Hair of the Dog: Identification of a Cis-Regulatory Module Predicted to Influence Canine Coat Composition



Abstract
:1. Introduction
2. Materials and Methods
2.1. Coat Number Assignment
2.2. Sample Dataset for Single Nucleotide Polymorphism-Based Analysis
2.3. Sample Dataset for Whole Genome Sequence-Based Analysis
2.4. Genome Wide Association and Linkage Disequilibrium
2.5. Structural Variant Prediction Analysis
2.6. Splice Site Prediction Analysis
2.7. Transcription Factor Binding Prediction
3. Results
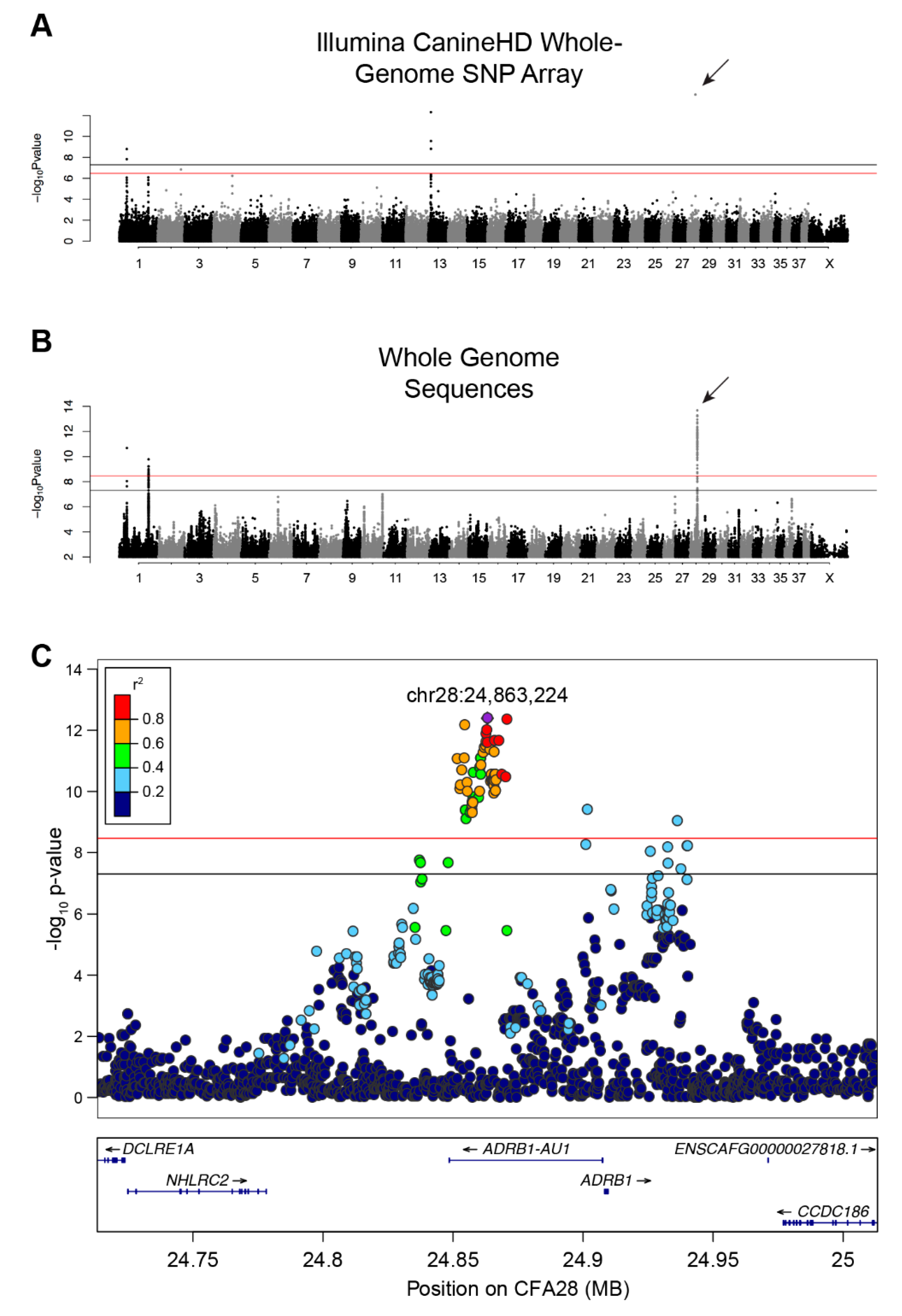
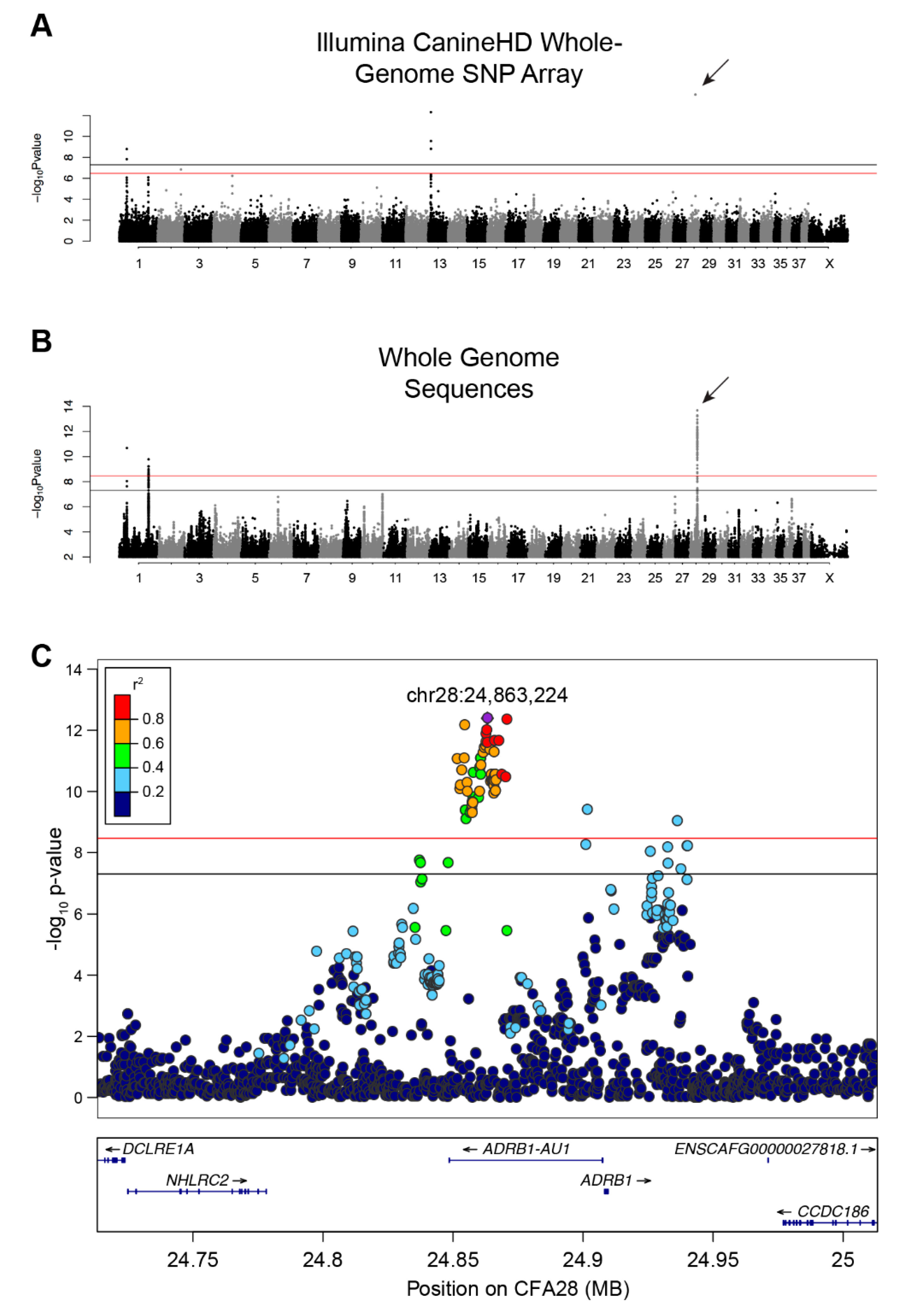
3.1. SNP Array-Based GWAS
3.2. Whole Genome Sequence-Based GWAS and Linkage Disequilibrium
3.3. Genomic Structural Variants
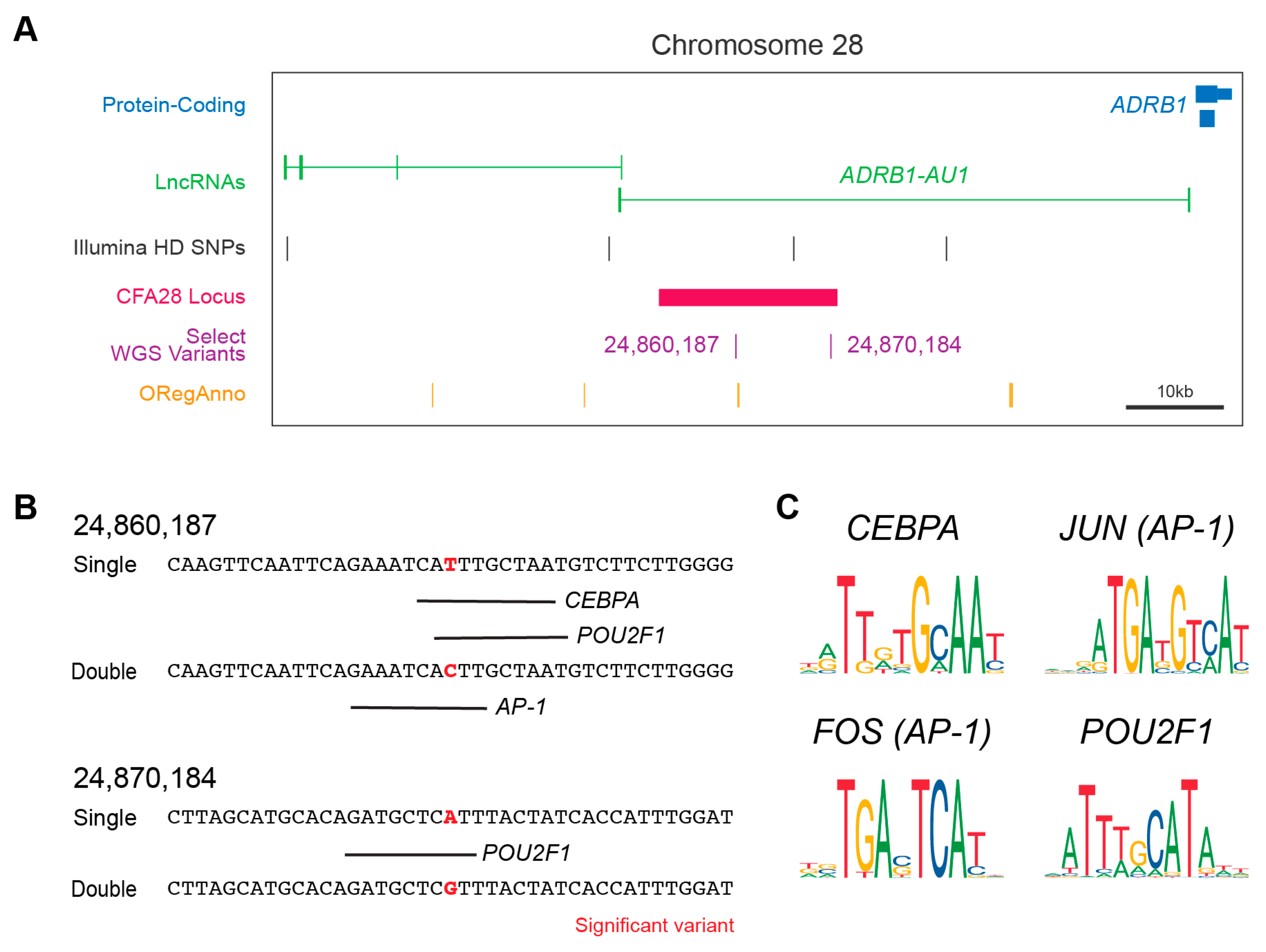
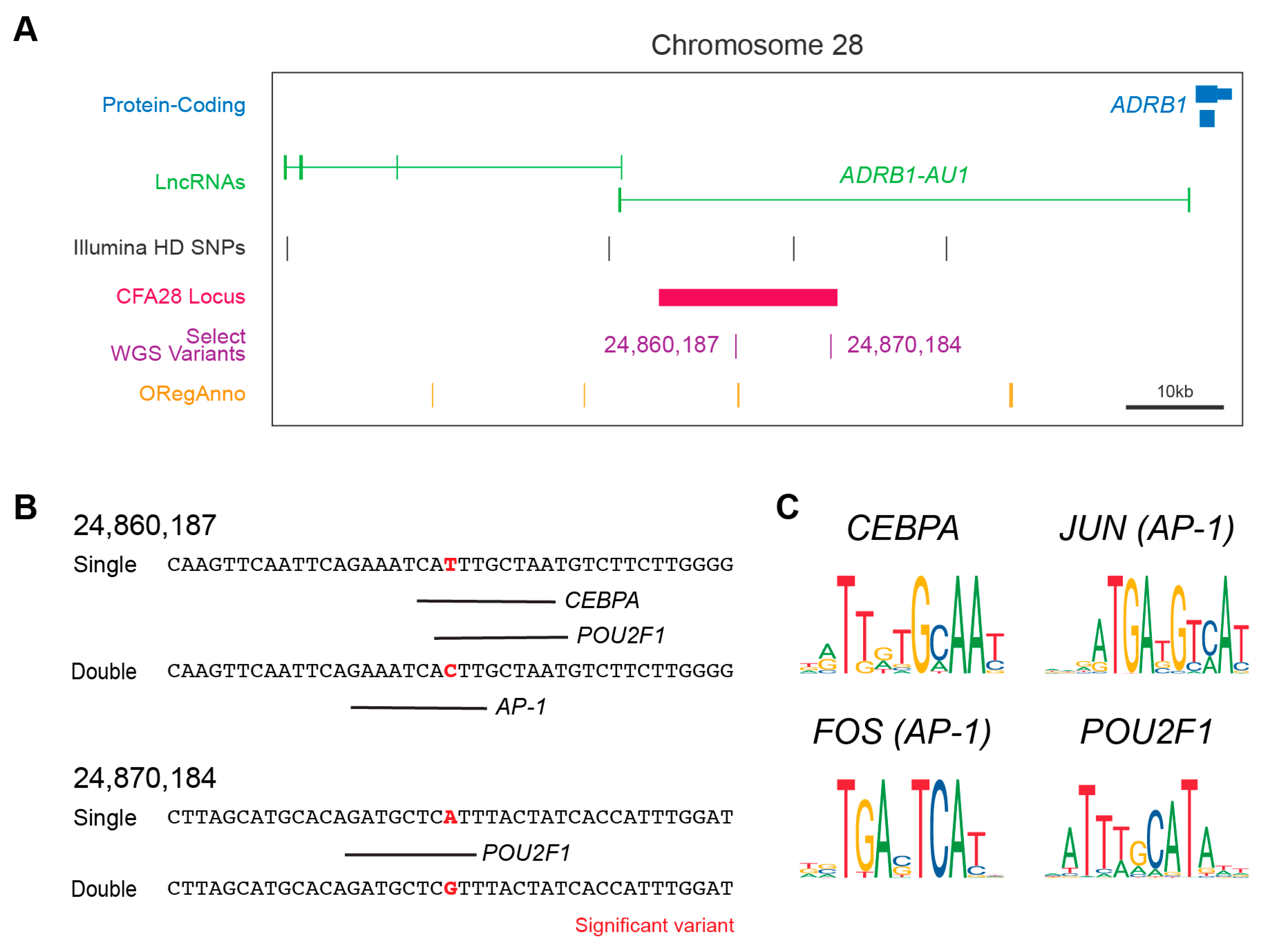
3.4. Fine Mapping of Small Variants
3.5. Impact of Variants on Gene Regulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrander, E.A.; Wayne, R.K.; Freedman, A.H.; Davis, B.W. Demographic history, selection and functional diversity of the canine genome. Nat. Rev. Genet. 2017, 18, 705–720. [Google Scholar] [CrossRef]
- Club, A.K. The Complete Dog Book; Random House Digital, Inc.: New York, NY, USA, 2006. [Google Scholar]
- Wilcox, B.; Walkowicz, C. Atlas of Dog Breeds of the World; TFH Publications: Neptune City, NJ, USA, 1995. [Google Scholar]
