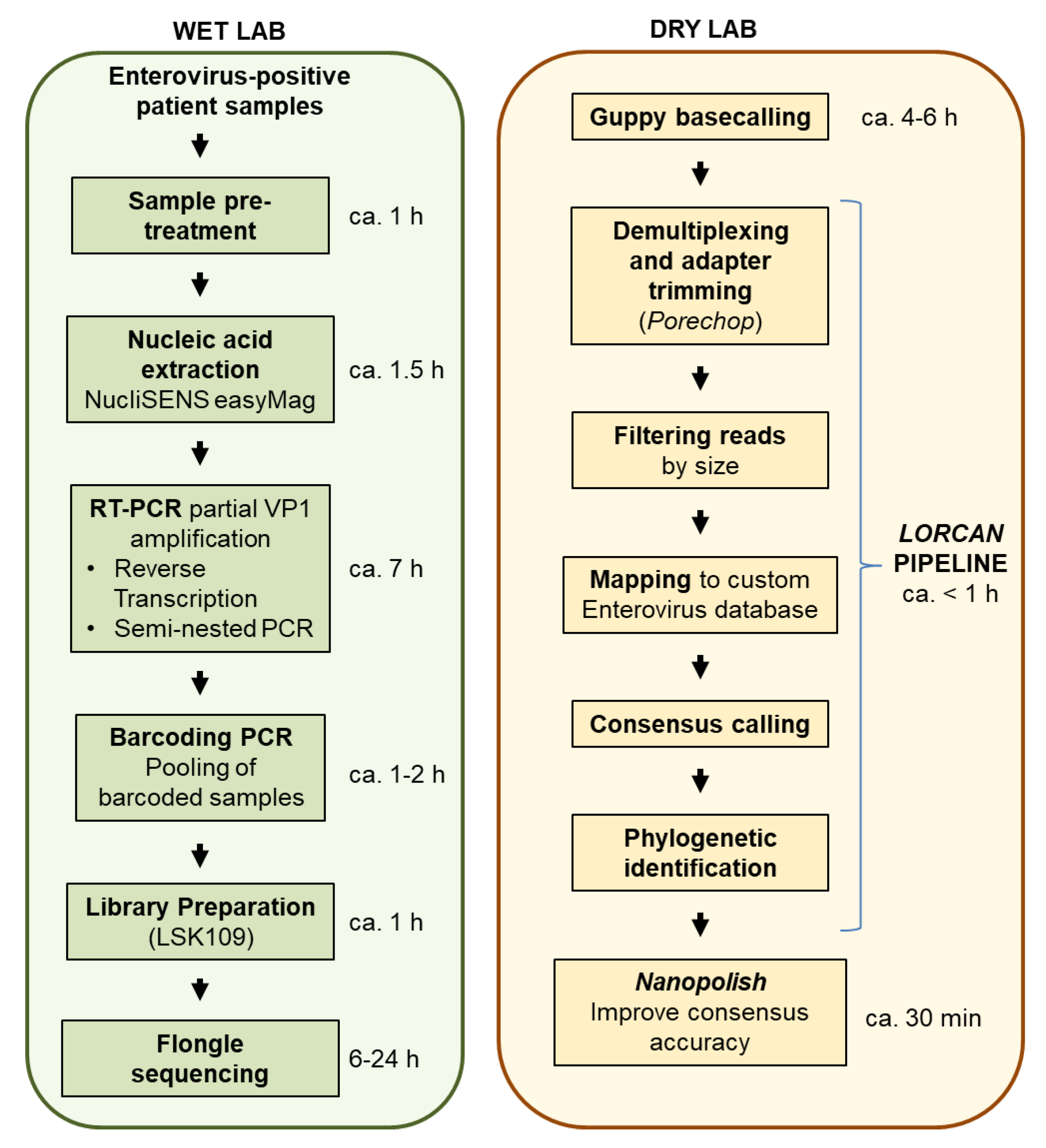
1. Introduction
Enteroviruses are RNA viruses and among the most common viruses infecting humans. Enterovirus infections are associated with a broad range of clinical symptoms including mild respiratory illness to severe neurological diseases [
1]. Their genomes have high variability due to frequent mutations and recombinations and more than 100 genotypes are known to infect humans [
2,
3]. The surveillance of circulating genotypes and detection of new emerging genotypes must be conducted at regular intervals. Molecular diagnostic tests for genotyping of enteroviruses usually target the
VP1 region, which is known to correlate with viral serotype [
4], by Sanger sequencing the PCR amplified gene. Sanger sequencing is still the standard for enterovirus amplicon sequencing as next generation sequencing (NGS) approaches tend to have longer turnaround times and generally require larger capital investments [
5].
