A (3D-Nuclear) Space Odyssey: Making Sense of Hi-C Maps


Abstract
{kind=link}
{kind=link}
1. Introduction
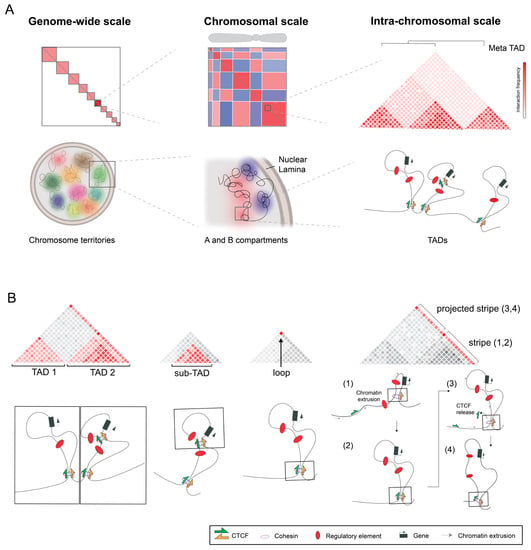
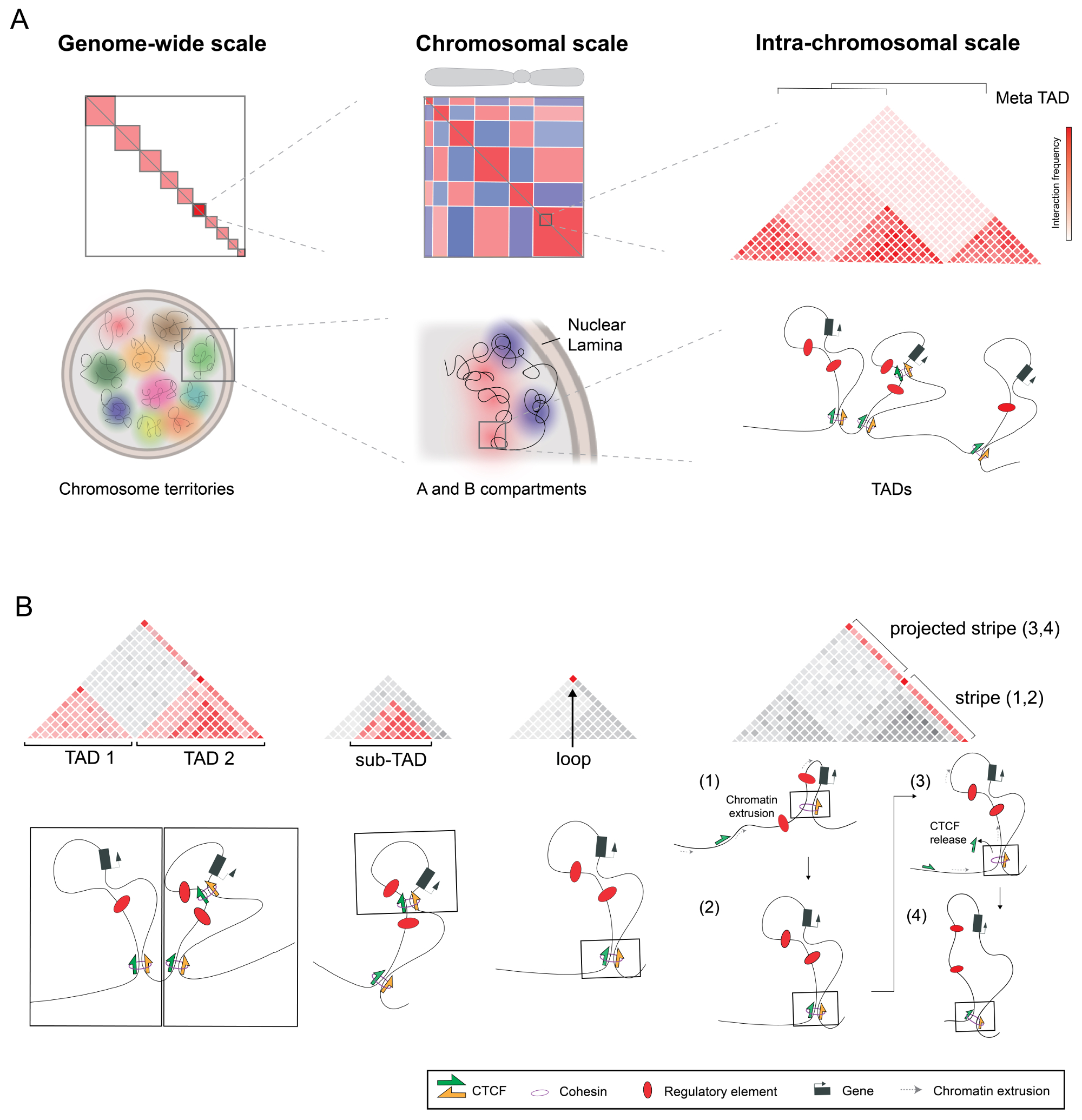
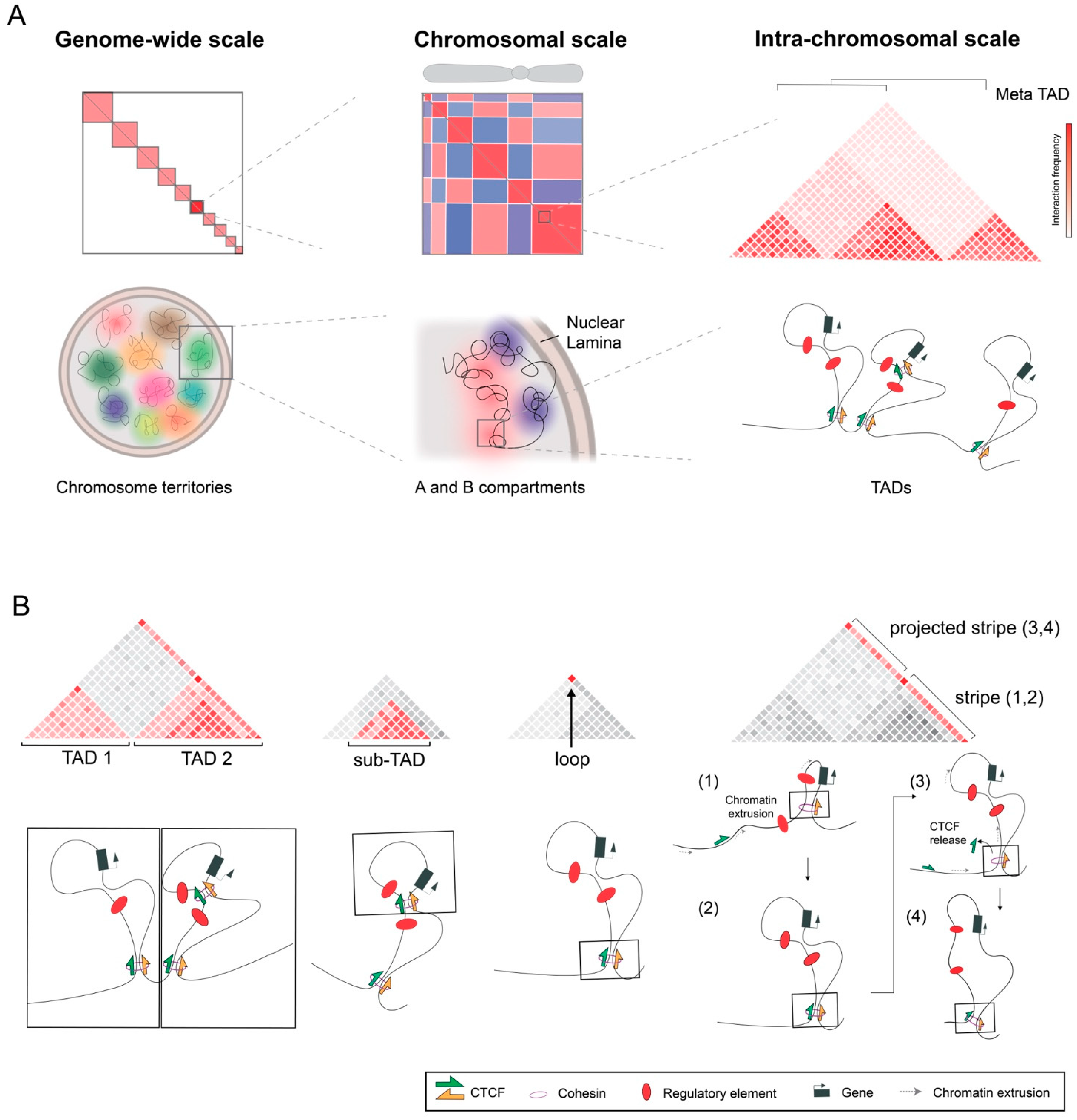
2. Prominent Structures and Mechanisms of 3D Nuclear Organization
2.1. Transcription-Based Compartments
2.2. Domains and Loops Formed by Chromatin Extrusion
2.3. Opposing Forces Organizing the 3D Chromatin Space
3. Additional Features of Hi-C Maps
3.1. Hierarchies between and within TADs
3.2. Stripes or Tracks
3.3. Assembly-Related Signatures
4. Bridging the Gap: From Population-Averaged to Single-Cell Interactions
5. Outlook
Acknowledgments
Conflicts of Interest
References
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef]
- Harmston, N.; Ing-Simmons, E.; Tan, G.; Perry, M.; Merkenschlager, M.; Lenhard, B. Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat. Commun. 2017, 8, 441. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Spielmann, M.; Mundlos, S. Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. TIG 2016, 32, 225–237. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Gómez-Skarmeta, J.L. An evolutionary perspective of regulatory landscape dynamics in development and disease. Curr. Opin. Cell Biol. 2018, 55, 24–29. [Google Scholar] [CrossRef]
- Bickmore, W.A. The spatial organization of the human genome. Annu. Rev. Genomics Hum. Genet. 2013, 14, 67–84. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, M. Chromosome Territories. Cold Spring Harb. Perspect. Biol. 2010, 2, a003889. [Google Scholar] [CrossRef]
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.-P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin architecture reorganization during stem cell differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.; Ferrai, C.; Chiariello, A.M.; Schueler, M.; Rito, T.; Laudanno, G.; Barbieri, M.; Moore, B.L.; Kraemer, D.C.A.; Aitken, S.; et al. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol. Syst. Biol. 2015, 11, 852. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E6697–E6706. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Bianco, S.; Lupiáñez, D.G.; Chiariello, A.M.; Annunziatella, C.; Kraft, K.; Schöpflin, R.; Wittler, L.; Andrey, G.; Vingron, M.; Pombo, A.; et al. Polymer physics predicts the effects of structural variants on chromatin architecture. Nat. Genet. 2018, 50, 662–667. [Google Scholar] [CrossRef]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef]