- Parker, H.G.; VonHoldt, B.M.; Quignon, P.; Margulies, E.H.; Shao, S.; Mosher, D.S.; Spady, T.C.; Elkahloun, A.; Cargill, M.; Jones, P.G.; et al. An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs. Science 2009, 325, 995–998. [Google Scholar]
- Vaysse, A.; Ratnakumar, A.; Derrien, T.; Axelsson, E.; Rosengren Pielberg, G.; Sigurdsson, S.; Fall, T.; Seppala, E.H.; Hansen, M.S.; Lawley, C.T.; et al. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 2011, 7, e1002316. [Google Scholar] [CrossRef]
- Boyko, A.R.; Quignon, P.; Li, L.; Schoenebeck, J.J.; Degenhardt, J.D.; Lohmueller, K.E.; Zhao, K.; Brisbin, A.; Parker, H.G.; von Holdt, B.M.; et al. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010, 8, e1000451. [Google Scholar] [CrossRef]
- Plassais, J.; Kim, J.; Davis, B.W.; Karyadi, D.M.; Hogan, A.N.; Harris, A.C.; Decker, B.; Parker, H.G.; Ostrander, E.A. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Cadieu, E.; Neff, M.W.; Quignon, P.; Walsh, K.; Chase, K.; Parker, H.G.; Vonholdt, B.M.; Rhue, A.; Boyko, A.; Byers, A.; et al. Coat variation in the domestic dog is governed by variants in three genes. Science 2009, 326, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.J.; Castelhano, M.G.; Oliveira, K.C.; Corey, E.; Balkman, C.; Baxter, T.L.; Casal, M.L.; Center, S.A.; Fang, M.; Garrison, S.J.; et al. Complex disease and phenotype mapping in the domestic dog. Nat. Commun. 2016, 7, 10460. [Google Scholar] [CrossRef]
