Computational Characterization of the mtORF of Pocilloporid Corals: Insights into Protein Structure and Function in Stylophora Lineages from Contrasting Environments

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Evolutionary Origin of the mtORF
2.2. Characterization of the mtORF Gene
2.3. Annotation of Protein Domains and Detection of Intrinsic Disorder
2.4. Structural and Functional Annotation
2.5. Signatures of Selection in a Family Framework and Analysis of Codon Usage
2.5.1. Selection
2.5.2. Codon Usage Bias
2.6. Signatures of Adaptive Evolution in the mtORF of Stylophora Inhabiting Different Environments
3. Results
3.1. The mtORF Occurs in All Pocilloporids and Does Not Exhibit Stop Codons
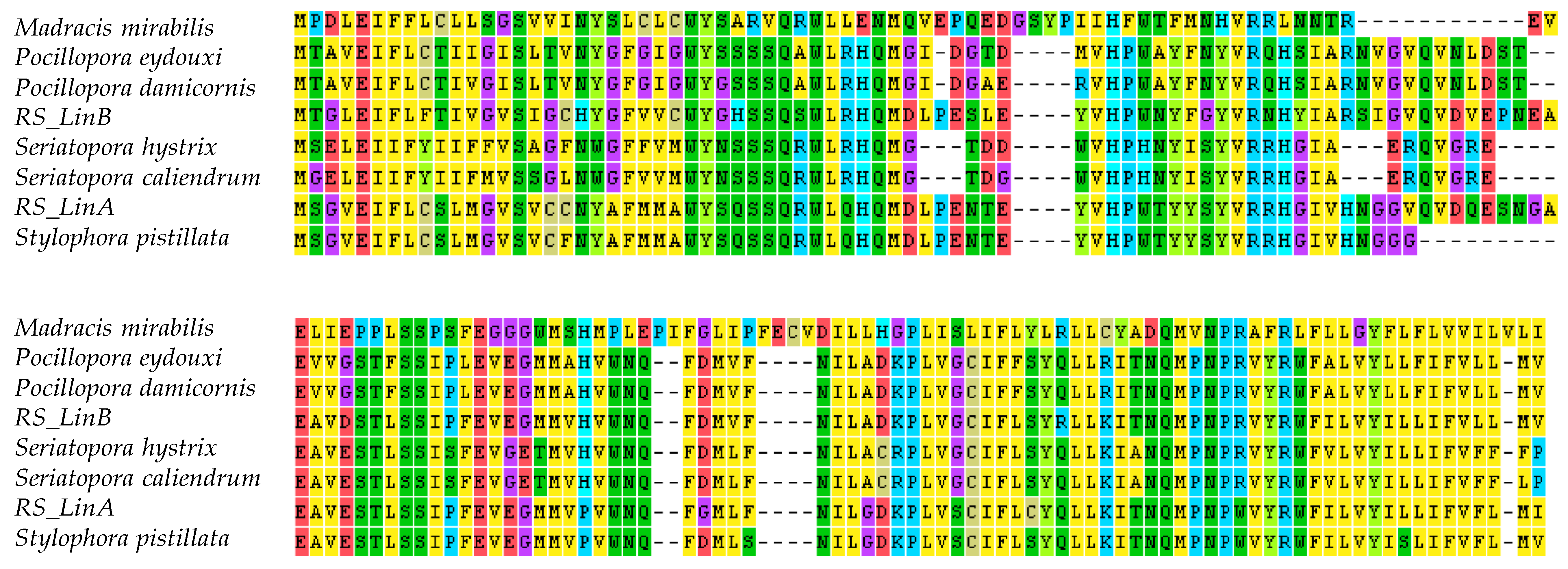
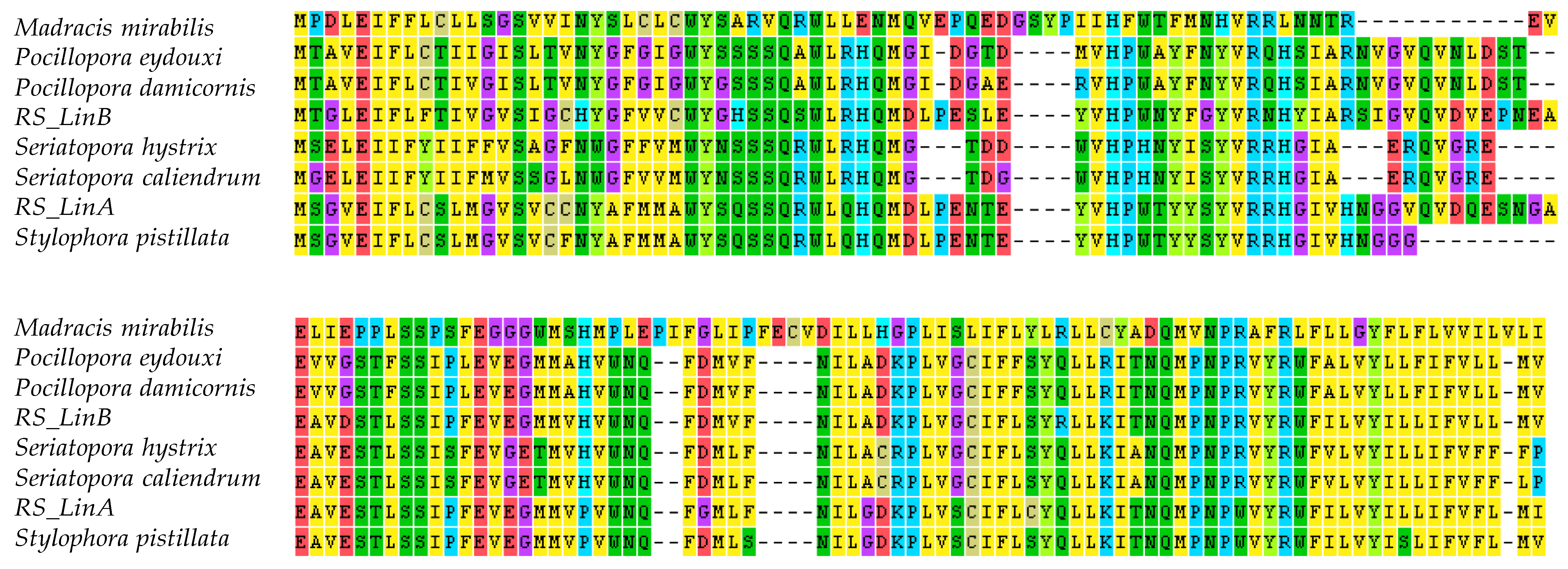
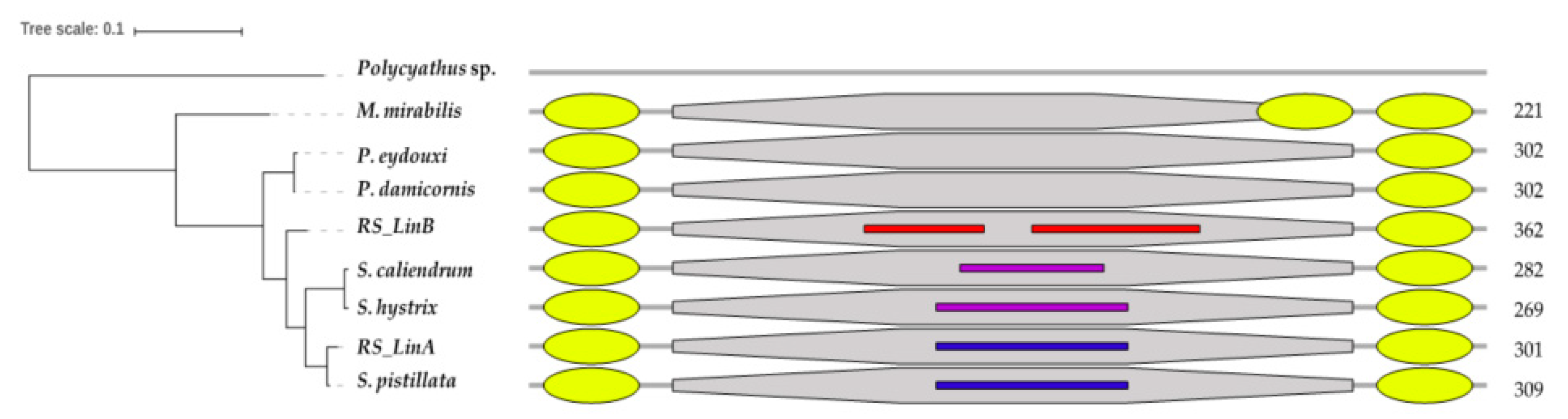
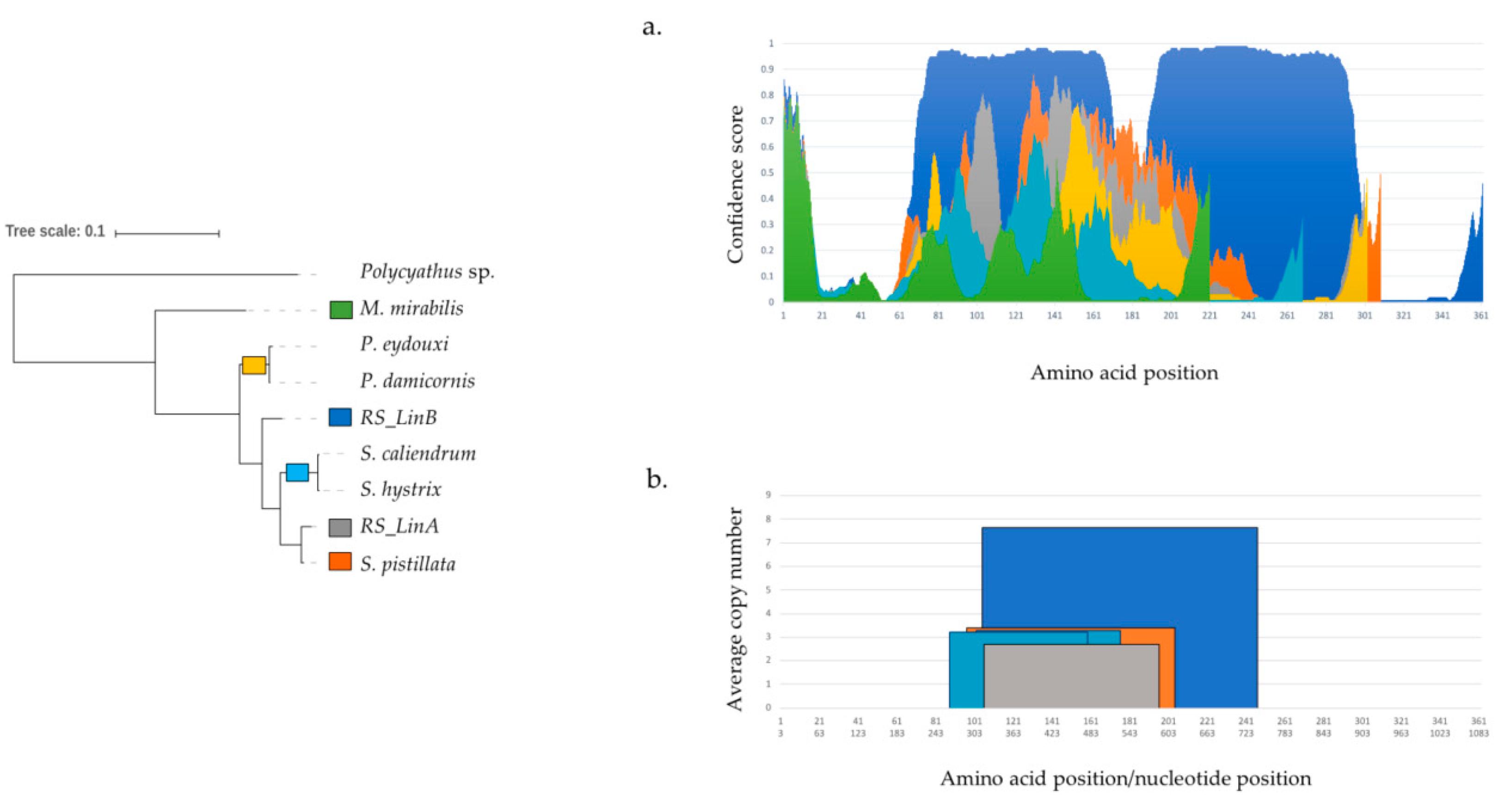
3.2. The mtORF-Encoded Proteins of Pocilloporids Vary in Length and in Aliphatic Indices
3.3. mtORF-Encoded Proteins Contain Transmembrane Domains and Intrinsically Disordered Regions
3.4. Remote Homologs Suggest that mtORF has a Hydrolase Domain
3.5. Biological Processes and Cellular Localization Support the Hypothesis of a Mitochondrial Transmembrane Protein
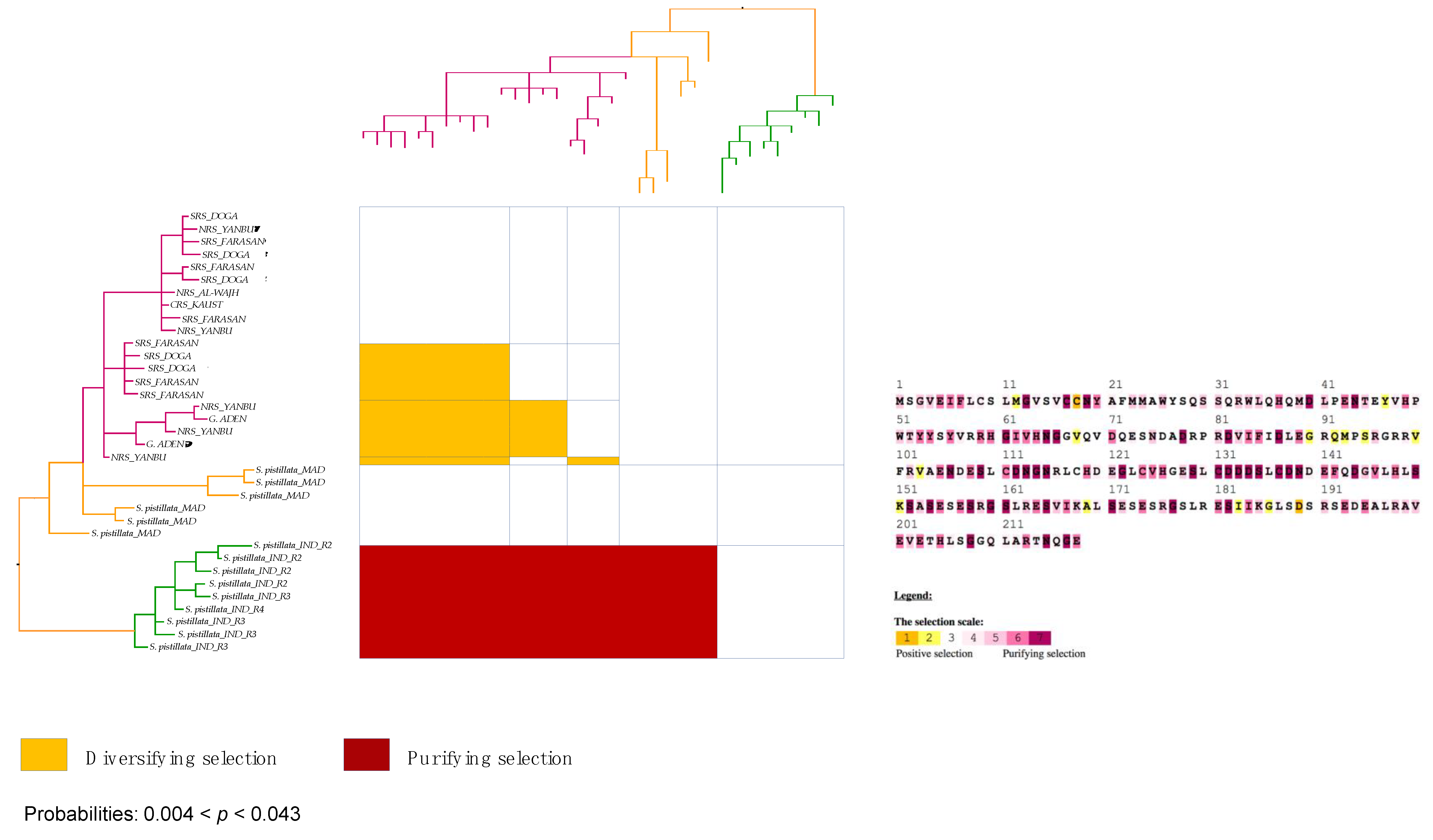
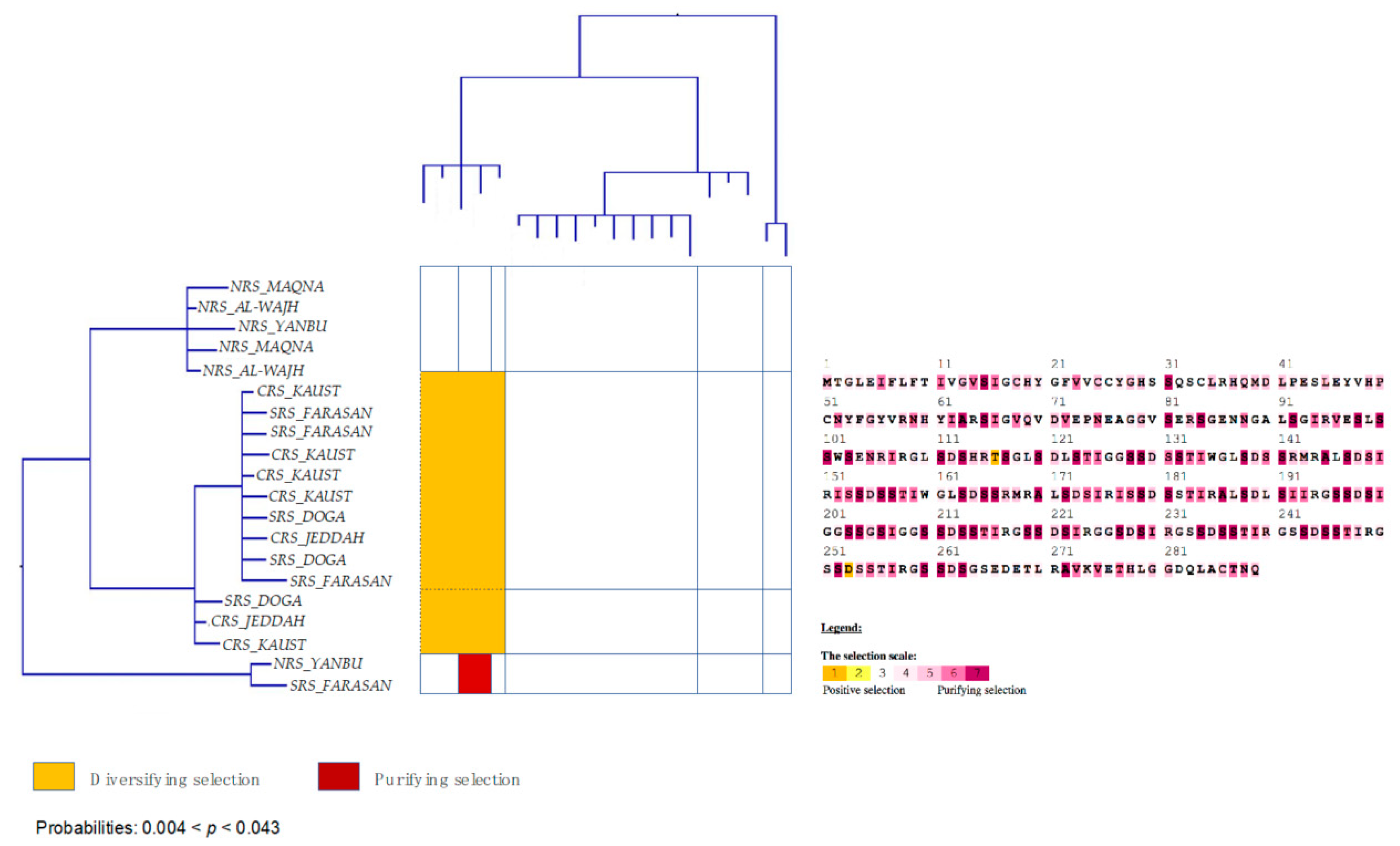
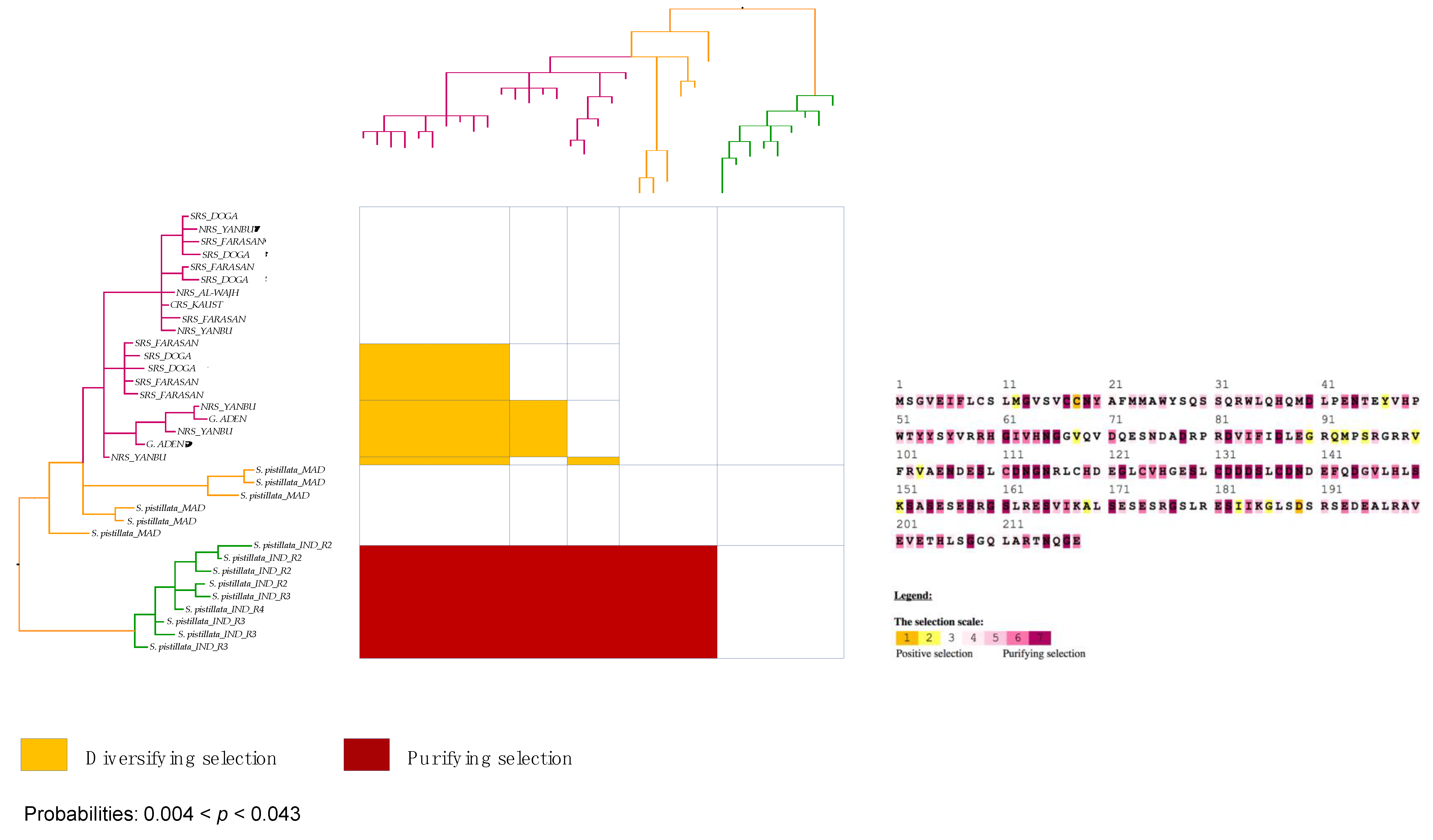
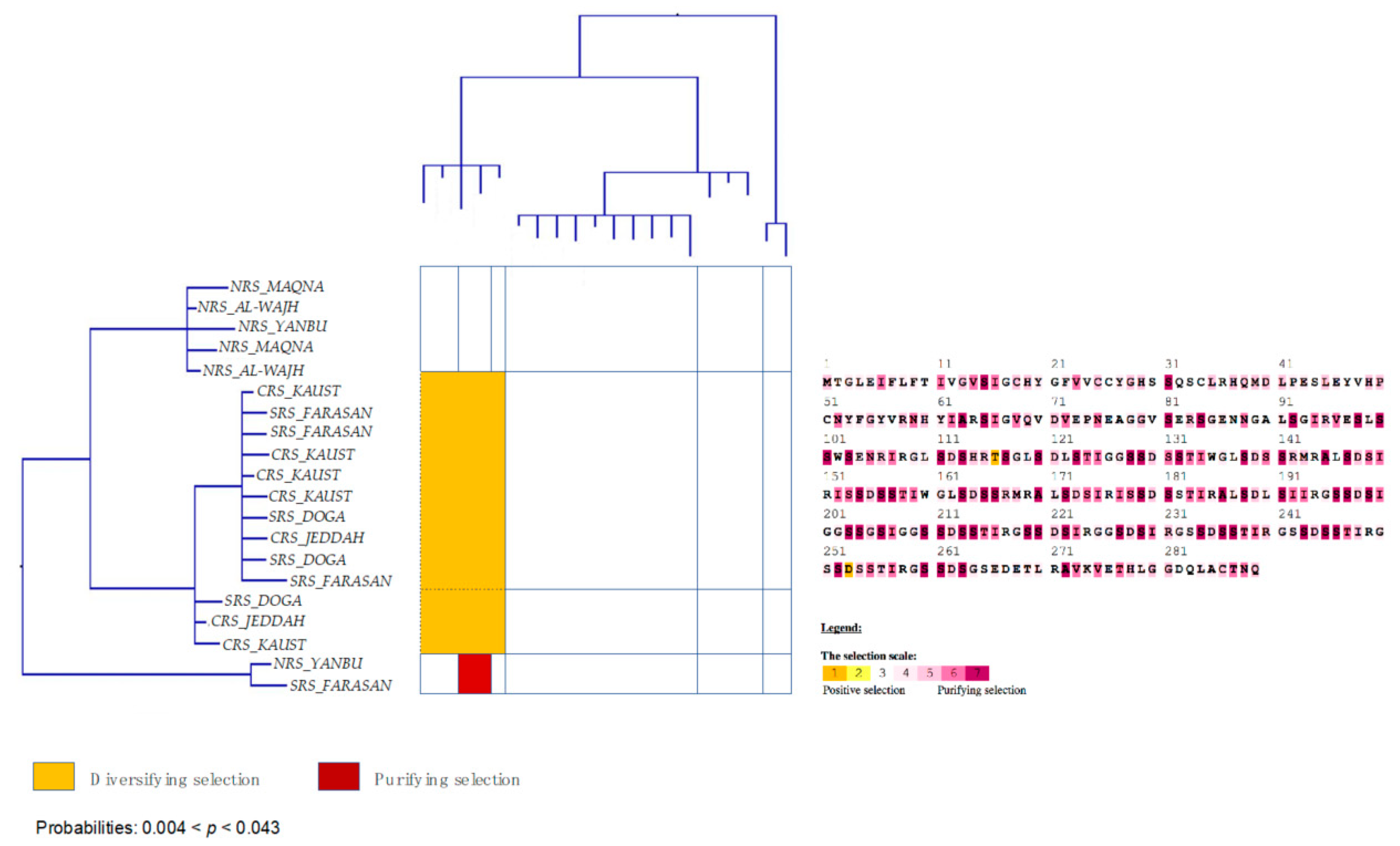
3.6. Strong Signatures for Positive and Negative Selection are Detected in the mtORF of Pocilloporid Corals
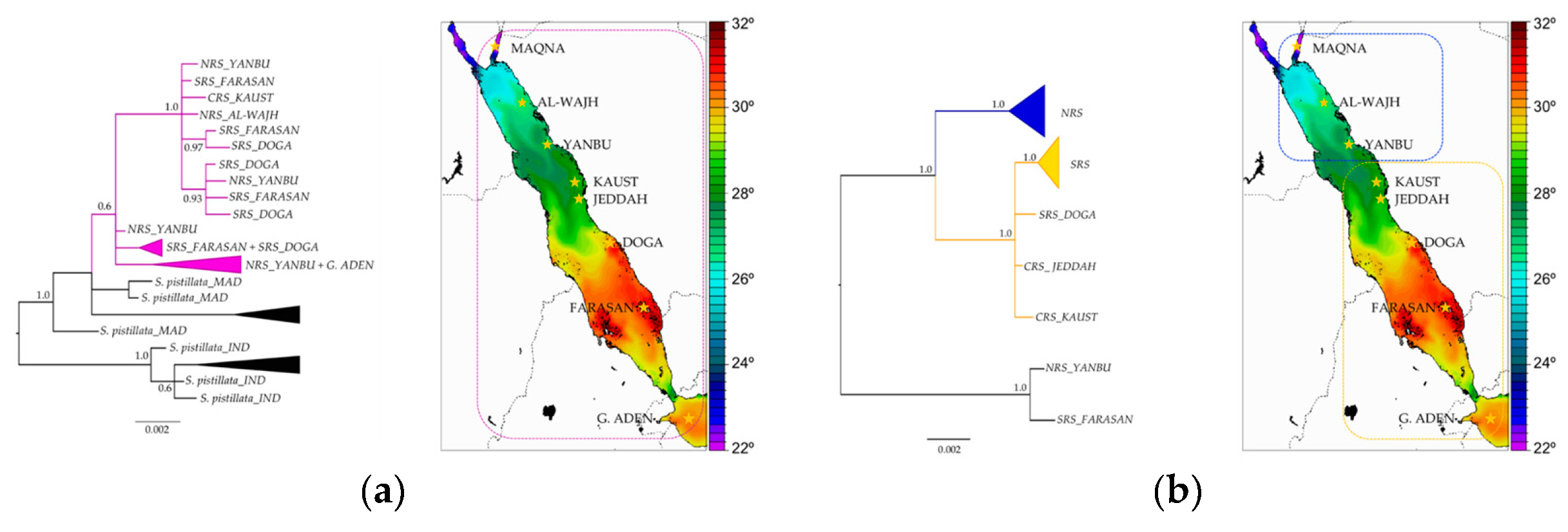
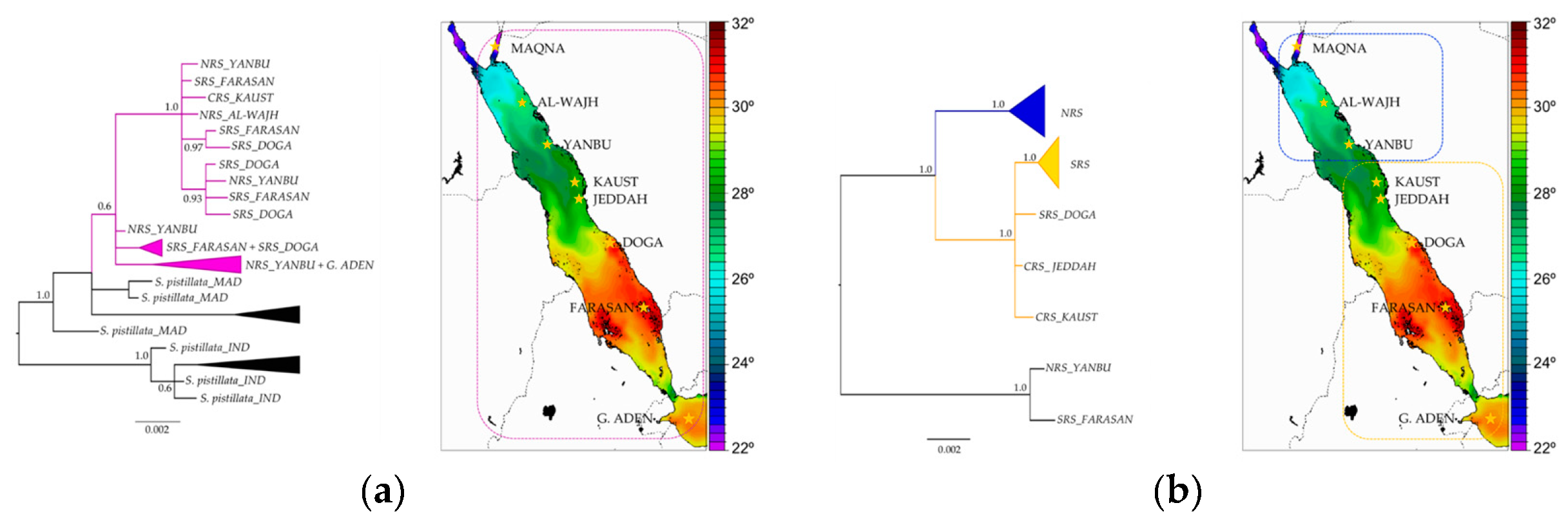
3.7. Stylophora Corals in the Red Sea Exhibit Signatures of Selection in the Their mtORF-Encoded Protein Along a Latitudinal Gradient
4. Discussion
4.1. Origin and Conservation of tmp362
4.2. Putative Homology with a Bacterial Hydrolase Domain
4.3. Putative Role of tmp362 in Environmental Adaptation
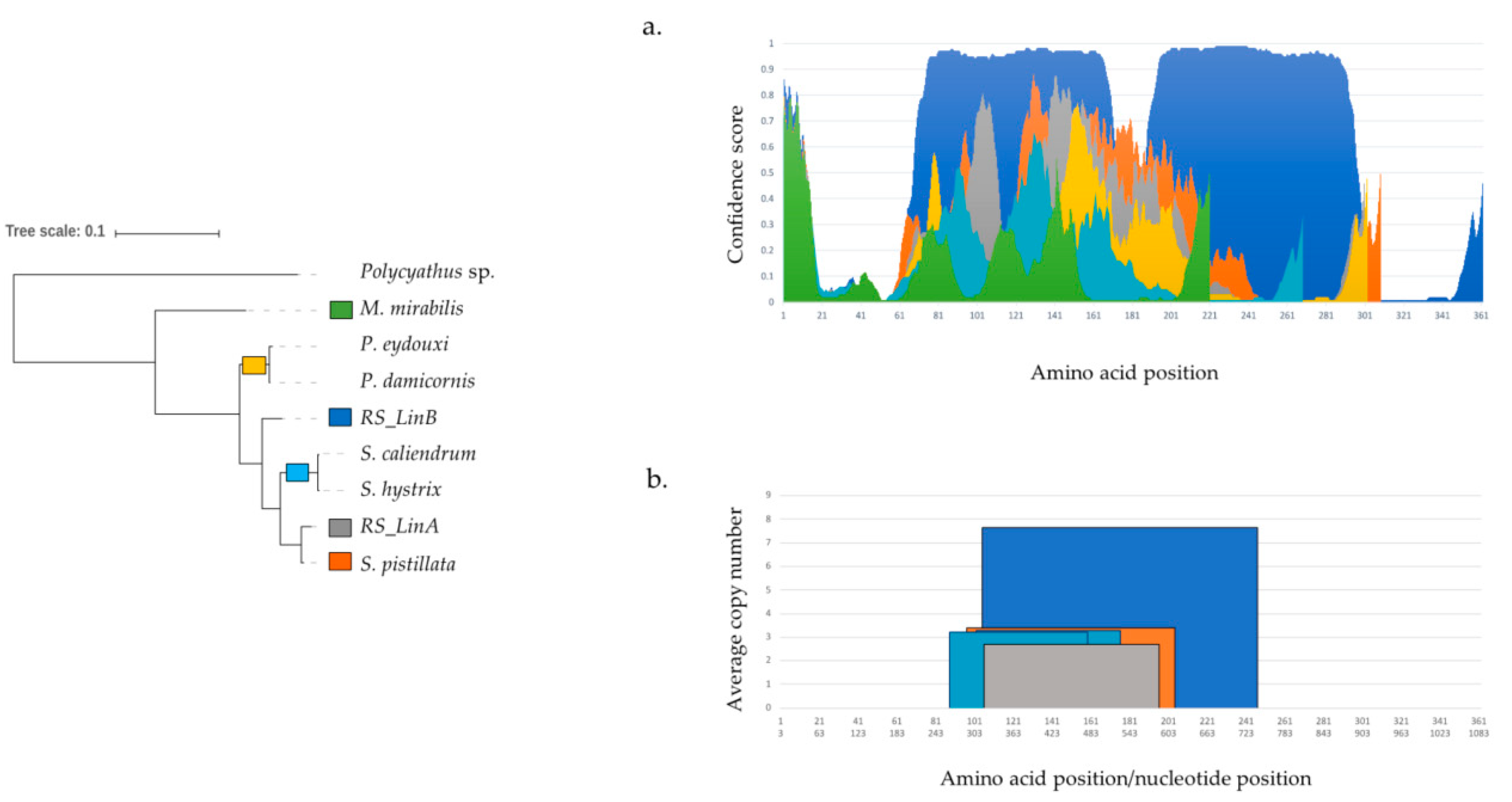
4.3.1. Tandem Repeats (TRs) and Intrinsically Disordered Regions (IDRs)
4.3.2. Structural Diversification of TMP362
4.3.3. Signatures of Selection in the TMP362 of Stylophora Lineages along Latitudinal and Environmental Gradients
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Szafranski, P. Evolutionarily recent, insertional fission of mitochondrial cox2 into complementary genes in bilaterian Metazoa. BMC Genomics 2017, 18, 269. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Noguchi, Y.; Ueshima, R.; Jacobs, H.T. Novel repetitive structures, deviant protein-encoding sequences and unidentified ORFs in the mitochondrial genome of the brachiopod Lingula anatina. J. Mol. Evol. 2005, 61, 36–53. [Google Scholar] [CrossRef]
