Viral Metagenomics in the Clinical Realm: Lessons Learned from a Swiss-Wide Ring Trial

 , ,
, ,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
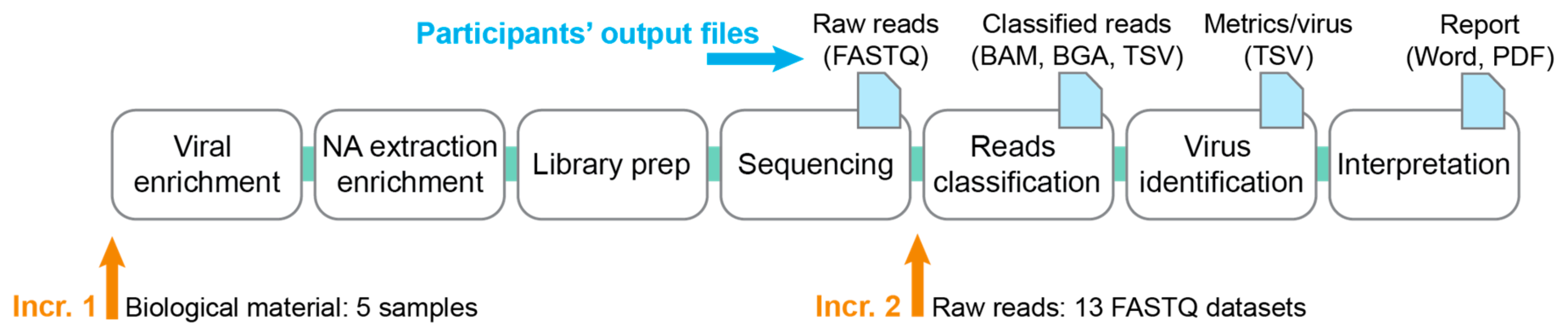
2.1. Design of the Ring Trial
2.1.1. Increment 1
- Questionnaire on methodology used (cf. Section 2.6).
- Five samples consisting of the human blood plasma spiked with known viruses (cf. Section 2.3).
- SIB common database (cf. Section 2.2).
- Filled questionnaire on the methodology that was used for each pipeline (done only once when first joining the RT, either at increment 1 or 2).
- Raw reads from the sequencer (FASTQ), before further pre-processing.
- Classified reads (e.g., BAM, BGA if aligning reads; or e.g., TSV if using a k-mer-based approach).
- The identified viruses with associated metrics (virus metrics file): Virus|database_ID|Number_of_reads|percent_coverage|genome_size
- A technical lab report containing the results interpretation and methodological details.
2.1.2. Increment 2
- Thirteen FASTQ datasets labeled with a random number, consisting of five samples from increment 1 sequenced by some of the participants, and eight in-silico generated FASTQ datasets (cf. Section 2.4).
- SIB common database (cf. Section 2.2).
2.2. Swiss Institute of Bioinformatics (SIB) Common Database
2.3. Viral Samples for Increment 1
- Human betaherpesvirus 5
- Human mastadenovirus B
- Enterovirus C
- Influenza A virus
2.4. FASTQ Datasets for Increment 2
2.4.1. Real Sequencing Reads
2.4.2. Artificial Sequencing Reads
- Human mastadenovirus A
- Human coronavirus HKU1