- Parker, H.G.; Harris, A.; Dreger, D.L.; Davis, B.W.; Ostrander, E.A. The bald and the beautiful: hairlessness in domestic dog breeds. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef]
- Parker, H.G.; Chase, K.; Cadieu, E.; Lark, K.G.; Ostrander, E.A. An insertion in the RSPO2 gene correlates with improper coat in the Portuguese water dog. J. Hered. 2010, 101, 612–617. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.P.; Johnson, G.S.; Schnabel, R.D.; Khan, S.; Coates, J.R.; Johnson, G.C.; Taylor, J.F. Genetic mapping of canine multiple system degeneration and ectodermal dysplasia loci. J. Hered. 2005, 96, 727–734. [Google Scholar] [CrossRef]
- Drogemuller, C.; Karlsson, E.K.; Hytonen, M.K.; Perloski, M.; Dolf, G.; Sainio, K.; Lohi, H.; Lindblad-Toh, K.; Leeb, T. A mutation in hairless dogs implicates FOXI3 in ectodermal development. Science 2008, 321, 1462. [Google Scholar] [CrossRef] [PubMed]
- Drogemuller, C.; Rufenacht, S.; Wichert, B.; Leeb, T. Mutations within the FGF5 gene are associated with hair length in cats. Anim. Genet. 2007, 38, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.A.; Petukhova, L.; Harel, S.; Ho, Y.Y.; Drill, E.; Shapiro, L.; Wajid, M.; Christiano, A.M. FGF5 is a crucial regulator of hair length in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 10648–10653. [Google Scholar] [CrossRef]
