Predicting miRNA-Disease Associations by Incorporating Projections in Low-Dimensional Space and Local Topological Information

Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Establishment of the miRNA-Disease Dual Heterogeneous Network
2.3. miRNA-Disease Association Prediction Model
2.4. Optimization
3. Performance Evaluation and Analysis
3.1. Performance Evaluation
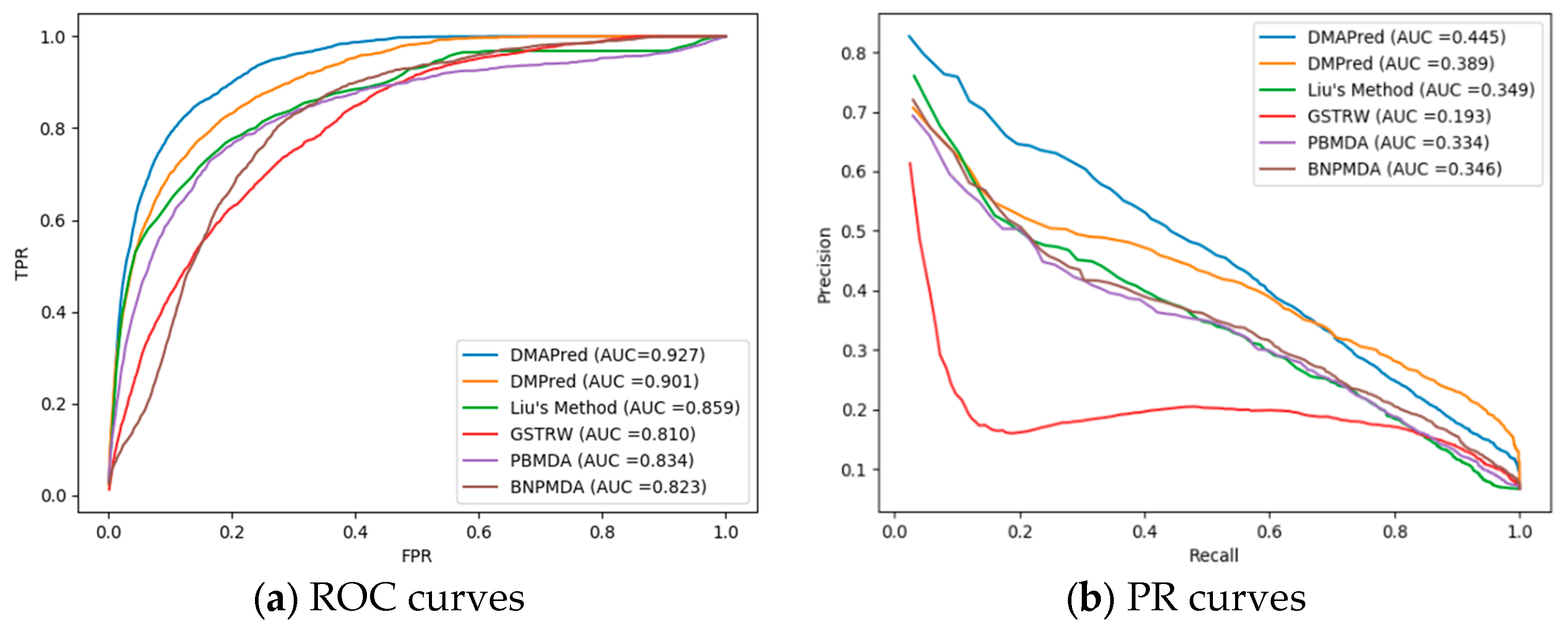
3.2. Comparison with Other Methods
3.3. Case Studies on Breast Neoplasms, Prostatic Neoplasms, and Lung Neoplasms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Calin, G.A.; Croce, C.M. MicroRNA-cancer connection: The beginning of a new tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Abdellatif, M. MicroRNAs in development and disease. Physiol. Rev. 2011, 91, 827–887. [Google Scholar] [CrossRef] [PubMed]
- Meola, N.; Gennarino, V.A.; Banfi, S. microRNAs and genetic diseases. Pathogenetics 2009, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, D.; Zhao, Q.; You, Z.-H. MicroRNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2017, 20, 515–539. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Jiang, Q.; Hao, Y.; Wang, G.; Juan, L.; Zhang, T.; Teng, M.; Liu, Y.; Wang, Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 2010, 4, S2. [Google Scholar] [CrossRef]
- Shi, H.; Xu, J.; Zhang, G.; Xu, L.; Li, C.; Wang, L.; Zhao, Z.; Jiang, W.; Guo, Z.; Li, X. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst. Biol. 2013, 7, 101. [Google Scholar] [CrossRef] [PubMed]
- Qabaja, A.; Alshalalfa, M.; Bismar, T.A.; Alhajj, R. Protein network-based Lasso regression model for the construction of disease-miRNA functional interactions. EURASIP J. Bioinform. Syst. Biol. 2013, 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ping, Y.; Li, X.; Zhao, H.; Wang, L.; Fan, H.; Xiao, Y.; Li, X. Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol. Biosyst. 2014, 10, 2800–2809. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Mitra, R.; Maulik, U.; Zhang, M.Q. Development of the human cancer microRNA network. Silence 2010, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and between score for MiRNA-disease association prediction. Sci. Rep. 2016, 6, 21106. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Q.; Rong, Z.-H.; Chen, X.; Yan, G.-Y.; You, Z.-H. MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget 2017, 8, 21187. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.; Wang, J.; Li, M.; Liu, J.; Wu, F.-X.; Pan, Y. Predicting microRNA-disease associations based on improved microRNA and disease similarities. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 1774–1782. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.-X.; Yan, G.-Y. RWRMDA: Predicting novel human microRNA–disease associations. Mol. Biosyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef]