- Severe acute respiratory syndrome-related coronavirus (SARS)
- Influenza B virus
- Human respirovirus 1 (HPIV-1)
- Human rubulavirus 4 (HPIV-4)
- Human orthopneumovirus (Human respiratory syncytial virus)
- Human metapneumovirus
- Norwalk virus
- Rotavirus A
- Hepacivirus C (HCV)
- Human betaherpesvirus 5 (CMV)
- Human alphaherpesvirus 1 (HSV-1)
- Human betaherpesvirus 6A (HHV-6A)
- Parechovirus A
- Human alphaherpesvirus 3 (VZV)
- JC virus
- BK virus
- 1 × 100 bp
- 1 × 150 bp
- 1 × 250 bp
- 2 × 100 bp
- 2 × 150 bp
- 2 × 250 bp
2.5. Implementation of the Ring Trial
2.6. Questionnaire
- Storage
- Sample preparation (enrichment)
- DNA/RNA extraction, quantification, quality assessment
- Library preparation
- Sequencing
- Bioinformatics (reads pre-processing, methodology)
2.7. Analysis of Results
2.7.1. Pre-Processing of the Metrics File
2.7.2. Virus Identification
2.7.3. Analysis of Results
3. Results
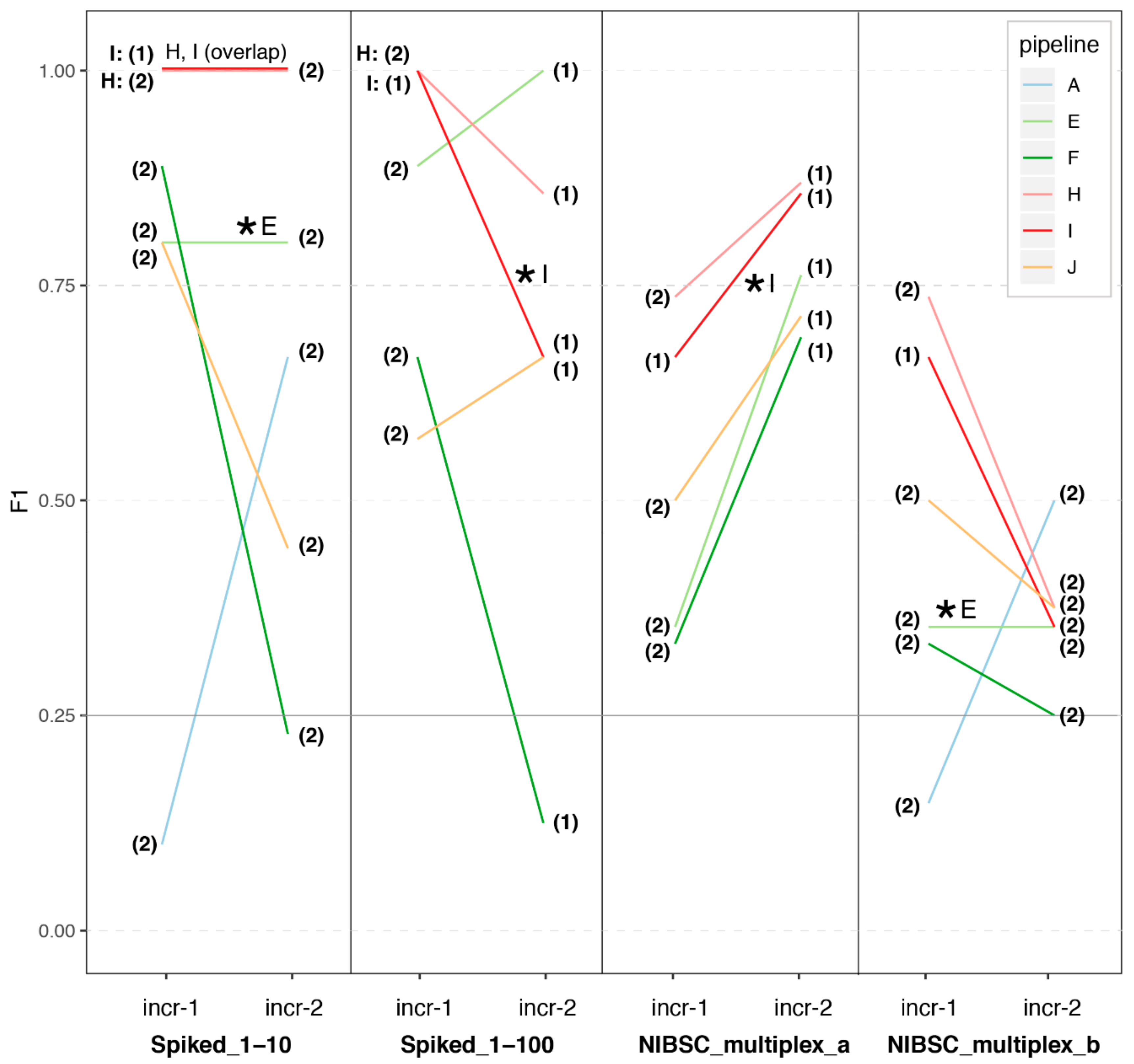
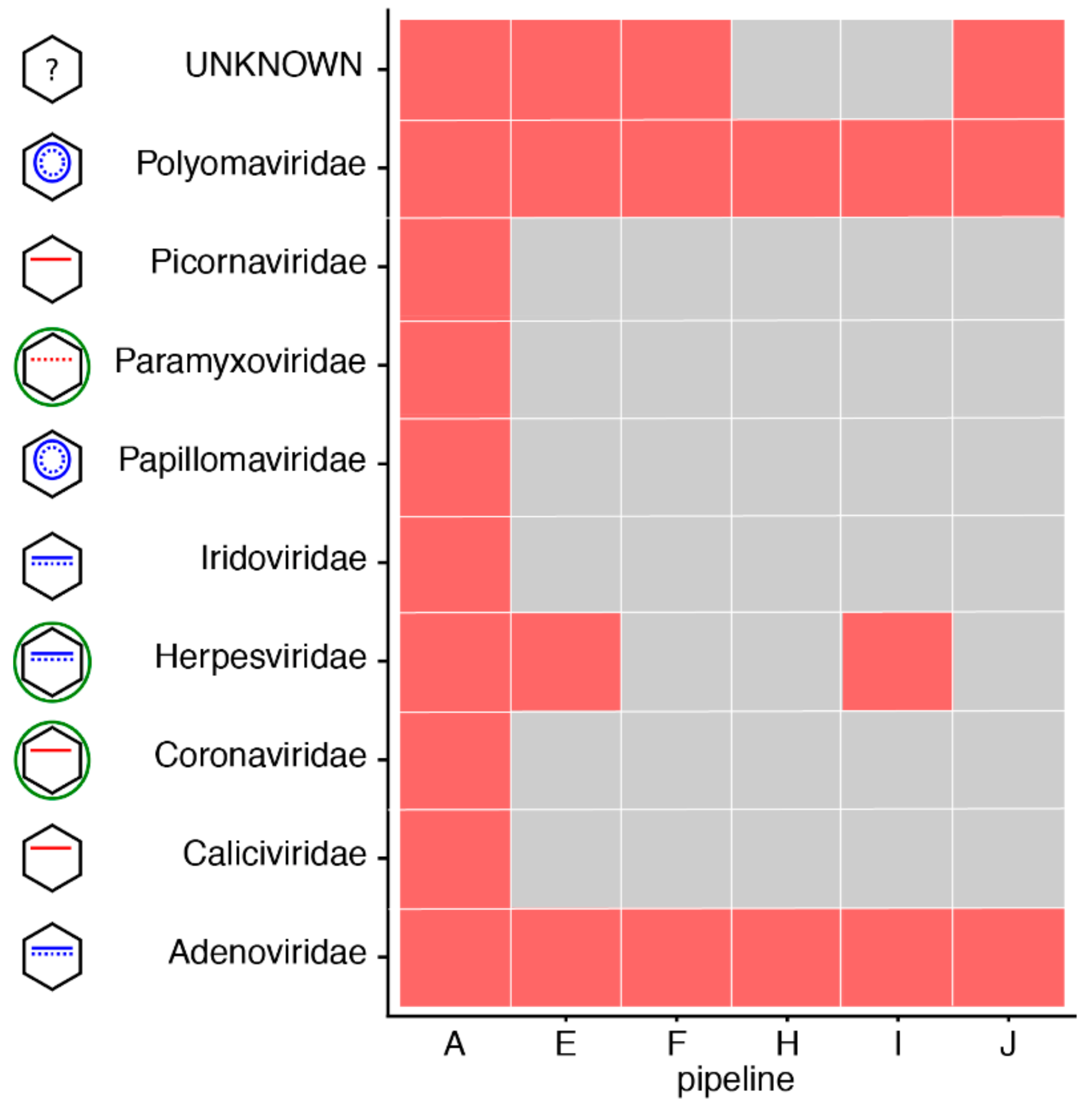
3.1. Great Variability in Overall Performance
3.2. Depth of Coverage per Virus Is Correlated with Performance
3.3. Impact of Sample Preparation
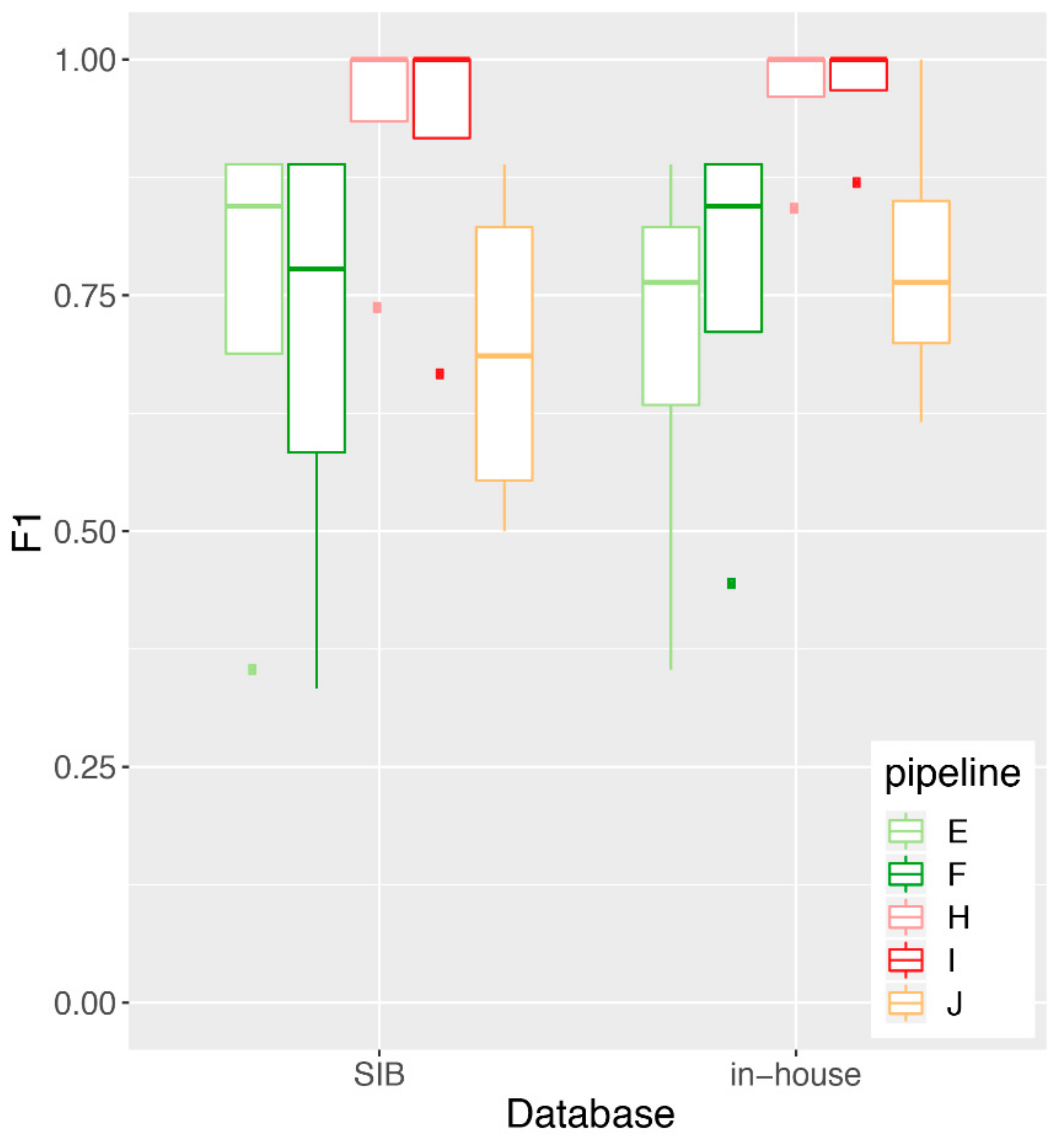
3.4. Impact of Database Quality and Size
4. Discussion
4.1. Lessons Learned and Recommendations
4.2. From Pilot Studies to Accredited External Quality Assessment (EQA)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Zanella, M.C.; Lenggenhager, L.; Schrenzel, J.; Cordey, S.; Kaiser, L. High-throughput sequencing for the aetiologic identification of viral encephalitis, meningoencephalitis, and meningitis. A narrative review and clinical appraisal. Clin. Microbiol. Infect. 