Two Years of Viral Metagenomics in a Tertiary Diagnostics Unit: Evaluation of the First 105 Cases

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Design
2.3. Viral Metagenomic Sequencing of Clinical Samples
2.4. Criteria for Positive Virus Hits
2.5. Evaluation of the Utility of Viral Metagenomic Sequencing Compared to Conventional Testing
2.6. Evaluation of the Clinical Impact
3. Results
3.1. Case Statistics of Samples Analyzed by Viral Metagenomics
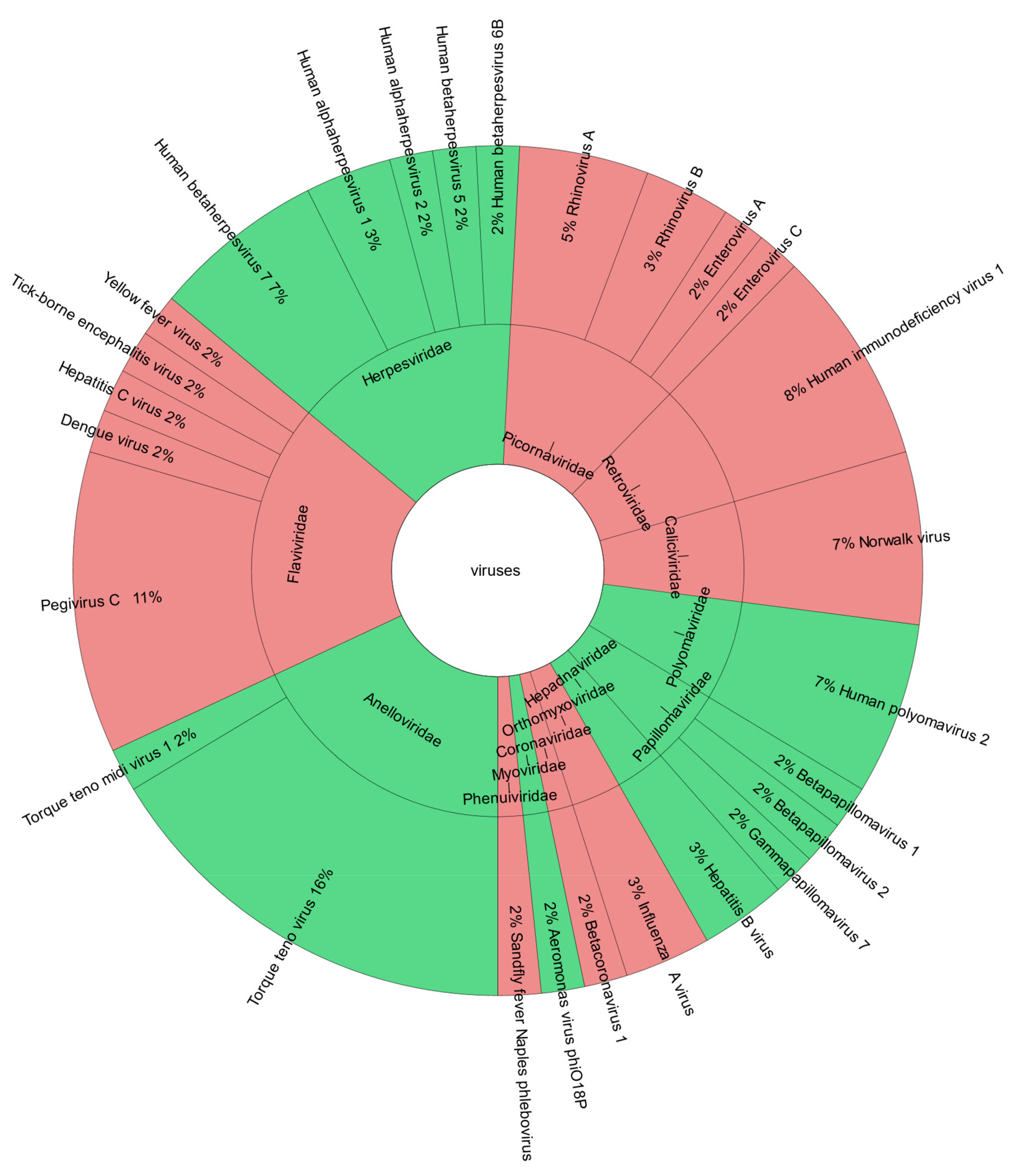
3.2. Viruses Detected by Viral Metagenomic Sequencing
3.3. Outcome of Viral Metagenomic Assay Versus Conventional Testing
3.4. Workload of Viral Metagenomic Assay Versus Conventional Testing
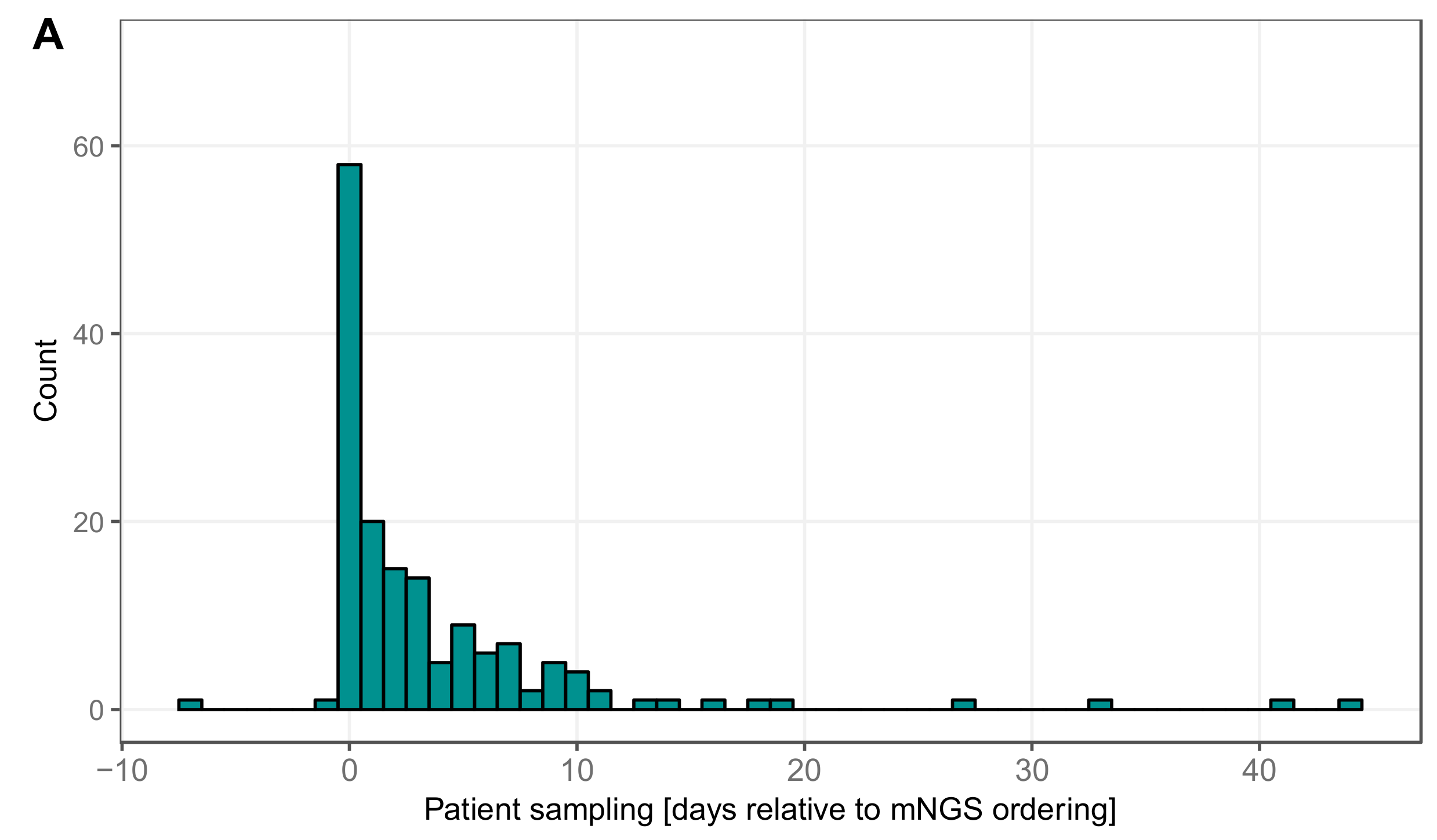
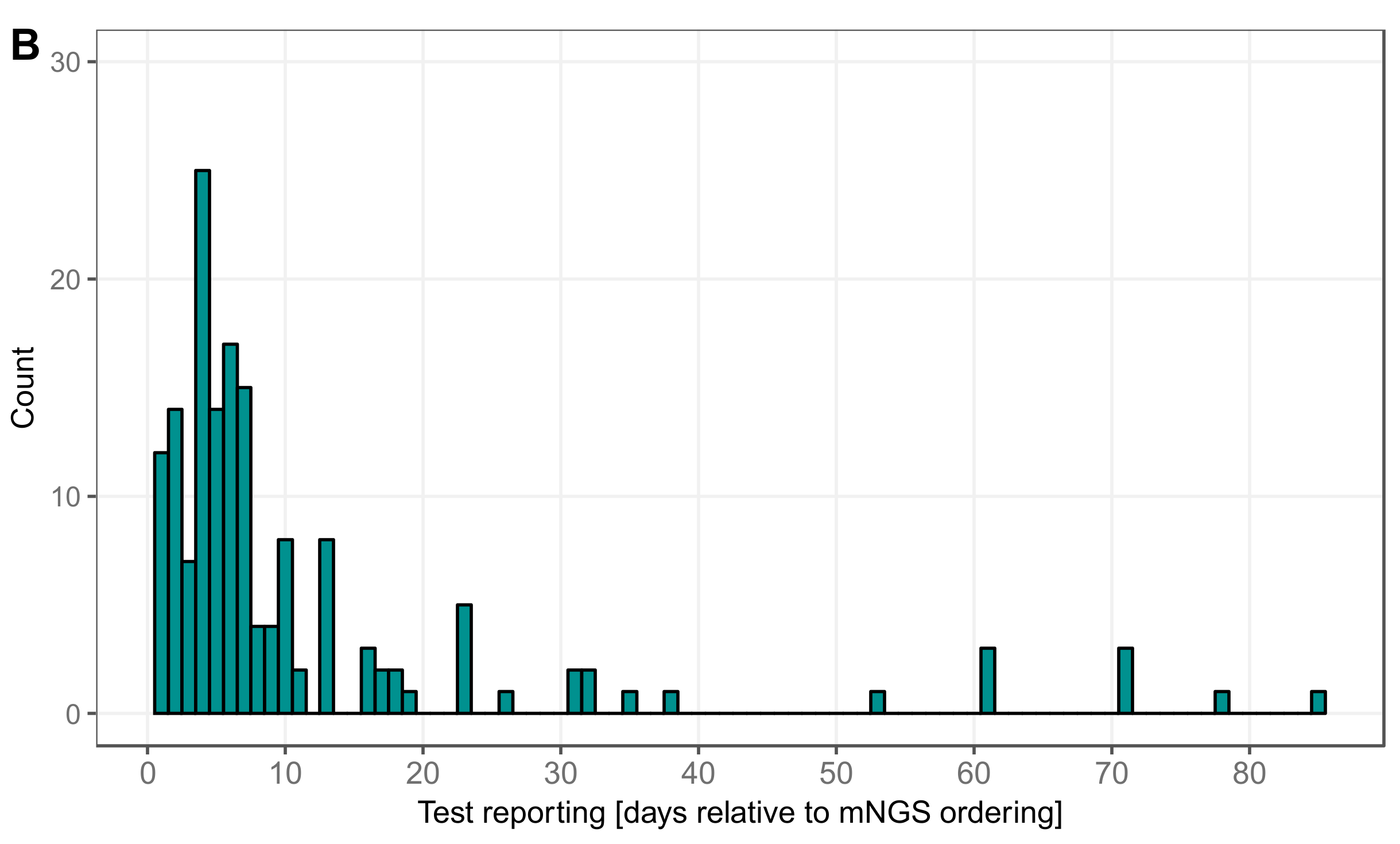
3.5. Timing Viral Metagenomic Assay Versus Conventional Testing
3.6. Patient Characteristics in the Study Subgroup
3.7. Clinical Impact of Viral Metagenomics Results in the Study Subgroup
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A (Supplemental Methods)
Appendix A.1. Metagenomic Sequencing v2.1.0
Appendix A.1.1. Sample Pre-Processing, Nucleic Acid Extraction and DNase Treatment
Appendix A.1.2. Random Reverse Transcription and Second Strand Synthesis
Appendix A.1.3. Viral Metagenomic High-Throughput Sequencing
Appendix A.1.4. Bioinformatic Analysis
Appendix A.2. Metagenomic Sequencing v2.2.1
Appendix B (Case vignettes)
Appendix B.1. Sandfly Fever Naples phlebovirus Associated Meningitis: Direct Impact of mNGS on Diagnosis and Antibiotic Treatment
Appendix B.2. Tick-Borne Encephalitis: Rapid Diagnosis of an Unexpected Virus in Encephalitis
Appendix B.3. Pegivirus C Associated Meningoencephalitis: A Possible New Encephalitis Pathogen