- Xuan, P.; Han, K.; Guo, Y.; Li, J.; Li, X.; Zhong, Y.; Zhang, Z.; Ding, J. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics 2015, 31, 1805–1815. [Google Scholar] [CrossRef]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zeng, X.; He, Z.; Zou, Q. Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 14, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xiao, Q. A novel approach for predicting microRNA-disease associations by unbalanced bi-random walk on heterogeneous network. J. Biomed. Inform. 2017, 66, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Luo, J.; Liang, C.; Cai, J.; Ding, P. A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics 2017, 34, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, L. LRSSLMDA: Laplacian regularized sparse subspace learning for MiRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005912. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.-N.; Li, J.-Q. Predicting miRNA–Disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef]
- Xuan, P.; Shen, T.; Wang, X.; Zhang, T.; Zhang, W. Inferring disease-associated microRNAs in heterogeneous networks with node attributes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018. [Google Scholar] [CrossRef]
- You, Z.-H.; Huang, Z.-A.; Zhu, Z.; Yan, G.-Y.; Li, Z.-W.; Wen, Z.; Chen, X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005455. [Google Scholar] [CrossRef]
- Zhang, X.; Zou, Q.; Rodriguez-Paton, A.; Zeng, X. Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 283–291. [Google Scholar] [CrossRef]
- Xuan, P.; Dong, Y.; Guo, Y.; Zhang, T.; Liu, Y. Dual convolutional neural network based method for predicting disease-related miRNAs. Int. J. Mol. Sci. 2018, 19, 3732. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, L.; Xie, D.; Zhao, Q. EGBMMDA: Extreme gradient boosting machine for MiRNA-disease association prediction. Cell Death Dis. 2018, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Sun, H.; Wang, X.; Zhang, T.; Pan, S. Inferring the disease-associated miRNAs based on network representation learning and convolutional neural networks. Int. J. Mol. Sci. 2019, 20, 3648. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2. 0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2013, 42, D1070–D1074. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Xuan, P.; Wang, X.; Zhang, T.; Li, J.; Liu, Y.; Zhang, W. A non-negative matrix factorization based method for predicting disease-associated miRNAs in miRNA-disease bilayer network. Bioinformatics 2017, 34, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liao, B.; Li, Z. Global similarity method based on a two-tier random walk for the prediction of microRNA–disease association. Sci. Rep. 2018, 8, 6481. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.-H.; Liu, H. BNPMDA: Bipartite network projection for MiRNA–disease association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef] [PubMed]
- Eichner, L.J.; Perry, M.-C.; Dufour, C.R.; Bertos, N.; Park, M.; St-Pierre, J.; Giguère, V. miR-378∗ mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. 2010, 12, 352–361. [Google Scholar] [CrossRef]
- Kang, H.; Kim, C.; Lee, H.; Rho, J.; Seo, J.; Nam, J.-W.; Song, W.; Nam, S.; Kim, W.; Lee, E. Downregulation of microRNA-362-3p and microRNA-329 promotes tumor progression in human breast cancer. Cell Death Differ. 2016, 23, 484. [Google Scholar] [CrossRef]
- Ma, T.; Yang, L.; Zhang, J. miRNA-542-3p downregulation promotes trastuzumab resistance in breast cancer cells via AKT activation. Oncol. Rep. 2015, 33, 1215–1220. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, M.; Sui, P.; Ding, L.; Yang, Q. Upregulation of microRNA-574-3p in a human gastric cancer cell line AGS by TGF-β1. Gene 2017, 605, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Ujihira, T.; Ikeda, K.; Suzuki, T.; Yamaga, R.; Sato, W.; Horie-Inoue, K.; Shigekawa, T.; Osaki, A.; Saeki, T.; Okamoto, K. MicroRNA-574-3p, identified by microRNA library-based functional screening, modulates tamoxifen response in breast cancer. Sci. Rep. 2015, 5, 7641. [Google Scholar] [CrossRef] [PubMed]
- Eichelser, C.; Stückrath, I.; Müller, V.; Milde-Langosch, K.; Wikman, H.; Pantel, K.; Schwarzenbach, H. Increased serum levels of circulating exosomal microRNA-373 in receptor-negative breast cancer patients. Oncotarget 2014, 5, 9650. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y. dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genomics 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Kowarsch, A.; Schmidl, D.; Buggenthin, F.; Brauner, B.; Dunger, I.; Fobo, G.; Frishman, G.; Montrone, C.; Theis, F.J. PhenomiR: A knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010, 11, R6. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases Name | AUC | |||||
|---|---|---|---|---|---|---|
| DMAPred | GSTRW | DMPred | PBMDA | Liu’s Method | BNPMDA | |
| Breast neoplasms | 0.966 | 0.822 | 0.938 | 0.852 | 0.863 | 0.905 |
| Hepatocellular carcinoma | 0.957 | 0.779 | 0.900 | 0.803 | 0.845 | 0.853 |
| Renal cell carcinoma | 0.926 | 0.816 | 0.903 | 0.813 | 0.832 | 0.845 |
| Squamous cell carcinoma | 0.942 | 0.817 | 0.908 | 0.881 | 0.890 | 0.877 |