2019, 25, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Bharucha, T.; Breuer, J. Encephalitis diagnosis using metagenomics: Application of next generation sequencing for undiagnosed cases. J. Infect. 2018, 76, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Greninger, A.L. The challenge of diagnostic metagenomics. Expert Rev. Mol. Diagn. 2018, 18, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-cobos, S.; Kooistra-smid, A.M.D.; Raangs, E.C.; Rosema, S.; Veloo, A.C.M.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, D.W.; Zagordi, O.; Zbinden, A.; Schuurmans, M.M.; Schreiber, P.; Geissberger, F.D.; Huder, J.B.; Böni, J.; Benden, C.; Mueller, N.J.; et al. Unbiased metagenomic sequencing complements specific routine diagnostic methods and increases chances to detect rare viral strains. Diagn. Microbiol. Infect. Dis. 2015, 83, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Petty, T.J.; Cordey, S.; Padioleau, I.; Docquier, M.; Turin, L.; Preynat-Seauve, O.; Zdobnov, E.M.; Kaiser, L. Comprehensive human virus screening using high-throughput sequencing with a user-friendly representation of bioinformatics analysis: A pilot study. J. Clin. Microbiol. 2014, 52, 3351–3361. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Schuele, L.; Raangs, E.C.; Machado, M.P.; Mendes, C.I.; Jesus, T.F.; Chlebowicz, M.; Rosema, S.; Ramirez, M.; Carriço, J.A.; et al. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Sci. Rep. 2018, 8, 13767. [Google Scholar] [CrossRef] [PubMed]