- Breton, S.; Beaupré, H.D.; Stewart, D.T.; Piontkivska, H.; Karmakar, M.; Bogan, A.E.; Blier, P.U.; Hoeh, W.R. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: unionoida) with doubly uniparental inheritance of mtDNA: gender-specific open reading frames and putative origins of replication. Genetics 2009, 183, 1575–1589. [Google Scholar] [CrossRef]
- Breton, S.; Milani, L.; Ghiselli, F.; Guerra, D.; Stewart, D.T.; Passamonti, M. A resourceful genome: updating the functional repertoire and evolutionary role of animal mitochondrial DNAs. Trends Genet. 2014, 30, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, X.; Li, L.; Xu, X.; Xia, J.; Yu, Z. New features of Asian Crassostrea oyster mitochondrial genomes: A novel alloacceptor tRNA gene recruitment and two novel ORFs. Gene 2012, 507, 112–118. [Google Scholar] [CrossRef]
- Kayal, E.; Bentlage, B.; Collins, A.G.; Kayal, M.; Pirro, S.; Lavrov, D.V. Evolution of linear mitochondrial genomes in medusozoan cnidarians. Genome Biol. Evol. 2012, 4, 1–12. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Higashi, A.; Nagai, S.; Salomon, P.S.; Ueki, S. A unique, highly variable mitochondrial gene with coding capacity of Heterosigma akashiwo, class Raphidophyceae. J. Appl. Phycol. 2017, 29, 2961–2969. [Google Scholar] [CrossRef]
- Flot, J.-F.; Tillier, S. The mitochondrial genome of Pocillopora (Cnidaria: Scleractinia) contains two variable regions: the putative D-loop and a novel ORF of unknown function. Gene 2007, 401, 80–87. [Google Scholar] [CrossRef]
- Flot, J.-F.; Magalon, H.; Cruaud, C.; Couloux, A.; Tillier, S. Patterns of genetic structure among Hawaiian corals of the genus Pocillopora yield clusters of individuals that are compatible with morphology. C. R. Biol. 2008, 331, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Johnston, E.C.; Forsman, Z.H.; Flot, J.-F.; Schmidt-Roach, S.; Pinzón, J.H.; Knapp, I.S.S.; Toonen, R.J. A genomic glance through the fog of plasticity and diversification in Pocillopora. Sci. Rep. 2017, 7, 5991. [Google Scholar] [CrossRef]
- Flot, J.-F.; Licuanan, W.Y.; Nakano, Y.; Payri, C.; Cruaud, C.; Tillier, S. Mitochondrial sequences of Seriatopora corals show little agreement with morphology and reveal the duplication of a tRNA gene near the control region. Coral Reefs 2008, 27, 789–794. [Google Scholar] [CrossRef]
- Warner, P.A.; Van Oppen, M.J.H.; Willis, B.L. Unexpected cryptic species diversity in the widespread coral Seriatopora hystrix masks spatial-genetic patterns of connectivity. Mol. Ecol. 2015, 24, 2993–3008. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Nishikawa, A.; Iguchi, A.; Nagata, T.; Uyeno, D.; Sakai, K.; Mitarai, S. Elucidating the multiple genetic lineages and population genetic structure of the brooding coral Seriatopora (Scleractinia: Pocilloporidae) in the Ryukyu Archipelago. Coral Reefs 2017, 36, 415–426. [Google Scholar] [CrossRef]