References
- Glaser, C.A.; Honarmand, S.; Anderson, L.J.; Schnurr, D.P.; Forghani, B.; Cossen, C.K.; Schuster, F.L.; Christie, L.J.; Tureen, J.H. Beyond viruses: clinical profiles and etiologies associated with encephalitis. Clin. Infect. Dis. 2006, 43, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Ewig, S.; Torres, A.; Angeles Marcos, M.; Angrill, J.; Rano, A.; de Roux, A.; Mensa, J.; Martinez, J.A.; de la Bellacasa, J.P.; Bauer, T. Factors associated with unknown aetiology in patients with community-acquired pneumonia. Eur. Respir. J. 2002, 20, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Bleeker-Rovers, C.P.; Vos, F.J.; de Kleijn, E.M.; Mudde, A.H.; Dofferhoff, T.S.; Richter, C.; Smilde, T.J.; Krabbe, P.F.; Oyen, W.J.; van der Meer, J.W. A prospective multicenter study on fever of unknown origin: The yield of a structured diagnostic protocol. Medicine (Baltimore) 2007, 86, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Self, W.H.; Wunderink, R.G.; Team, C.E.S. Community-acquired pneumonia requiring hospitalization. N. Engl. J. Med. 2015, 373, 2382. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. The challenge of diagnostic metagenomics. Expert Rev. Mol. Diagn. 2018, 18, 605–615. [Google Scholar] [CrossRef]
- Delwart, E.L. Viral metagenomics. Rev. Med. Virol. 2007, 17, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Murkey, J.A.; Chew, K.W.; Carlson, M.; Shannon, C.L.; Sirohi, D.; Sample, H.A.; Wilson, M.R.; Vespa, P.; Humphries, R.M.; Miller, S.; et al. Hepatitis E virus-associated meningoencephalitis in a lung transplant recipient diagnosed by clinical metagenomic sequencing. Open Forum Infect. Dis. 2017, 4, ofx121. [Google Scholar] [CrossRef]