- Schlaberg, R.; Chiu, C.Y.; Miller, S.; Procop, G.W.; Weinstock, G. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch. Pathol. Lab. Med. 2017, 141, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) to Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, e00069. [Google Scholar] [CrossRef]
- NIBSC Viral Multiplex Control. Available online: http://www.nibsc.org/documents/ifu/15-130-xxx.pdf (accessed on 27 August 2019).
- NIBSC Negative Control. Available online: http://www.nibsc.org/documents/ifu/11-B606.pdf (accessed on 27 August 2019).
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Asplund, M.; Kjartansdóttir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, in press. [Google Scholar] [CrossRef]
- Brinkmann, A.; Andrusch, A.; Belka, A.; Wylezich, C.; Höper, D.; Pohlmann, A.; Petersen, T.N.; Lucas, P.; Blanchard, Y.; Papa, A.; et al. Proficiency testing of virus diagnostics based on bioinformatics analysis of simulated in silico high-throughput sequencing datasets. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset Name | Label | Pipeline | Settings | |
|---|---|---|---|---|
| Incr-1 | Incr-2 | Incr-2 | Incr-2 | |
| plasma_spike_1_1 | 5 | NA | NA | NA |
| plasma_spike_1_10 | 2 | 11 | E | 2 × 150 |
| plasma_spike_1_100 | 1 | 13 | I | 1 × 150 |
| NIBSC_viral_control a | 3 | 2 | I | 1 × 150 |
| NIBSC_viral_control b | 3 | 8 | E | 2 × 150 |
| NIBSC_negative_control | 4 | 6 | A | 2 × 150 |
| II_1-1 | NA | 9 | in-silico | various |
| II_1-40 | NA | 3 | in-silico | various |
| II_1-40 high | NA | 4 | in-silico | various |
| II_1-400 | NA | 5 | in-silico | various |
| III_1-1 | NA | 12 | in-silico | various |