- Symmons, O.; Uslu, V.V.; Tsujimura, T.; Ruf, S.; Nassari, S.; Schwarzer, W.; Ettwiller, L.; Spitz, F. Functional and topological characteristics of mammalian regulatory domains. Genome Res. 2014, 24, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, Q.; Canzio, D.; Shou, J.; Li, J.; Gorkin, D.U.; Jung, I.; Wu, H.; Zhai, Y.; Tang, Y.; et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015, 162, 900–910. [Google Scholar] [CrossRef] [PubMed]
- de Wit, E.; Vos, E.S.M.; Holwerda, S.J.B.; Valdes-Quezada, C.; Verstegen, M.J.A.M.; Teunissen, H.; Splinter, E.; Wijchers, P.J.; Krijger, P.H.L.; de Laat, W. CTCF Binding Polarity Determines Chromatin Looping. Mol. Cell 2015, 60, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Marín, C.; Tena, J.J.; Acemel, R.D.; López-Mayorga, M.; Naranjo, S.; de la Calle-Mustienes, E.; Maeso, I.; Beccari, L.; Aneas, I.; Vielmas, E.; et al. Evolutionary comparison reveals that diverging CTCF sites are signatures of ancestral topological associating domains borders. Proc. Natl. Acad. Sci. 2015, 112, 7542–7547. [Google Scholar] [CrossRef] [PubMed]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Krietenstein, N.; Abraham, S.; Venev, S.V.; Abdennur, N.; Gibcus, J.; Hsieh, T.-H.S.; Parsi, K.M.; Yang, L.; Maehr, R.; Mirny, L.A.; et al. Ultrastructural details of mammalian chromosome architecture. bioRxiv 2019, 639922. [Google Scholar]
- Hsieh, T.-H.S.; Slobodyanyuk, E.; Hansen, A.S.; Cattoglio, C.; Rando, O.J.; Tjian, R.; Darzacq, X. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. bioRxiv 2019, 638775. [Google Scholar]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef]
- Sanborn, A.L.; Rao, S.S.P.; Huang, S.-C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [PubMed]
- Narendra, V.; Rocha, P.P.; An, D.; Raviram, R.; Skok, J.A.; Mazzoni, E.O.; Reinberg, D. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 2015, 347, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Goloborodko, A.; Valton, A.-L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef] [PubMed]
- Wutz, G.; Várnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; H Haering, C.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huang, S.-C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.-R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef]
- Gassler, J.; Brandão, H.B.; Imakaev, M.; Flyamer, I.M.; Ladstätter, S.; Bickmore, W.A.; Peters, J.; Mirny, L.A.; Tachibana, K. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J. 2017, 36, 3600–3618. [Google Scholar] [CrossRef]