- Hu, R.; Fan, Z.Y.; Wang, B.Y.; Deng, S.L.; Zhang, X.S.; Zhang, J.L.; Han, H.B.; Lian, Z.X. Rapid Communication: Generation of FGF5 knockout sheep via the CRISPR/Cas9 system. J. Anim. Sci. 2017, 95, 2019–2024. [Google Scholar] [PubMed]
- Rencz, F.; Gulacsi, L.; Pentek, M.; Wikonkal, N.; Baji, P.; Brodszky, V. Alopecia areata and health-related quality of life: A systematic review and meta-analysis. Br. J. Dermatol. 2016, 175, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Tabolli, S.; Sampogna, F.; di Pietro, C.; Mannooranparampil, T.J.; Ribuffo, M.; Abeni, D. Health status, coping strategies, and alexithymia in subjects with androgenetic alopecia: A questionnaire study. Am. J. Clin. Dermatol. 2013, 14, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Pinkus, H. Multiple hairs (Flemming-Giovannini; report of two cases of pili multigemini and discussion of some other anomalies of the pilary complex. J. Investig. Dermatol. 1951, 17, 291–301. [Google Scholar] [CrossRef]
- Jimenez, F.; Ruifernandez, J.M. Distribution of human hair in follicular units. A mathematical model for estimating the donor size in follicular unit transplantation. Dermatol. Surg. 1999, 25, 294–298. [Google Scholar] [CrossRef]