The MinION sequencer from Oxford Nanopore Technologies (ONT) is a portable device that allows real-time sequencing and low capital cost investment. Thousands of single nanopores are arrayed on individual synthetic polymer membranes in a single flow cell, and nucleic acid molecules loaded onto the flow cell migrate to and through nanopores by following an electric potential [
6]. While single nucleic molecules pass through individual nanopores at a rate determined by an engineered motor protein, the current at each nanopore is modified as a function of the actual molecule composition [
7], and those current fluctuations are recorded and converted into base sequences by using a recurrent neural network algorithm [
8]. Noticeably, for amplicon sequencing applications, sequencing cost can be further reduced by multiplexing several samples together, i.e., by adding short indexed adaptors (i.e., barcodes) specifically to sequences of each sample, so that the sample origin of each read can be identified. MinION sequencing has been used for sequencing viral amplicons before [
9,
10,
11]. A standard MinION flow cells can reach a total output of 10–20 Gb of data within 48 h and enough sequencing depth can be achieved within a few minutes of sequencing to produce a consensus sequence whose accuracy is then >99% nt identical to its Sanger sequenced counterpart [
10]. Recently, ONT has released a new type of flow cell called “Flongle” (R9.4.1 nanopores), which is smaller and cheaper than the current flow cell (SpotON flow cell Mk I, R9.4.1 nanopores). As a small consumable flow cell, a Flongle is mounted onto a Flongle adapter that contains the ONT proprietary sensor array and is compatible with MinION and GridION sequencing devices. A Flongle harbors 126 sequencing channels instead of the standard 512 channels and is typically designed for applications which require less sequencing output than standard flow cells.
The goal of this study was to evaluate whether amplicon sequencing using Flongles was suitable for enterovirus genotyping from diagnostic samples. We tested whether several tens of clinical samples could be multiplexed and sequenced on single Flongle flow cells. We compared nanopore sequencing accuracy to that of Sanger sequencing using repeated assays, and established turnover time and cost of the assays in order to provide first insights on potential clinical applications of Flongle-based sequencing.
3. Results
We assessed the suitability of the ONT flow cell, “Flongle”, for enterovirus genotyping based on multiplexed amplicons produced from samples of clinical origin. Samples were obtained randomly from enterovirus positive samples referred to IFIK from May 2016 to July 2018. We worked directly from patient material for all samples but two, for which the virus was first propagated in cell culture (
Table S1). In order to obtain homogeneous starting material for each sample, PCR amplicons were produced once for each sample. We then associated those amplicons to different barcodes, as follows: The first Flongle sequencing run included 26 barcoded samples. The second Flongle run included the same 26 samples, but this time in duplicates, i.e., using two different barcodes per sample. Therefore, each amplicon sample was sequenced three times and the variability within and between sequencing runs assessed. The first sequencing run contained four additional barcoded samples which were not part of this study and were excluded from further analysis. All amplicons were also sequenced with the Sanger method for comparative purposes. Based on Sanger sequencing, the samples set encompassed 10 different genotypes belonging to enterovirus species A and B (
Table S1).