- Haarhuis, J.H.I.; van der Weide, R.H.; Blomen, V.A.; Yáñez-Cuna, J.O.; Amendola, M.; van Ruiten, M.S.; Krijger, P.H.L.; Teunissen, H.; Medema, R.H.; van Steensel, B.; et al. The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 2017, 169, 693–707.e14. [Google Scholar] [CrossRef]
- Fishman, V.; Battulin, N.; Nuriddinov, M.; Maslova, A.; Zlotina, A.; Strunov, A.; Chervyakova, D.; Korablev, A.; Serov, O.; Krasikova, A. 3D organization of chicken genome demonstrates evolutionary conservation of topologically associated domains and highlights unique architecture of erythrocytes’ chromatin. Nucleic Acids Res. 2019, 47, 648–665. [Google Scholar] [CrossRef]
- Kaaij, L.J.T.; van der Weide, R.H.; Ketting, R.F.; de Wit, E. Systemic Loss and Gain of Chromatin Architecture throughout Zebrafish Development. Cell Rep. 2018, 24, 1–10.e4. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef]
- Van Bortle, K.; Nichols, M.H.; Li, L.; Ong, C.-T.; Takenaka, N.; Qin, Z.S.; Corces, V.G. Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol. 2014, 15, R82. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.J.; Nichols, M.H.; Lyu, X.; Ando-Kuri, M.; Rivera, I.S.M.; Hermetz, K.; Wang, P.; Ruan, Y.; Corces, V.G. Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol. Cell 2017, 67, 837–852.e7. [Google Scholar] [CrossRef] [PubMed]
- Eagen, K.P.; Aiden, E.L.; Kornberg, R.D. Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc. Natl. Acad. Sci. 2017, 114, 8764–8769. [Google Scholar] [CrossRef]
- Moore, J.M.; Rabaia, N.A.; Smith, L.E.; Fagerlie, S.; Gurley, K.; Loukinov, D.; Disteche, C.M.; Collins, S.J.; Kemp, C.J.; Lobanenkov, V.V.; et al. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS ONE 2012, 7, e34915. [Google Scholar] [CrossRef] [PubMed]
- Gambetta, M.C.; Furlong, E.E.M. The Insulator Protein CTCF Is Required for Correct Hox Gene Expression, but Not for Embryonic Development in Drosophila. Genetics 2018, 210, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-H.S.; Weiner, A.; Lajoie, B.; Dekker, J.; Friedman, N.; Rando, O.J. Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell 2015, 162, 108–119. [Google Scholar] [CrossRef]
- Le, T.B.K.; Imakaev, M.V.; Mirny, L.A.; Laub, M.T. High-resolution mapping of the spatial organization of a bacterial chromosome. Science 2013, 342, 731–734. [Google Scholar] [CrossRef]
- Liu, C.; Wang, C.; Wang, G.; Becker, C.; Zaidem, M.; Weigel, D. Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res. 2016, 26, 1057–1068. [Google Scholar] [CrossRef]