- Banguera-Hinestroza, E.; Sawall, Y.; Al-Sofyani, A.; Mardulyn, P.; Fuertes-Aguilar, J.; Cardenas-Henao, H.; Jimenez-Infante, F.; Voolstra, C.R.; Flot, J.-F. mtDNA recombination indicative of hybridization suggests a role of the mitogenome in the adaptation of reef building corals to extreme environments. bioRxiv 2018, 462069. [Google Scholar]
- Flot, J.-F.; Blanchot, J.; Charpy, L.; Cruaud, C.; Licuanan, W.Y.; Nakano, Y.; Payri, C.; Tillier, S. Incongruence between morphotypes and genetically delimited species in the coral genus Stylophora: phenotypic plasticity, morphological convergence, morphological stasis or interspecific hybridization? BMC Ecol. 2011, 11, 22. [Google Scholar] [CrossRef]
- Shearer, T.L.; van Oppen, M.J.H.; Romano, S.L.; Wörheide, G. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol. Ecol. 2002, 11, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Shearer, T.L.; Coffroth, M.A. DNA BARCODING: Barcoding corals: limited by interspecific divergence, not intraspecific variation. Mol. Ecol. Resour. 2008, 8, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.E. Mitonuclear coevolution as the genesis of speciation and the mitochondrial DNA barcode gap. Ecol. Evol. 2016, 6, 5831–5842. [Google Scholar] [CrossRef] [PubMed]
- Cheviron, Z.A.; Brumfield, R.T. Genomic insights into adaptation to high-altitude environments. Heredity 2012, 108, 354–361. [Google Scholar] [CrossRef]
- Morales, H.E.; Pavlova, A.; Joseph, L.; Sunnucks, P. Positive and purifying selection in mitochondrial genomes of a bird with mitonuclear discordance. Mol. Ecol. 2015, 24, 2820–2837. [Google Scholar] [CrossRef]
- Scott, G.R.; Schulte, P.M.; Egginton, S.; Scott, A.L.M.; Richards, J.G.; Milsom, W.K. Molecular evolution of cytochrome c oxidase underlies high-altitude adaptation in the bar-headed goose. Mol. Biol. Evol. 2011, 28, 351–363. [Google Scholar] [CrossRef]
- Silva, G.; Lima, F.P.; Martel, P.; Castilho, R. Thermal adaptation and clinal mitochondrial DNA variation of European anchovy. Proc. R. Soc. B Biol. Sci. 2014, 281, 20141093. [Google Scholar] [CrossRef]
- Saccone, C.; Lanave, C.; De Grassi, A. Metazoan OXPHOS gene families: evolutionary forces at the level of mitochondrial and nuclear genomes. Biochim. Biophys. Acta 2006, 1757, 1171–1178. [Google Scholar] [CrossRef]