3.1. Flongle Sequencing Run Statistics
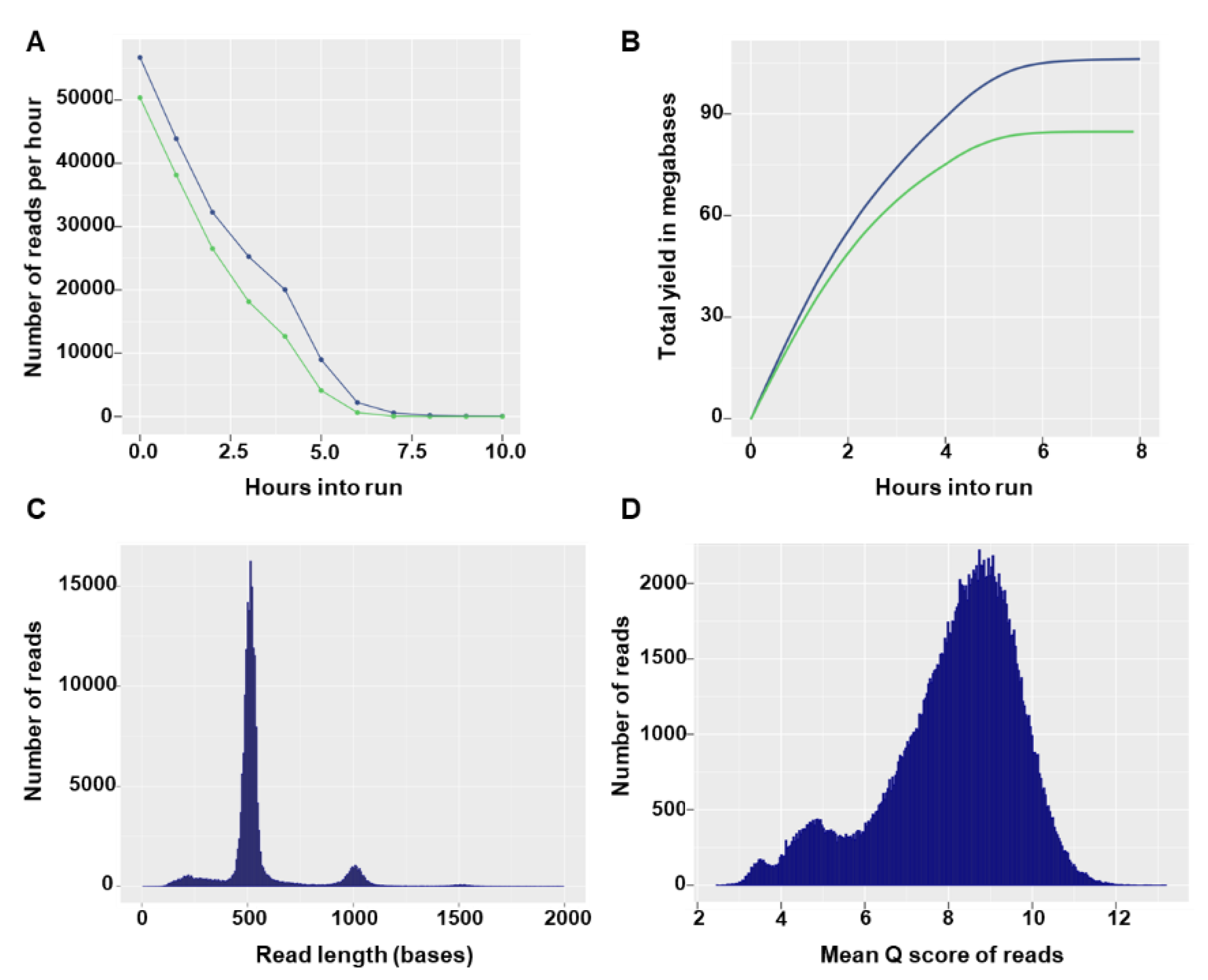
Flongle flow cells can have a maximum of 126 available nanopores, with a minimum of 60 pores guaranteed by the manufacturer. In the two sequencing runs, the Flongles had 55 and 60 available pores and after loading of the prepared library the sequencing run started with 27 and 42 active pores, respectively. In both runs, the number of reads per hour decreased rapidly (
Figure 2A) and no more notable output was produced after 7 h of sequencing (
Figure 2B). The total number of reads generated in the sequencing runs was 190.29 K (135.55 Mbases) and 267.38 K (172.4 Mbases). Read size distributions showed specific modal peak that fit the expected barcoded input amplicon size (
Figure 2C). The majority of reads had good quality scores, with 162,217 (85%) and 205,829 (77%) reads having a quality score equal to or higher than 7 in the first and second run, respectively (
Figure 2D). Overall, high similarity in yield and run statistics were observed between the two runs (data not shown) and only the first run was used for illustrative purposes.
3.2. Sequencing Accuracy Compared to Sanger
FASTQ files containing sequences with quality scores ≥ 7 were processed using the LORCAN pipeline. The resulting consensus sequences were polished using the nanopolish software. Sequence identities of the raw consensus sequences produced by LORCAN and of their polished counterparts were established in comparison to Sanger sequences produced from the same starting amplicons. The average sequence identity of all samples of the first run was 99.85% (standard deviation SD = 0.41). In the second run, the first barcode set had an average of 99.75% identity (SD = 0.46), and the second set an average of 99.80% (SD = 0.23) identity to their Sanger counterparts. The most common differences between the nanopore consensuses and Sanger sequences were a gap or unassigned base (N) in a short homopolymer sequence (4-mer) and these errors were generally observed in different samples associated with the same genotype (data not shown).