- Crane, E.; Bian, Q.; McCord, R.P.; Lajoie, B.R.; Wheeler, B.S.; Ralston, E.J.; Uzawa, S.; Dekker, J.; Meyer, B.J. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 2015, 523, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Hug, C.B.; Grimaldi, A.G.; Kruse, K.; Vaquerizas, J.M. Chromatin Architecture Emerges during Zygotic Genome Activation Independent of Transcription. Cell 2017, 169, 216–228.e19. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lyu, X.; Hou, C.; Takenaka, N.; Nguyen, H.Q.; Ong, C.-T.; Cubeñas-Potts, C.; Hu, M.; Lei, E.P.; Bosco, G.; et al. Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol. Cell 2015, 58, 216–231. [Google Scholar] [CrossRef]
- Battulin, N.; Fishman, V.S.; Mazur, A.M.; Pomaznoy, M.; Khabarova, A.A.; Afonnikov, D.A.; Prokhortchouk, E.B.; Serov, O.L. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 2015, 16, 77. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zheng, H.; Huang, B.; Ma, R.; Wu, J.; Zhang, X.; He, J.; Xiang, Y.; Wang, Q.; Li, Y.; et al. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 2017, 547, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Sauria, M.E.G.; Lyu, X.; Cheema, M.S.; Ausio, J.; Taylor, J.; Corces, V.G. Chromatin States in Mouse Sperm Correlate with Embryonic and Adult Regulatory Landscapes. Cell Rep. 2017, 18, 1366–1382. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.; Ali, T.M.L.; Nekrasov, M.; Delbarre, E.; Baudement, M.-O.; Kurscheid, S.; Tremethick, D.; Collas, P. Long-range interactions between topologically associating domains shape the four-dimensional genome during differentiation. Nat. Genet. 2019, 51, 835. [Google Scholar] [CrossRef]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of Cell Identity Genes Occurs in Insulated Neighborhoods in Mammalian Chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef]
- Ji, X.; Dadon, D.B.; Powell, B.E.; Fan, Z.P.; Borges-Rivera, D.; Shachar, S.; Weintraub, A.S.; Hnisz, D.; Pegoraro, G.; Lee, T.I.; et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 2016, 18, 262–275. [Google Scholar] [CrossRef]
- Phillips-Cremins, J.E.; Sauria, M.E.G.; Sanyal, A.; Gerasimova, T.I.; Lajoie, B.R.; Bell, J.S.K.; Ong, C.-T.; Hookway, T.A.; Guo, C.; Sun, Y.; et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013, 153, 1281–1295. [Google Scholar] [CrossRef]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Vian, L.; Pękowska, A.; Rao, S.S.P.; Kieffer-Kwon, K.-R.; Jung, S.; Baranello, L.; Huang, S.-C.; El Khattabi, L.; Dose, M.; Pruett, N.; et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 2018, 173, 1165–1178.e20. [Google Scholar] [CrossRef] [PubMed]