- Wilson, M.R.; Suan, D.; Duggins, A.; Schubert, R.D.; Khan, L.M.; Sample, H.A.; Zorn, K.C.; Rodrigues Hoffman, A.; Blick, A.; Shingde, M.; et al. A novel cause of chronic viral meningoencephalitis: Cache Valley virus. Ann. Neurol. 2017, 82, 105–114. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Peretti, A.; Welch, N.L.; Borgogna, C.; Olivero, C.; Badolato, R.; Notarangelo, L.D.; Gariglio, M.; FitzGerald, P.C.; McIntosh, C.E.; et al. Metagenomic discovery of 83 new human papillomavirus types in patients with immunodeficiency. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Eibach, D.; Hogan, B.; Sarpong, N.; Winter, D.; Struck, N.S.; Adu-Sarkodie, Y.; Owusu-Dabo, E.; Schmidt-Chanasit, J.; May, J.; Cadar, D. Viral metagenomics revealed novel betatorquevirus species in pediatric inpatients with encephalitis/meningoencephalitis from Ghana. Sci. Rep. 2019, 9, 2360. [Google Scholar] [CrossRef]
- McMullan, L.K.; Folk, S.M.; Kelly, A.J.; MacNeil, A.; Goldsmith, C.S.; Metcalfe, M.G.; Batten, B.C.; Albarino, C.G.; Zaki, S.R.; Rollin, P.E.; et al. A new phlebovirus associated with severe febrile illness in Missouri. N. Engl. J. Med. 2012, 367, 834–841. [Google Scholar] [CrossRef]
- Brown, J.R.; Bharucha, T.; Breuer, J. Encephalitis diagnosis using metagenomics: application of next generation sequencing for undiagnosed cases. J. Infect. 2018, 76, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.D.; Raangs, E.C.; Rosema, S.; Veloo, A.C.M.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Zanella, M.C.; Lenggenhager, L.; Schrenzel, J.; Cordey, S.; Kaiser, L. High-throughput sequencing for the aetiologic identification of viral encephalitis, meningoencephalitis, and meningitis. A narrative review and clinical appraisal. Clin. Microbiol. Infect. 