| III_1-10 | NA | 7 | in-silico | various |
| III_1-10 high | NA | 1 | in-silico | various |
| III_1-100 | NA | 10 | in-silico | various |
| Dilution | Mutation Rate | Nb Reads/Virus | |
|---|---|---|---|
| II_1-1 | 1:1 | Normal | ~2000/virus |
| II_1-40 | 1:40 | Normal | ~50/virus |
| II_1-40_high | 1:40 | high | ~50/virus |
| II_1-400 | 1:400 | normal | ~6/virus |
| III_1-1 | 1:1 | normal | ~500/virus |
| III_1-10 | 1:10 | normal | ~50/virus |
| III_1-10_high | 1:10 | high | ~50/virus |
| III_1-100 | 1:100 | normal | ~6/virus |
| A | B | C | E | F | J | I | H | |
|---|---|---|---|---|---|---|---|---|
| Storage | −20 °C | −20 °C | NA | −20 °C | −20 °C | −20 °C | −20 °C | −20 °C |
| Enrichment prior extraction | None | None | NA | homogenization, centrifugation, filtration, nuclease tx | Centrifugation, filtration | Homogenization, centrifugation, DNase tx | ||
| DNA extraction | Trizol LS/Phasemaker | Trizol LS/Phasemaker | NA | QIAamp Viral RNA Mini Kit | easyMAG | easyMAG | ||
| RNA extraction | Trizol LS/Phasemaker | Not extracted | NA | Trizol | ||||
| Enrichment after extraction | QuantiTect Whole Transcriptome Kit, Qiagen | None | NA | None | None | None | None | None |
| Library preparation DNA | Nextera XT DNA Library v2 | NA | NEBNext® Ultra™ II DNA Library Prep Kit | Nextera XT | Nextera XT | |||
| Library preparation RNA | NA | TruSeq | ||||||
| Sequencer | MiSeq (2 × 150 bp) | NA | NextSeq (2 × 150 bp) | MiSeq (1 × 150 bp) | HiSeq 2500 & 4000 (2 × 100 bp) | |||
| Reads filtering | adapter trimming (bbduk.sh) | FastQC | Trimmomatic | Trimmomatic | PrinSeq, seqtk trimfq | Trimmo-matic | ||