- Kraft, K.; Magg, A.; Heinrich, V.; Riemenschneider, C.; Schöpflin, R.; Markowski, J.; Ibrahim, D.M.; Acuna-Hidalgo, R.; Despang, A.; Andrey, G.; et al. Serial genomic inversions induce tissue-specific architectural stripes, gene misexpression and congenital malformations. Nat. Cell Biol. 2019, 21, 305. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Xu, J.; Dileep, V.; Zhan, Y.; Song, F.; Le, V.T.; Yardımcı, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, O.; Shamim, M.S.; Batra, S.; Durand, N.C.; Musial, N.T.; Mostofa, R.; Pham, M.; Hilaire, B.G.S.; Yao, W.; Stamenova, E.; et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. bioRxiv 2018, 254797. [Google Scholar]
- Giorgetti, L.; Galupa, R.; Nora, E.P.; Piolot, T.; Lam, F.; Dekker, J.; Tiana, G.; Heard, E. Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell 2014, 157, 950–963. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandão, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef]
- Tan, L.; Xing, D.; Chang, C.-H.; Li, H.; Xie, X.S. Three-dimensional genome structures of single diploid human cells. Science 2018, 361, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Bintu, B.; Mateo, L.J.; Su, J.-H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef] [PubMed]
- Mateo, L.J.; Murphy, S.E.; Hafner, A.; Cinquini, I.S.; Walker, C.A.; Boettiger, A.N. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 2019, 568, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Q.; Jost, D.; Chang, J.-M.; Cattoni, D.I.; Papadopoulos, G.L.; Bonev, B.; Sexton, T.; Gurgo, J.; Jacquier, C.; Nollmann, M.; et al. TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 2018, 4, eaar8082. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mota-Gómez, I.; Lupiáñez, D.G. A (3D-Nuclear) Space Odyssey: Making Sense of Hi-C Maps. Genes 2019, 10, 415. https://doi.org/10.3390/genes10060415
Mota-Gómez I, Lupiáñez DG. A (3D-Nuclear) Space Odyssey: Making Sense of Hi-C Maps. Genes. 2019; 10(6):415. https://doi.org/10.3390/genes10060415
Chicago/Turabian StyleMota-Gómez, Irene, and Darío G. Lupiáñez. 2019. "A (3D-Nuclear) Space Odyssey: Making Sense of Hi-C Maps" Genes 10, no. 6: 415. https://doi.org/10.3390/genes10060415
APA StyleMota-Gómez, I., & Lupiáñez, D. G. (2019). A (3D-Nuclear) Space Odyssey: Making Sense of Hi-C Maps. Genes, 10(6), 415. https://doi.org/10.3390/genes10060415