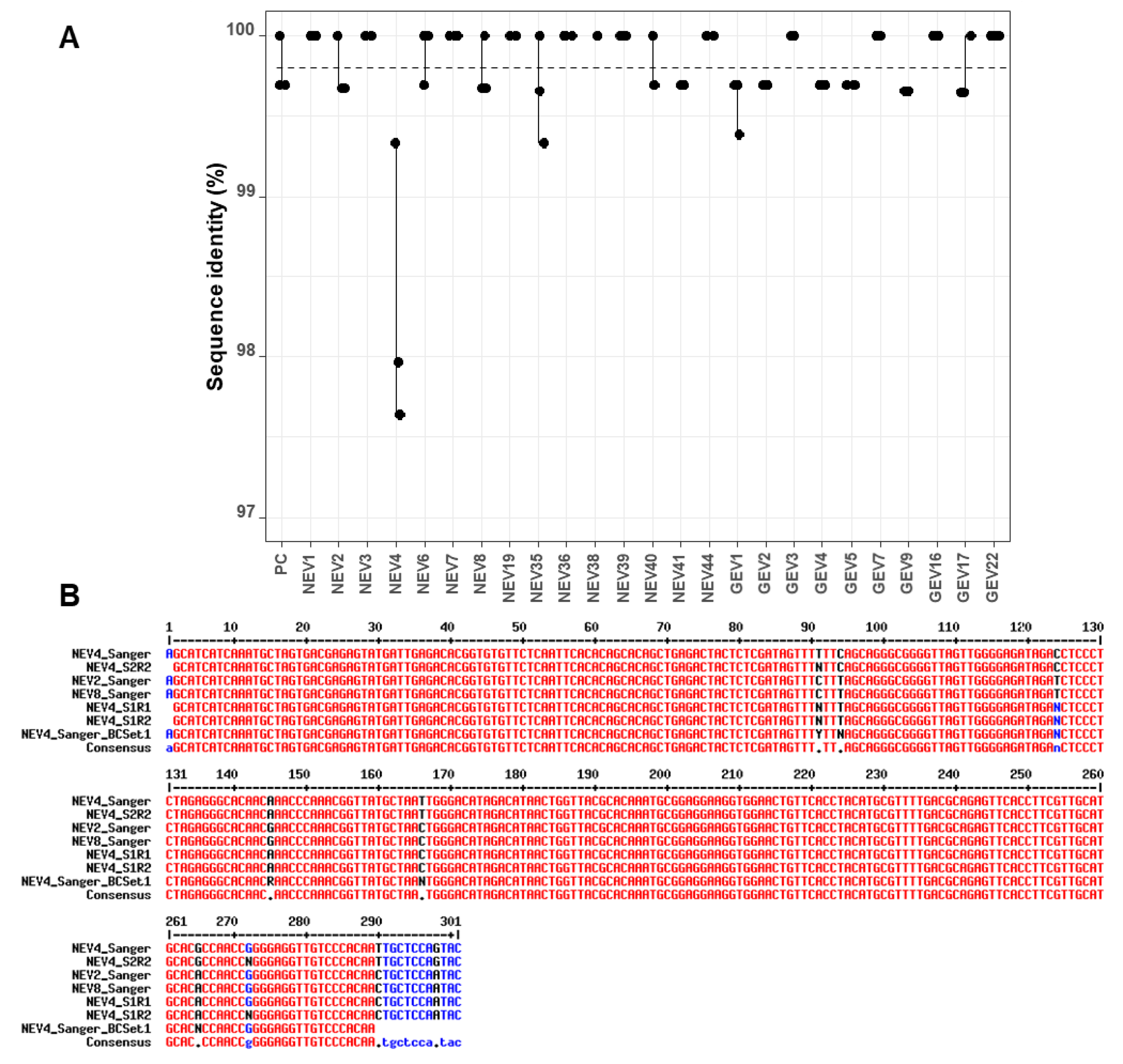
The analysis of sequence identity between the three replicates of each sample indicated little variability, with values >99% for all replicates across sequencing runs and barcode sets (
Figure 3A). There was, however, one clear outlier (NEV4) within the first set of barcodes, which had several mismatches and unassigned bases compared to the corresponding Sanger sequence (
Figure 3B). Additionally, the
LORCAN consensuses of NEV4 in set 1 in both runs were also the only cases where
nanopolish correction appeared to introduce more errors rather than improving the sequences (drop of sequence identity from 98.0% to 97.3% and 97.6% to 97.0% for the two runs;
Table S2) by introducing additional gaps and a mismatch. These results were consistent between runs of the first set of barcodes, but not observed in the second set of barcodes.