- Welle, M.M.; Wiener, D.J. The Hair Follicle: A Comparative Review of Canine Hair Follicle Anatomy and Physiology. Toxicol. Pathol. 2016, 44, 564–574. [Google Scholar] [CrossRef]
- Thalmann, O.; Shapiro, B.; Cui, P.; Schuenemann, V.J.; Sawyer, S.K.; Greenfield, D.L.; Germonpre, M.B.; Sablin, M.V.; Lopez-Giraldez, F.; Domingo-Roura, X.; et al. Complete mitochondrial genomes of ancient canids suggest a European origin of domestic dogs. Science 2013, 342, 871–874. [Google Scholar] [CrossRef]
- Parker, H.G.; Dreger, D.L.; Rimbault, M.; Davis, B.W.; Mullen, A.B.; Carpintero-Ramirez, G.; Ostrander, E.A. Genomic Analyses Reveal the Influence of Geographic Origin, Migration, and Hybridization on Modern Dog Breed Development. Cell Rep. 2017, 19, 697–708. [Google Scholar] [CrossRef]
- American Kennel Club. Available online: https://www.akc.org/ (accessed on 25 April 2019).
- Fédération Cynologique Internationale. Available online: http://www.fci.be/en/ (accessed on 25 April 2019).
- United Kennel Club. Available online: https://www.ukcdogs.com/ (accessed on 25 April 2019).
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Risch, N.; Merikangas, K. The future of genetic studies of complex human diseases. Science 1996, 273, 1516–1517. [Google Scholar] [CrossRef] [PubMed]
- Bland, J.M.; Altman, D.G. Multiple significance tests: the Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef]