- Saraste, M. Oxidative phosphorylation at the fin de siècle. Science. 1999, 283, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Ben Slimen, H.; Schaschl, H.; Knauer, F.; Suchentrunk, F. Selection on the mitochondrial ATP synthase 6 and the NADH dehydrogenase 2 genes in hares (Lepus capensis L., 1758) from a steep ecological gradient in North Africa. BMC Evol. Biol. 2017, 17, 46. [Google Scholar] [CrossRef]
- Gershoni, M.; Templeton, A.R.; Mishmar, D. Mitochondrial bioenergetics as a major motive force of speciation. BioEssays 2009, 31, 642–650. [Google Scholar] [CrossRef]
- Lajbner, Z.; Pnini, R.; Camus, M.F.; Miller, J.; Dowling, D.K. Experimental evidence that thermal selection shapes mitochondrial genome evolution. Sci. Rep. 2018, 8, 9500. [Google Scholar] [CrossRef] [PubMed]
- Sunnucks, P.; Morales, H.E.; Lamb, A.M.; Pavlova, A.; Greening, C. Integrative approaches for studying mitochondrial and nuclear genome co-evolution in oxidative phosphorylation. Front. Genet. 2017, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Baris, T.Z.; Wagner, D.N.; Dayan, D.I.; Du, X.; Blier, P.U.; Pichaud, N.; Oleksiak, M.F.; Crawford, D.L. Evolved genetic and phenotypic differences due to mitochondrial-nuclear interactions. PLoS Genet. 2017, 13, e1006517. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Korkin, D.; Finch, T.M.; Frederick, K.H.; Eggert, L.S. Evidence of positive selection in mitochondrial complexes I and V of the African elephant. PLoS One 2014, 9, e92587. [Google Scholar]
- Rand, D.M.; Haney, R.A.; Fry, A.J. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol. Evol. 2004, 19, 645–653. [Google Scholar] [CrossRef]
- Hill, G.E. Mitonuclear ecology. Mol. Biol. Evol. 2015, 32, 1917–1927. [Google Scholar] [CrossRef]
- Pavlova, A.; Amos, J.N.; Joseph, L.; Loynes, K.; Austin, J.J.; Keogh, J.S.; Stone, G.N.; Nicholls, J.A.; Sunnucks, P. Perched at the mito-nuclear crossroads: divergent mitochondrial lineages correlate with environment in the face of ongoing nuclear gene flow in an Australian bird. Evolution 2013, 67, 3412–3428. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.E. The mitonuclear compatibility species concept. Auk 2017, 134, 393–409. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef]
- Sørensen, J.G.; Kristensen, T.N.; Loeschcke, V. The evolutionary and ecological role of heat shock proteins. Ecol. Lett. 2003, 6, 1025–1037. [Google Scholar] [CrossRef]
- Kvitt, H.; Rosenfeld, H.; Tchernov, D. The regulation of thermal stress induced apoptosis in corals reveals high similarities in gene expression and function to higher animals. Sci. Rep. 2016, 6, 30359. [Google Scholar] [CrossRef]