2019, 25, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Ruppé, E.; Schrenzel, J. Messages from the third International Conference on Clinical Metagenomics (ICCMg3). Microbes Infect. 2019, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Victoria, J.G.; Kapoor, A.; Li, L.; Blinkova, O.; Slikas, B.; Wang, C.; Naeem, A.; Zaidi, S.; Delwart, E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J. Virol. 2009, 83, 4642–4651. [Google Scholar] [CrossRef]
- Greninger, A.L.; Naccache, S.N.; Federman, S.; Yu, G.; Mbala, P.; Bres, V.; Stryke, D.; Bouquet, J.; Somasekar, S.; Linnen, J.M.; et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 2015, 6, e01491-15. [Google Scholar] [CrossRef]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef]
- Luk, K.-C.; Berg, M.G.; Naccache, S.N.; Kabre, B.; Federman, S.; Mbanya, D.; Kaptué, L.; Chiu, C.Y.; Brennan, C.A.; Hackett, J. Utility of metagenomic next-generation sequencing for characterization of HIV and human Pegivirus diversity. PLoS ONE 2015, 10, e0141723. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Simner, P.J.; Miller, S.; Carroll, K.C. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clin. Infect. Dis 2017, 380, 2095. [Google Scholar] [CrossRef] [PubMed]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, S.A.; Deveson, I.W.; Mercer, T.R. Reference standards for next-generation sequencing. Nat. Rev. Genet. 2017, 18, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, D.W.; Zagordi, O.; Geissberger, F.-D.; Kufner, V.; Schmutz, S.; Böni, J.; Metzner, K.J.; Trkola, A.; Huber, M. Optimization and validation of sample preparation for metagenomic sequencing of viruses in clinical samples. Microbiome 2017, 5, 1–13. [Google Scholar] [CrossRef]