- Pruim, R.J.; Welch, R.P.; Sanna, S.; Teslovich, T.M.; Chines, P.S.; Gliedt, T.P.; Boehnke, M.; Abecasis, G.R.; Willer, C.J. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 2010, 26, 2336–2337. [Google Scholar] [CrossRef] [PubMed]
- UCSC Genome Browser. Available online: https://genome.ucsc.edu/index.html (accessed on 25 April 2019).
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef]
- Wang, M.; Marin, A. Characterization and prediction of alternative splice sites. Gene 2006, 366, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Grabe, N. AliBaba2: Context specific identification of transcription factor binding sites. Silico Biol. 2002, 2, S1–S15. [Google Scholar]
- Wingender, E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 2008, 9, 326–332. [Google Scholar] [CrossRef]
- Wright, M.W. A short guide to long non-coding RNA gene nomenclature. Hum. Genomics 2014, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Lesurf, R.; Cotto, K.C.; Wang, G.; Griffith, M.; Kasaian, K.; Jones, S.J.; Montgomery, S.B.; Griffith, O.L. The Open Regulatory Annotation Consortium. ORegAnno 3.0: A community-driven resource for curated regulatory annotation. Nucleic Acids Res. 2016, 44, D126–D132.45. [Google Scholar] [CrossRef]
- Plassais, J.; (National Human Genome Research Institute, Bethesda, MD, USA). Personal communication, 2019.