| Colorectal neoplasms | 0.895 | 0.737 | 0.842 | 0.826 | 0.857 | 0.801 |
| Glioblastoma | 0.928 | 0.814 | 0.904 | 0.803 | 0.842 | 0.817 |
| Heart failure | 0.965 | 0.817 | 0.987 | 0.791 | 0.828 | 0.891 |
| Acute myeloid leukemia | 0.967 | 0.788 | 0.890 | 0.844 | 0.874 | 0.845 |
| Lung neoplasms | 0.973 | 0.791 | 0.948 | 0.905 | 0.920 | 0.912 |
| Melanoma | 0.907 | 0.789 | 0.913 | 0.836 | 0.860 | 0.889 |
| Ovarian neoplasms | 0.939 | 0.830 | 0.929 | 0.889 | 0.897 | 0.725 |
| Pancreatic neoplasms | 0.933 | 0.838 | 0.916 | 0.891 | 0.904 | 0.829 |
| Prostatic neoplasms | 0.958 | 0.822 | 0.951 | 0.843 | 0.855 | 0.894 |
| Stomach neoplasms | 0.935 | 0.762 | 0.908 | 0.821 | 0.836 | 0.784 |
| Urinary bladder neoplasms | 0.860 | 0.816 | 0.919 | 0.854 | 0.865 | 0.901 |
| Average AUC for the 326 diseases | 0.927 | 0.810 | 0.901 | 0.834 | 0.859 | 0.823 |
| Disease Name | AUPR | |||||
|---|---|---|---|---|---|---|
| DMAPred | Liu’s Method | GSTRW | DMPred | PBMDA | BNPMDA | |
| Breast neoplasms | 0.761 | 0.573 | 0.322 | 0.699 | 0.574 | 0.254 |
| Hepatocellular carcinoma | 0.719 | 0.498 | 0.279 | 0.501 | 0.454 | 0.618 |
| Renal cell carcinoma | 0.485 | 0.186 | 0.150 | 0.293 | 0.181 | 0.334 |
| Squamous cell carcinoma | 0.299 | 0.208 | 0.109 | 0.213 | 0.211 | 0.214 |
| Colorectal neoplasms | 0.340 | 0.371 | 0.141 | 0.186 | 0.367 | 0.197 |
| Glioblastoma | 0.517 | 0.243 | 0.151 | 0.219 | 0.217 | 0.227 |
| Heart failure | 0.786 | 0.189 | 0.191 | 0.700 | 0.168 | 0.178 |
| Acute myeloid leukemia | 0.317 | 0.236 | 0.140 | 0.211 | 0.191 | 0.190 |
| Lung neoplasms | 0.740 | 0.503 | 0.147 | 0.511 | 0.537 | 0.547 |
| Melanoma | 0.342 | 0.397 | 0.171 | 0.389 | 0.363 | 0.334 |
| Ovarian neoplasms | 0.441 | 0.361 | 0.169 | 0.404 | 0.361 | 0.357 |
| Pancreatic neoplasms | 0.303 | 0.354 | 0.137 | 0.329 | 0.364 | 0.357 |
| Prostatic neoplasms | 0.532 | 0.264 | 0.166 | 0.463 | 0.282 | 0.345 |
| Stomach neoplasms | 0.469 | 0.346 | 0.220 | 0.446 | 0.344 | 0.284 |
| Urinary bladder neoplasms | 0.118 | 0.280 | 0.163 | 0.315 | 0.252 | 0.242 |
| Average AUPR for the 326 diseases | 0.445 | 0.349 | 0.193 | 0.389 | 0.334 | 0.346 |
| DMPred | Liu’s Method | GSTRW | PBMDA | BNPMDA | |
|---|---|---|---|---|---|
| p-value of AUCs | 0.00247 | 5.0135 × 10−7 | 2.4835 × 10−9 | 2.3143 × 10−6 | 9.5824 × 10−6 |
| p-value of AUPRs | 0.00168 | 0.00199 | 3.6475 × 10−6 | 0.00289 | 0.00182 |
| Rank | MiRNA Name | Description | Rank | MiRNA Name | Description |
|---|---|---|---|---|---|
| 1 | hsa-mir-15b | dbDEMC2,PhenomiR | 26 | hsa-mir-184 | dbDEMC2,PhenomiR |
| 2 | hsa-mir-142 | PhenomiR | 27 | hsa-mir-363 | dbDEMC2 |
| 3 | hsa-mir-192 | PhenomiR | 28 | hsa-mir-30e | PhenomiR |