- Lewandowska, D.W.; Zagordi, O.; Zbinden, A.; Schuurmans, M.M.; Schreiber, P.; Geissberger, F.-D.; Huder, J.B.; Böni, J.; Benden, C.; Mueller, N.J.; et al. Unbiased metagenomic sequencing complements specific routine diagnostic methods and increases chances to detect rare viral strains. Diagn. Microbiol. Infect. Dis. 2015, 83, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, D.W.; Capaul, R.; Prader, S.; Zagordi, O.; Geissberger, F.-D.; Kügler, M.; Knorr, M.; Berger, C.; Güngör, T.; Reichenbach, J.; et al. Persistent mammalian orthoreovirus, coxsackievirus and adenovirus co-infection in a child with a primary immunodeficiency detected by metagenomic sequencing: A case report. BMC Infect. Dis. 2018, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tschumi, F.; Schmutz, S.; Kufner, V.; Heider, M.; Pigny, F.; Schreiner, B.; Capaul, R.; Achermann, Y.; Huber, M. Meningitis and epididymitis caused by Toscana virus infection imported to Switzerland diagnosed by metagenomic sequencing: A case report. BMC Infect. Dis. 2019, 19, 1–4. [Google Scholar] [CrossRef]
- Moustafa, A.; Xie, C.; Kirkness, E.; Biggs, W.; Wong, E.; Turpaz, Y.; Bloom, K.; Delwart, E.; Nelson, K.E.; Venter, J.C.; et al. The blood DNA virome in 8,000 humans. PLoS Pathog. 2017, 13, e1006292. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Kaiser, L. Virome and Transplantation. 23 April 2018. Available online: https://www.eccmidlive.org/#resources/virome-and-transplantation. (accessed on 31 July 2019).