- Balmer, P.; Bauer, A.; Pujar, S.; McGarvey, K.M.; Welle, M.; Galichet, A.; Muller, E.J.; Pruitt, K.D.; Leeb, T.; Jagannathan, V. A curated catalog of canine and equine keratin genes. PLoS ONE 2017, 12, e0180359. [Google Scholar] [CrossRef]
- Rezza, A.; Wang, Z.; Sennett, R.; Qiao, W.; Wang, D.; Heitman, N.; Mok, K.W.; Clavel, C.; Yi, R.; Zandstra, P.; et al. Signaling Networks among Stem Cell Precursors, Transit-Amplifying Progenitors, and their Niche in Developing Hair Follicles. Cell Rep. 2016, 14, 3001–3018. [Google Scholar] [CrossRef]
- Sennett, R.; Wang, Z.; Rezza, A.; Grisanti, L.; Roitershtein, N.; Sicchio, C.; Mok, K.W.; Heitman, N.J.; Clavel, C.; Ma’ayan, A.; et al. An Integrated Transcriptome Atlas of Embryonic Hair Follicle Progenitors, Their Niche, and the Developing Skin. Dev. Cell 2015, 34, 577–591. [Google Scholar] [CrossRef]
- Rimbault, M.; Beale, H.C.; Schoenebeck, J.J.; Hoopes, B.C.; Allen, J.J.; Kilroy-Glynn, P.; Wayne, R.K.; Sutter, N.B.; Ostrander, E.A. Derived variants at six genes explain nearly half of size reduction in dog breeds. Genome Res. 2013, 23, 1985–1995. [Google Scholar] [CrossRef]
- Yang, Z.; Cui, K.; Zhang, Y.; Deng, X. Transcriptional regulation analysis and the potential transcription regulator site in the extended KAP6.1 promoter in sheep. Mol. Biol. Rep. 2014, 41, 6089–6096. [Google Scholar] [CrossRef]
- Hoeppner, M.P.; Lundquist, A.; Pirun, M.; Meadows, J.R.; Zamani, N.; Johnson, J.; Sundstrom, G.; Cook, A.; FitzGerald, M.G.; Swofford, R.; et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS ONE 2014, 9, e91172. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, D.K.; Desai, K.H.; Jasper, J.R.; Stevens, M.E.; Regula, D.P., Jr.; Barsh, G.S.; Bernstein, D.; Kobilka, B.K. Targeted disruption of the mouse beta1-adrenergic receptor gene: Developmental and cardiovascular effects. Proc. Natl. Acad. Sci. USA 1996, 93, 7375–7380. [Google Scholar] [CrossRef]
- Pak, Y.; Pham, N.; Rotin, D. Direct binding of the beta1 adrenergic receptor to the cyclic AMP-dependent guanine nucleotide exchange factor CNrasGEF leads to Ras activation. Mol. Cell. Biol. 2002, 22, 7942–7952. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Torres, D.; Marone, R.; Feng, X.; Martin, D.; Simaan, M.; Chen, M.; Weinstein, L.S.; Taylor, S.S.; Molinolo, A.A.; et al. Inactivation of a Galpha(s)-PKA tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat. Cell. Biol. 2015, 17, 793–803. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| CFA28 Position | Ancestral Allele | Derived Allele | Wolf Derived | Double Derived | Single Derived | Wald p-Value * |
|---|---|---|---|---|---|---|
| 24,860,187 | C | T | 0.0000 | 0.3731 | 0.91011 | 1.01 × 10−10 |
| 24,870,184 | G | A | 0.0571 | 0.3769 | 0.91573 | 3.37 × 10−11 |
| 24,851,582 | G | C | 0.1429 | 0.3918 | 0.9382 | 8.79 × 10−12 |
| 24,865,942 | T | C | 0.2286 | 0.3657 | 0.92135 | 2.23 × 10−12 |
