UUCCACAGCUUUCUUGAACUG | −3.76 | GRF12, ARF11, RDR3, FDM1, KEG, GRF4, ROPGEF14, GRF10, GRF2, RGA3, TFIID |
| R | UUCCACAGCUUUCUUGAACUU | −3.7 | TPP2, ICR2, GRF12, RDR3, FDM1, KEG, GRF4, ROPGEF14, PUB34, GRF10, GRF2, RGA3 |
| S | UUCCACAGCUUUCUUGAACUU | −3.7 | TPP2, ICR2, GRF12, RDR3, FDM1, KEG, GRF4, ROPGEF14, PUB34, GRF10, GRF2 |
| T | UUCCACAGCUUUCUUGAACUU | −3.7 | TPP2, ICR2, GRF12, RDR3, FDM1, KEG, GRF14, ROPGEF1, PUB34, GRF10, GRF4, GRF2 |
| U | UUCCACAGCUUUCUUGAACUU | −3.7 | TPP2, ICR2, GRF12, RDR3, FDM1, KEG, GRF4, ROPGEF14, PUB34, GRF10, GRF2, RGA3 |
| V | UUCCACAGCUUUCUUGAACUG | −3.76 | GRF12, ARF11, RDR3, FDM1, KEG, GRF4, ROPGEF14, GRF10, GRF2, RGA3, TFIID |
| Y | UUCCACAGCUUUCUUGAACUG | −3.76 | GRF12, ARF11, RDR3, FDM1, KEG, GRF4, ROPGEF14, GRF10, GRF2, RGA3, TFIID |
| AD | CCUGUUGAGCUUGACCCC | 2.41 | CESA7, CYP71A1, LOGL10, F3L12.7, At1g68400, OSJNBa0006L06.10, GDPDL2, RPL28, At5g67360, At5g16590, T17J13.110, OJ1123_G04.23, F13M22.21, CYP93A2, XTH8, FL2, YSL16, Os11g0156000, At3g14460, CYP93A3, CYP72A15, HT1, T26J14.10, FL1, TBL38, Ole10, FSD1, MAP65-6, ST3, OsJ_09915, T1D16.11, B1088C09.5, MJG19.3, RBOHC, FAD7, CEBIP, bHLH61, LACS9, T7N9.10, tsf, BAG3, EIL3, F13G24.190, F27K19.170, At4g16820, KCS12, AOC4, APL, F17I23.50, F5D14.23, CYP711A1, Os01g0267300, PAO, KCS11, PUB11, NRPE1, UGT85A23, FSD1, BGLU7, CYP93A1, BGLU28, FSD2, RPP13L4, LECRKS4 |
| AJ | UCGGACCAGGCUUCAUUCCUU | 2.02 | HOX29 |
| AL | UCGGACCAGGCUUCAUUCCUU | 2.02 | HOX29, HOX32 |
| AM | UCGGACCAGGCUUCAUUCCUU | 2.02 | HOX29, HOX32 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Jiao, Z.; He, R.; Sun, Y.; Xu, Q.; Zhang, J.; Jiang, Y.; Li, Q.; Niu, J. Gene Expression Profiles and microRNA Regulation Networks in Tiller Primordia, Stem Tips, and Young Spikes of Wheat Guomai 301. Genes 2019, 10, 686. https://doi.org/10.3390/genes10090686
Li J, Jiao Z, He R, Sun Y, Xu Q, Zhang J, Jiang Y, Li Q, Niu J. Gene Expression Profiles and microRNA Regulation Networks in Tiller Primordia, Stem Tips, and Young Spikes of Wheat Guomai 301. Genes. 2019; 10(9):686. https://doi.org/10.3390/genes10090686
Chicago/Turabian StyleLi, Junchang, Zhixin Jiao, Ruishi He, Yulong Sun, Qiaoqiao Xu, Jing Zhang, Yumei Jiang, Qiaoyun Li, and Jishan Niu. 2019. "Gene Expression Profiles and microRNA Regulation Networks in Tiller Primordia, Stem Tips, and Young Spikes of Wheat Guomai 301" Genes 10, no. 9: 686. https://doi.org/10.3390/genes10090686
APA StyleLi, J., Jiao, Z., He, R., Sun, Y., Xu, Q., Zhang, J., Jiang, Y., Li, Q., & Niu, J. (2019). Gene Expression Profiles and microRNA Regulation Networks in Tiller Primordia, Stem Tips, and Young Spikes of Wheat Guomai 301. Genes, 10(9), 686. https://doi.org/10.3390/genes10090686