- Narum, S.R.; Campbell, N.R.; Meyer, K.A.; Miller, M.R.; Hardy, R.W. Thermal adaptation and acclimation of ectotherms from differing aquatic climates. Mol. Ecol. 2013, 22, 3090–3097. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M. Heat acclimation: phenotypic plasticity and cues to the underlying molecular mechanisms. J. Therm. Biol. 2001, 26, 357–363. [Google Scholar] [CrossRef]
- Veron, C.; Stafford-Smith, M.; Turak, E.; DeVantier, L. Corals of the World. Version 0.01 Beta. Available online: http://www.coralsoftheworld.org/page/home/ (accessed on February 2019).
- Kürten, B.; Al-Aidaroos, A.M.; Struck, U.; Khomayis, H.S.; Gharbawi, W.Y.; Sommer, U. Influence of environmental gradients on C and N stable isotope ratios in coral reef biota of the Red Sea, Saudi Arabia. J. Sea Res. 2014, 85, 379–394. [Google Scholar] [CrossRef]
- Osman, E.O.; Smith, D.J.; Ziegler, M.; Kürten, B.; Conrad, C.; El-Haddad, K.M.; Voolstra, C.R.; Suggett, D.J. Thermal refugia against coral bleaching throughout the northern Red Sea. Glob. Chang. Biol. 2018, 24, e474–e484. [Google Scholar] [CrossRef] [PubMed]
- Sawall, Y.; Al-Sofyani, A.; Hohn, S.; Banguera-Hinestroza, E.; Voolstra, C.R.; Wahl, M. Extensive phenotypic plasticity of a Red Sea coral over a strong latitudinal temperature gradient suggests limited acclimatization potential to warming. Sci. Rep. 2015, 5, 8940. [Google Scholar] [CrossRef]
- Rasul, N.M.A.; Stewart, I.C.F.; Nawab, Z.A. Introduction to the Red Sea: its origin, structure, and environment. In The Red Sea: The formation, morphology, oceanography and environment of a young ocean basin; Rasul, N.M.A., Stewart, I.C.F., Eds.; Springer: Heidelberg, Germany, 2015; pp. 1–28. [Google Scholar]
- Bruckner, A.; Rowlands, G.; Riegl, B.; Purkis, S.; Williams, A.; Renaud, P.; Khaled Bin Sultan Living Oceans Foundation. Atlas of Saudi Arabian Red Sea Marine Habitats; Panoramic Press: Phoenix, AZ, USA, 2012; ISBN 9780983561118. [Google Scholar]
- DiBattista, J.D.; Roberts, M.B.; Bouwmeester, J.; Bowen, B.W.; Coker, D.J.; Lozano-Cortés, D.F.; Howard Choat, J.; Gaither, M.R.; Hobbs, J.-P.A.; Khalil, M.T.; et al. A review of contemporary patterns of endemism for shallow water reef fauna in the Red Sea. J. Biogeogr. 2016, 43, 423–439. [Google Scholar] [CrossRef]
- DiBattista, J.D.; Howard Choat, J.; Gaither, M.R.; Hobbs, J.-P.A.; Lozano-Cortés, D.F.; Myers, R.F.; Paulay, G.; Rocha, L.A.; Toonen, R.J.; Westneat, M.W.; et al. On the origin of endemic species in the Red Sea. J. Biogeogr. 2016, 43, 13–30. [Google Scholar] [CrossRef]
- Siddall, M.; Smeed, D.A.; Hemleben, C.; Rohling, E.J.; Schmelzer, I.; Peltier, W.R. Understanding the Red Sea response to sea level. Earth Planet. Sci. Lett. 2004, 225, 421–434. [Google Scholar] [CrossRef]