| 4 | hsa-mir-378a | Literature [38] | 29 | hsa-mir-208a | dbDEMC2,PhenomiR |
| 5 | hsa-mir-106a | dbDEMC2,PhenomiR | 30 | hsa-mir-449b | dbDEMC2 |
| 6 | hsa-mir-99a | dbDEMC2,PhenomiR | 31 | hsa-mir-491 | PhenomiR |
| 7 | hsa-mir-130a | dbDEMC2,PhenomiR | 32 | hsa-mir-494 | dbDEMC2,PhenomiR |
| 8 | hsa-mir-150 | dbDEMC2,PhenomiR | 33 | hsa-mir-186 | dbDEMC2,PhenomiR |
| 9 | hsa-mir-196b | dbDEMC2,PhenomiR | 34 | hsa-mir-362 | Literature [39] |
| 10 | hsa-mir-130b | dbDEMC2,PhenomiR | 35 | hsa-mir-424 | dbDEMC2,PhenomiR |
| 11 | hsa-mir-98 | dbDEMC2,PhenomiR | 36 | hsa-mir-370 | dbDEMC2,PhenomiR |
| 12 | hsa-mir-1266 | dbDEMC2 | 37 | hsa-mir-542 | Literature [40] |
| 13 | hsa-mir-92b | dbDEMC2 | 38 | hsa-mir-32 | dbDEMC2,PhenomiR |
| 14 | hsa-mir-372 | dbDEMC2,PhenomiR | 39 | hsa-mir-181d | dbDEMC2,PhenomiR |
| 15 | hsa-mir-138 | dbDEMC2,PhenomiR | 40 | hsa-mir-483 | PhenomiR |
| 16 | hsa-mir-574 | Literature [41,42] | 41 | hsa-mir-302e | dbDEMC2 |
| 17 | hsa-mir-144 | dbDEMC2,PhenomiR | 42 | hsa-mir-302f | dbDEMC2 |
| 18 | hsa-mir-28 | dbDEMC2,PhenomiR | 43 | hsa-mir-208b | dbDEMC2 |
| 19 | hsa-mir-212 | dbDEMC2,PhenomiR | 44 | hsa-mir-134d | dbDEMC2 |
| 20 | hsa-mir-181c | dbDEMC2,PhenomiR | 45 | hsa-mir-330 | dbDEMC2,PhenomiR |
| 21 | hsa-mir-371a | Literature [43] | 46 | hsa-mir-381 | dbDEMC2,PhenomiR |
| 22 | hsa-mir-449a | dbDEMC2,PhenomiR | 47 | hsa-mir-198 | dbDEMC2,PhenomiR |
| 23 | hsa-mir-185 | dbDEMC2,PhenomiR | 48 | hsa-mir-548a | dbDEMC2 |
| 24 | hsa-mir-211 | dbDEMC2,PhenomiR | 49 | hsa-mir-154 | dbDEMC2,PhenomiR |
| 25 | hsa-mir-99b | dbDEMC2,PhenomiR | 50 | hsa-mir-503 | dbDEMC2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, P.; Zhang, Y.; Zhang, T.; Li, L.; Zhao, L. Predicting miRNA-Disease Associations by Incorporating Projections in Low-Dimensional Space and Local Topological Information. Genes 2019, 10, 685. https://doi.org/10.3390/genes10090685
Xuan P, Zhang Y, Zhang T, Li L, Zhao L. Predicting miRNA-Disease Associations by Incorporating Projections in Low-Dimensional Space and Local Topological Information. Genes. 2019; 10(9):685. https://doi.org/10.3390/genes10090685
Chicago/Turabian StyleXuan, Ping, Yan Zhang, Tiangang Zhang, Lingling Li, and Lianfeng Zhao. 2019. "Predicting miRNA-Disease Associations by Incorporating Projections in Low-Dimensional Space and Local Topological Information" Genes 10, no. 9: 685. https://doi.org/10.3390/genes10090685
APA StyleXuan, P., Zhang, Y., Zhang, T., Li, L., & Zhao, L. (2019). Predicting miRNA-Disease Associations by Incorporating Projections in Low-Dimensional Space and Local Topological Information. Genes, 10(9), 685. https://doi.org/10.3390/genes10090685