- Parras-Molto, M.; Rodriguez-Galet, A.; Suarez-Rodriguez, P.; Lopez-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Bukowska-Osko, I.; Perlejewski, K.; Pawelczyk, A.; Rydzanicz, M.; Pollak, A.; Popiel, M.; Cortes, K.C.; Paciorek, M.; Horban, A.; Dzieciatkowski, T.; et al. Human Pegivirus in patients with encephalitis of unclear etiology, Poland. Emerg. Infect. Dis. 2018, 24, 1785–1794. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Respective Conventional Testing | ||||||
|---|---|---|---|---|---|---|
| + | – | |||||
| All Samples | OPA = 81/94 1 | PPA = 65/92% 1 | mNGS | + | 22 | 2 |
| NPA = 95% | - | 2 pos 10 low pos 1 | 39 | |||
| CSF | OPA = 81/91% 2 | PPA = 64/88% 2 | mNGS | + | 7 | 1 |
| NPA = 93% | - | 1 pos 3 low pos 2 | 14 | |||
| Blood | OPA = 68/100% 3 | PPA = 46/100% 3 | mNGS | + | 5 | 0 |
| NPA = 100% | - | 0 6 low pos 3 | 8 | |||
| Throat swab | OPA = 91% | PPA = 100% | mNGS | + | 4 | 1 |
| NPA = 86% | - | 0 | 6 | |||
| Age: Median (Range) | 53 (17–88 Years) |
| Male gender | 43 (64.2%) |
| Patients immunocompromised | 24 (35.8%) |
| Post SOT | 7 (29.2%) |
| Malignancy | 5 (20.8%) |
| HIV | 5 (20.8%) |
| Autoimmune disorder | 7 (29.2%) |
| Patients by department | |
| Internal medicine and subspecialties | 35 (52.2%) |
| General internal medicine | 15 (22.4%) |
| Cardiology | 7 (10.4%) |
| Infectious diseases | 7 (10.4%) |
| Pulmonolgy | 3 (4.5%) |
| Rheumatology | 2 (3%) |
| Hematology/Oncology | 1 (1.5%) |
| Neurology/Neurosurgery | 28 (41.8%) |
| Neurology | 26 (38.8%) |
| Neurosurgery | 2 (3%) |
| Other | 4 (6%) |
| Emergency department | 1 (1.5%) |
| Otorhinolaryngology | 1 (1.5%) |
| Dermatology | 2 (3%) |
| Clinical Samples: | 101 |
|---|---|
| CSF | 35 (34.7%) |
| Blood | 32 (31.7%) |
| Throat swab | 11 (11%) |
| Biopsy | 6 (6%) |
| Stool | 4 (4%) |
| Urine | 3 (3%) |
| Punctures | 3 (3%) |
| BAL | 2 (2%) |
| Others | 5 (5%) |
| Disease | Number of Cases |
|---|---|
| Neurological disorders | |
| Meningitis and/or encephalitis | 17 |
| Other central nervous system disorders 1 | 11 |
| Cerebral lesion/abscess | 3 |
| Peripheral nervous system disorders | 2 |
| PML | 1 |
| Other diseases, disorders & syndromes | |
| Pericarditis and/or myocarditis | 8 |
| Febrile syndromes (including FUO) | 8 |
| Respiratory tract infections | 4 |
| Allograft dysfunction after lung transplantation | 3 |
| Diarrhea | 3 |
| Sepsis in neutropenia | 1 |
| Cytokine-Release-Syndrome | 1 |
| Unspecific polyarthritis and lymphadenopathy | 1 |
| Constitutional symptoms unknown etiology | 1 |
| Unspecific myalgia syndrome | 1 |
| Unspecific cutaneous lesions | 1 |
| Chronic sinusitis | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kufner, V.; Plate, A.; Schmutz, S.; Braun, D.L.; Günthard, H.F.; Capaul, R.; Zbinden, A.; Mueller, N.J.; Trkola, A.; Huber, M. Two Years of Viral Metagenomics in a Tertiary Diagnostics Unit: Evaluation of the First 105 Cases. Genes 2019, 10, 661. https://doi.org/10.3390/genes10090661
Kufner V, Plate A, Schmutz S, Braun DL, Günthard HF, Capaul R, Zbinden A, Mueller NJ, Trkola A, Huber M. Two Years of Viral Metagenomics in a Tertiary Diagnostics Unit: Evaluation of the First 105 Cases. Genes. 2019; 10(9):661. https://doi.org/10.3390/genes10090661
Chicago/Turabian StyleKufner, Verena, Andreas Plate, Stefan Schmutz, Dominique L. Braun, Huldrych F. Günthard, Riccarda Capaul, Andrea Zbinden, Nicolas J. Mueller, Alexandra Trkola, and Michael Huber. 2019. "Two Years of Viral Metagenomics in a Tertiary Diagnostics Unit: Evaluation of the First 105 Cases" Genes 10, no. 9: 661. https://doi.org/10.3390/genes10090661
APA StyleKufner, V., Plate, A., Schmutz, S., Braun, D. L., Günthard, H. F., Capaul, R., Zbinden, A., Mueller, N. J., Trkola, A., & Huber, M. (2019). Two Years of Viral Metagenomics in a Tertiary Diagnostics Unit: Evaluation of the First 105 Cases. Genes, 10(9), 661. https://doi.org/10.3390/genes10090661