- Siddall, M.; Rohling, E.J.; Almogi-Labin, A.; Hemleben, C.; Meischner, D.; Schmelzer, I.; Smeed, D. Sea-level fluctuations during the last glacial cycle. Nature 2003, 423, 853–858. [Google Scholar] [CrossRef]
- Moustafa, M.Z.; Moustafa, M.S.; Moustafa, Z.D.; Moustafa, S.E. Survival of high latitude fringing corals in extreme temperatures: Red Sea oceanography. J. Sea Res. 2014, 88, 144–151. [Google Scholar] [CrossRef]
- Arrigoni, R.; Benzoni, F.; Terraneo, T.I.; Caragnano, A.; Berumen, M.L. Recent origin and semi-permeable species boundaries in the scleractinian coral genus Stylophora from the Red Sea. Sci. Rep. 2016, 6, 34612. [Google Scholar] [CrossRef]
- Stefani, F.; Benzoni, F.; Yang, S.Y.; Pichon, M.; Galli, P.; Chen, C.A. Comparison of morphological and genetic analyses reveals cryptic divergence and morphological plasticity in Stylophora (Cnidaria, Scleractinia). Coral Reefs 2011, 30, 1033–1049. [Google Scholar] [CrossRef]
- Pohl, M.; Theiβen, G.; Schuster, S. GC content dependency of open reading frame prediction via stop codon frequencies. Gene 2012, 511, 441–446. [Google Scholar] [CrossRef]
- Tutar, Y. Pseudogenes. Comp. Funct. Genomics 2012, 2012, 1–4. [Google Scholar] [CrossRef]
- Xiao, J.; Sekhwal, M.K.; Li, P.; Ragupathy, R.; Cloutier, S.; Wang, X.; You, F.M. Pseudogenes and their genome-wide prediction in plants. Int. J. Mol. Sci. 2016, 17, 1991. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Nugent, T.; Jones, D.T. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics 2009, 10, 159. [Google Scholar] [CrossRef]
- JEN, V.; Pichon, M. Scleractinia of eastern Australia. Part I: Families Thamnasteriidae, Astrocoeniidae, Pocilloporidae. Aust. Gov. Publ. Serv. 1976, 208. [Google Scholar]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, The Pennsylvania State University, University Park, PA, USA, 2007. [Google Scholar] [CrossRef]
- Ankenbrand, M.J.; Hohlfeld, S.; Hackl, T.; Förster, F. AliTV — interactive visualization of whole genome comparisons. PeerJ. Comput. Sci. 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Audic, S.; Claverie, J.-M.M.; Blanc, G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 2010, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.; Kitahara, M.; Fukami, H.; Tracey, D.; Miller, D.J.; Chen, C.A. Loss and gain of group I introns in the mitochondrial cox1 gene of the Scleractinia (Cnidaria; Anthozoa). Zool. Stud. 2017, 56. [Google Scholar]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Robitzch, V.; Banguera-Hinestroza, E.; Sawall, Y.; Al-Sofyani, A.; Voolstra, C.R. Absence of genetic differentiation in the coral Pocillopora verrucosa along environmental gradients of the Saudi Arabian Red Sea. Front. Mar. Sci. 2015, 2. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.A.; Ward, S.M.; Lobley, A.E.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010, 38, W563–W568. [Google Scholar] [CrossRef] [PubMed]