To investigate this discrepancy in nanopore consensuses, Sanger sequencing of the NEV4 barcoded amplicon after PCR barcoding in the ONT protocol was also performed. Unassigned bases were also evidenced at the same positions (
Figure 3B), suggesting the variability might originate from the barcoding PCR step rather than the sequencing run. Another explanation for the discrepancy may be a contamination with NEV2 or NEV8 samples during the barcoding PCR step, because those latter sequences displayed the same sequence variability as in NEV4 nanopore consensuses (comparison of NEV2_Sanger and NEV8_Sanger, and NEV4_S1R1 and NEV4_S1R2 at positions 94 and 166 in
Figure 3B). If this scenario is true, lower sequence identity in
Figure 3A for NEV4 nanopore consensuses would be explained by the fact that the NEV4 nanopore consensuses were compared to a wrong Sanger reference sequence due to a probable cross-contamination during the barcoding PCR step of Set 1. A closer look at the data indicated that, even though the
LORCAN generated consensuses from NEV4 displayed an average sequence identity to NEV2 (or NEV8) Sanger sequences of 98.84% for both sequencing runs and only of 97.805% to NEV4 Sanger sequence, the NEV4 nanopore consensuses did not perfectly match any of the NEV2 or NEV8 Sanger reference sequences (
Figure 3B). Therefore, the cross-contamination hypothesis for NEV4 is likely, but not completely established at this stage.
Overall, there was a significant difference in sequence identity when the factors “Run number”, “barcode set”, and “genotype” were evaluated (multivariable linear regression, ANOVA, F = 3.236, P = 0.001). Significant differences were uniquely due to the factor “genotype” (F = 3.7577, P = 0.0005), while the factors “barcode set” (F = 0.007, P = 0.978) and “Run number” (F = 1.2579, P = 0.2661) were not significant. Although nanopolish was able to improve the consensus sequences in a few cases, overall it was not able to make a significant improvement in sequence identity for any of the replicated samples (two-sided t-test, P > 0.05 for each set). There was also no significant relationship between the number of reads that were used to build the consensus and the sequence identity (ANOVA, F = 1.5188, P = 0.2216; Pearson correlation coefficient 0.1399).
3.3. Phylogenetic Relatedness of Consensus Sequences
We further evaluated whether the sequences generated by nanopore sequencing using Flongle or by Sanger sequencing produced evolutionary related clusters. A phylogenetic tree including all three Flongle sequencing replicates and the Sanger sequences was constructed (
Figure 4). All sequences clustered together with their corresponding Sanger sequence and the results were consistent across all three replicated sample sets (
Figure 4).
3.4. Sequencing Costs, Turnover Time
We estimated the sequencing cost on Flongle at around $10 per sample when multiplexing 26 amplicon samples. Those costs include costs for the flow cell, quality control (DNA concentration and fragment sizing), and library preparation with plasticware, reagents, and barcoding tailed primers. We demonstrated that sequence quality and taxonomic positioning were not affected by doubling the number of samples (i.e., up to 52) multiplexed together per Flongle sequencing run. Therefore, our estimated sequencing cost may be in the range of $7 per sample, or even lower if more samples are combined together.
Alternatively, if the use of tailed primers and barcoding PCR is not possible, the native barcoding kits from ONT with a ligation based barcoding approach can be used. Then, cost estimation would be around
$12 per sample, with the limitation that only up to 24 samples maximum may be multiplexed together in the same library, which is enabled by using the two native barcoding expansion kits (ONT EXP-NBD104, EXP-NBD114). The turnover time from patient samples to final output (including RNA extraction, reverse transcription and PCR, library preparation, sequencing time, automatic sequence analyses, and reporting) may be estimated at around 22.5 h for each multiplexed Flongle run (
Figure 1).
4. Discussion
In this proof-of-concept study, we describe the successful application of the new Flongle flow cell for enterovirus genotyping via nanopore sequencing. We were able to correctly identify enterovirus genotypes in all 26 randomly selected clinical samples known to be enterovirus positive using multiplexed amplicon sequencing on Flongles. We observed a high sequencing accuracy when comparing the sequence identity of the resulting consensus sequences with Sanger sequencing, which ranged from 97% to 100%, and the average of the 26 samples was >99.7% identical when considering all replicates. The phylogenetic analysis of the resulting consensus sequences indicated that the Flongle nanopore sequencing provides sufficiently accurate results for correct clustering of sequences, as all sequences grouped together with their Sanger counterparts. These results were also achieved when combining the set twice using different barcodes and therefore combining 52 samples on one flow cell.