| Taxonomic classification | k-mer | k-mer + mapping | mapping | mapping | k-mer | mapping | mapping | |
| Tool | bbmap bbsplit | Kraken2 + Bowtie2 | SeqMan NGen software v14 (DNAStar, Lasergene) | bowtie2 | Kraken | bwa mem, blastn | SNAP | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Junier, T.; Huber, M.; Schmutz, S.; Kufner, V.; Zagordi, O.; Neuenschwander, S.; Ramette, A.; Kubacki, J.; Bachofen, C.; Qi, W.; et al. Viral Metagenomics in the Clinical Realm: Lessons Learned from a Swiss-Wide Ring Trial. Genes 2019, 10, 655. https://doi.org/10.3390/genes10090655
Junier T, Huber M, Schmutz S, Kufner V, Zagordi O, Neuenschwander S, Ramette A, Kubacki J, Bachofen C, Qi W, et al. Viral Metagenomics in the Clinical Realm: Lessons Learned from a Swiss-Wide Ring Trial. Genes. 2019; 10(9):655. https://doi.org/10.3390/genes10090655
Chicago/Turabian StyleJunier, Thomas, Michael Huber, Stefan Schmutz, Verena Kufner, Osvaldo Zagordi, Stefan Neuenschwander, Alban Ramette, Jakub Kubacki, Claudia Bachofen, Weihong Qi, and et al. 2019. "Viral Metagenomics in the Clinical Realm: Lessons Learned from a Swiss-Wide Ring Trial" Genes 10, no. 9: 655. https://doi.org/10.3390/genes10090655
APA StyleJunier, T., Huber, M., Schmutz, S., Kufner, V., Zagordi, O., Neuenschwander, S., Ramette, A., Kubacki, J., Bachofen, C., Qi, W., Laubscher, F., Cordey, S., Kaiser, L., Beuret, C., Barbié, V., Fellay, J., & Lebrand, A. (2019). Viral Metagenomics in the Clinical Realm: Lessons Learned from a Swiss-Wide Ring Trial. Genes, 10(9), 655. https://doi.org/10.3390/genes10090655