- Mulder, N.; Apweiler, R. InterPro and InterProScan: Tools for protein sequence classification and comparison. Methods Mol Biol. 2007, 396, 59–70. [Google Scholar]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 2007, 23, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 1994, 33, 3038–3049. [Google Scholar] [CrossRef]
- Nielsen, H. Predicting secretory proteins with SignalP. Methods Mol Biol. 2017, 1611, 59–73. [Google Scholar]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Jones, D.T.; Ward, J.J. Prediction of disordered regions in proteins from position specific score matrices. Proteins Struct. Funct. Genet. 2003, 53, 573–578. [Google Scholar] [CrossRef]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdös, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Tabaro, F.; Paladin, L.; Necci, M.; Mieti, I.; Camilloni, C.; Davey, N.; Dosztányi, Z.; Mészáros, B.; Monzon, A.M.; et al. MobiDB 3.0: More annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018, 46, D471–D476. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Jaroszewski, L.; Li, Z.; Cai, X.-H.; Weber, C.; Godzik, A. FFAS server: novel features and applications. Nucleic Acids Res. 2011, 39, W38–W44. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: the protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Grishin, N.V. PROMALS: Towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 2007, 23, 802–808. [Google Scholar] [CrossRef]
- Bhagwat, M.; Aravind, L. PSI-BLAST Tutorial. 2007, 395, 177–186.
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007, 36, D13–D21. [Google Scholar] [CrossRef]
- Jaroszewski, L.; Rychlewski, L.; Li, Z.; Li, W.; Godzik, A. FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res. 2005, 33, W284–W288. [Google Scholar] [CrossRef]
- Minneci, F.; Piovesan, D.; Cozzetto, D.; Jones, D.T. FFPred 2.0: improved homology-independent prediction of gene ontology terms for eukaryotic protein sequences. PLoS One 2013, 8, e63754. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef]
- Lobley, A.; Sadowski, M.I.; Jones, D.T. pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 2009, 25, 1761–1767. [Google Scholar] [CrossRef]