It has been shown previously that nanopore sequencing using regular flow cells can be a useful method for sequencing short amplicons [
10,
26], and many of the same advantages also apply to Flongles, namely the low capital cost investment for the sequencing device and its portability, making the approach well-suited for diagnostic applications. For instance, nanopore sequencing has been applied for the genotyping of Newcastle disease virus, where >98% identity to Illumina MiSeq was achieved within 7 mins of sequencing and was able to resolve mixed infections of two genotypes from clinical samples [
10]. As the regular flow cells have many nanopores available and are designed for larger scale experiment, sufficient sequencing depth of amplicons can be achieved very quickly and due to the real-time nature of nanopore sequencing the run can be stopped after a short time if the desired sequencing depth has been obtained. The flow cell can be reused with a different set of barcodes for another run. The Flongle, in contrast, currently about five times cheaper than a regular flow cell, is designed for single use and has fewer nanopores available, which may therefore increase the sequencing run time substantially. Although a Flongle may be suited for smaller sequencing projects, we demonstrated here that multiplexing on a Flongle may further present significant cost reduction for simple assays, such as amplicon sequencing, without deterioration of the sequencing quality. The turnover time from patient samples to final report was estimated at around 22.5 h for each batch of samples. Since we privileged raw data acquisition over fast turnaround time in this study, we performed basecalling offline using the high accuracy option of the basecaller. Shorter turnaround time could still be obtained by performing live basecalling of the sequencing signal generated by the Flongle.
Using the PCR barcoding kit, the number of samples can in theory be scaled up to 96 per flow cell. The disadvantage, however, can be that it requires another (although short) PCR step for barcoding, thus increasing the chances of PCR cross-contamination and adding to the duration of the overall procedure, which would be better addressed by using automation instead of manual library preparation. Additionally, tailed primers need to be used to generate the amplicons, which may require the PCR conditions to be optimized. In our hands, tailed primers often demonstrate lower sensitivity as compared to the original, unmodified primers (data not shown). On the other hand, the Flongle protocol that we used required very little input, and weak positive samples could also be included. Another source of error may originate from cross-contamination between barcodes, with an estimated amount of up to 0.056% of total reads with incorrect barcodes in multiplex sequencing data [
27]. In our approach, we limited the analysis to the 3000 reads whose lengths were closest to the length distribution mode and further required that a least 100 reads mapped to a reference sequence to further derive a consensus sequence from the reads. We also required that
porechop be used with the more stringent option "require_two_barcodes" for demultiplexing. Those conditions overall may help address the issue with low amount of cross-barcode contamination that may be present.
One of the main limitations of nanopore sequencing is its still relatively high error rate per base in raw reads as compared to second generation sequencing techniques or to Sanger sequencing. Despite sequence consensus generation using
LORCAN, which should remove random errors provided enough reads were used to generate the consensus (i.e., at least 100 reads per consensus; data not shown), we did not always achieve 100% identity to the corresponding Sanger sequence, and the main source of error was found to be located in short homopolymer regions: a systematic error known for sequencing using R9.4.1 nanopores [
28].
Nanopolish is sometimes able to correct this kind of error, although for this dataset it did not make a significant difference in terms of sequence accuracy or phylogenetic positioning. Therefore, the consensus sequences produced by the
LORCAN pipeline alone were found to be already of sufficient high quality for genotyping applications, even when multiplexing several amplicon samples on the same Flongle flow cell. We did not perform real-time processing of the sequencing data being accumulated on the sequencing computer, and as such, faster turnover time may easily be attained in the future by using simultaneous data acquisition and bioinformatic treatment of the information (e.g., [
29]).
With the introduction of the Flongle, Oxford Nanopore Technologies provides a new flow cell that allows scaling down experiments, and which may be ideally suited for fast and cheap amplicon sequencing. Flongle nanopores offer sufficient sequencing accuracy to pool together multiple samples while allowing the possibility of resolving mixed amplicons via the further bioinformatic analyses of each single sequenced molecule, which is not feasible via Sanger sequencing. Consensus sequences obtained via nanopore sequencing may also be longer than their Sanger counterparts, as the latter typically lack tens of bases at the 5’ and 3’ end due to lower base quality, especially when sequencing long amplicons. Further experiments to evaluate the sensitivity of the approach are still required.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}