| 24,862,630 | A | G | 0.2714 | 0.3657 | 0.91011 | 1.29 × 10−12 |
| 24,864,369 | T | G | 0.2857 | 0.4403 | 0.9382 | 4.75 × 10−11 |
| 24,866,307 | T | G | 0.3000 | 0.4403 | 0.9382 | 9.35 × 10−11 |
| 24,863,224 | G | A | 0.3143 | 0.3619 | 0.91011 | 4.04 × 10−13 |
| 24,865,980 | C | T | 0.3143 | 0.4366 | 0.9382 | 2.77 × 10−11 |
| 24,864,985 | T | C | 0.3429 | 0.444 | 0.94382 | 4.37 × 10−11 |
| 24,865,626 | A | G | 0.3429 | 0.444 | 0.9382 | 1.15 × 10−10 |
| 24,865,654 | G | T | 0.3571 | 0.4403 | 0.9382 | 6.42 × 10−11 |
| 24,852,776 | A | AAGTCTTCAT | 0.3714 | 0.4142 | 0.9382 | 6.11 × 10−11 |
| 24,853,325 | T | TA | 0.3714 | 0.3993 | 0.9382 | 1.99 × 10−11 |
| 24,866,155 | C | A | 0.3714 | 0.4366 | 0.9382 | 2.77 × 10−11 |
| 24,866,296 | T | C | 0.3714 | 0.4403 | 0.9382 | 9.35 × 10−11 |
| 24,865,525 | A | G | 0.3857 | 0.4403 | 0.9382 | 4.67 × 10−11 |
| 24,865,800 | A | G | 0.3857 | 0.4366 | 0.9382 | 4.45 × 10−11 |
| 24,866,114 | C | A | 0.3857 | 0.4366 | 0.9382 | 2.77 × 10−11 |
| 24,866,181 | C | G | 0.3857 | 0.4366 | 0.9382 | 2.77 × 10−11 |
| 24,866,484 | G | A | 0.3857 | 0.4478 | 0.9382 | 4.28 × 10−11 |
| 24,867,588 | C | T | 0.3857 | 0.3657 | 0.92135 | 2.23 × 10−12 |
| 24,854,456 | A | ATTTGT | 0.4143 | 0.3993 | 0.94944 | 6.77 × 10−13 |
| 24,864,154 | A | T | 0.4286 | 0.4403 | 0.9382 | 4.23 × 10−12 |
| 24,864,658 | A | G | 0.4286 | 0.4366 | 0.9382 | 2.77 × 10−11 |
| 24,863,186 | C | CT | 0.4429 | 0.4067 | 0.90449 | 2.54 × 10−12 |
| 24,865,062 | A | AACAAC | 0.4429 | 0.4403 | 0.9382 | 4.67 × 10−11 |
| 24,868,735 | G | A | 0.4429 | 0.3843 | 0.92135 | 2.87 × 10−11 |
| Dog | E14.5 Mouse | P5 Mouse | ||||
|---|---|---|---|---|---|---|
| Gene Name | Hair Follicle | Placode | Dermal Condensate | Bulge Stem Cells | Transit Amplifying Cells | Dermal Papilla |
| CEBPA | 32.9 | 22.6 | 2.5 | 87.1 | 16.4 | 38.2 |
| FOS | 37.5 | 4.2 | 6.1 | 444.2 | 237.9 | 425.0 |
| JUN | 88.0 | 7.7 | 17.6 | 374.5 | 286.4 | 797.1 |
| POU2F1 | 0.42 | 14.3 | 6.5 | 12.9 | 14.3 | 9.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitaker, D.T.; Ostrander, E.A. Hair of the Dog: Identification of a Cis-Regulatory Module Predicted to Influence Canine Coat Composition. Genes 2019, 10, 323. https://doi.org/10.3390/genes10050323
Whitaker DT, Ostrander EA. Hair of the Dog: Identification of a Cis-Regulatory Module Predicted to Influence Canine Coat Composition. Genes. 2019; 10(5):323. https://doi.org/10.3390/genes10050323
Chicago/Turabian StyleWhitaker, D. Thad, and Elaine A. Ostrander. 2019. "Hair of the Dog: Identification of a Cis-Regulatory Module Predicted to Influence Canine Coat Composition" Genes 10, no. 5: 323. https://doi.org/10.3390/genes10050323
APA StyleWhitaker, D. T., & Ostrander, E. A. (2019). Hair of the Dog: Identification of a Cis-Regulatory Module Predicted to Influence Canine Coat Composition. Genes, 10(5), 323. https://doi.org/10.3390/genes10050323