- Knudsen, M.; Wiuf, C. The CATH database. Hum. Genomics 2010, 4, 207. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.-H. The codon adaptation index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Jia, W.; Higgs, P.G. Codon usage in mitochondrial genomes: distinguishing context-dependent mutation from translational selection. Mol. Biol. Evol. 2008, 25, 339–351. [Google Scholar] [CrossRef]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallvé, S. E-CAI: a novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinformatics 2008, 9, 65. [Google Scholar] [CrossRef]
- Delport, W.; Poon, A.; Frost, S.D.W.; Kosakovsky Pond, S.L. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Murrell, B.; Fourment, M.; Frost, S.D.W.; Delport, W.; Scheffler, K. A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 2011, 28, 3033–3043. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Doron-Faigenboim, A.; Pupko, T.A. Combined empirical and mechanistic codon model. Mol. Biol. Evol. 2006, 24, 388–397. [Google Scholar] [CrossRef]
- Doron-Faigenboim, A.; Stern, A.; Mayrose, I.; Bacharach, E.; Pupko, T.A. Selecton: a server for detecting evolutionary forces at a single amino-acid site. Bioinformatics 2005, 21, 2101–2103. [Google Scholar] [CrossRef]
- Stern, A.; Doron-Faigenboim, A.; Erez, E.; Martz, E.; Bacharach, E.; Pupko, T.A. Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 2007, 35, W506–W511. [Google Scholar] [CrossRef]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Hughes, A.L.; Friedman, R. Codon-based tests of positive selection, branch lengths, and the evolution of mammalian immune system genes. Immunogenetics 2008, 60, 495–506. [Google Scholar] [CrossRef]
- Merkler, D.J.; Farrington, G.K.; Wedler, F.C. Protein thermostability. Correlations between calculated macroscopic parameters and growth temperature for closely related thermophilic and mesophilic bacilli. Int. J. Pept. Protein Res. 1981, 18, 430–442. [Google Scholar] [CrossRef]
- Jones, D.T.; Cozzetto, D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Natale, D.A.; Aravind, L.; Koonin, E.V. A specialized version of the HD hydrolase domain implicated in signal transduction. J. Mol. Microbiol. Biotechnol. 1999, 1, 303–305. [Google Scholar] [PubMed]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef]
- Galtier, N.; Kitazaki, K.; Kubo, T.; Ballard, J.; Whitlock, M.; Davila, J.; Arrieta-Montiel, M.; Wamboldt, Y.; Cao, J.; Hagmann, J.; et al. The intriguing evolutionary dynamics of plant mitochondrial DNA. BMC Biol. 2011, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Raitsos, D.E.; Hoteit, I.; Prihartato, P.K.; Chronis, T.; Triantafyllou, G.; Abualnaja, Y. Abrupt warming of the Red Sea. Geophys. Res. Lett. 2011, 38. [Google Scholar] [CrossRef]
- Weeks, S.J.; Anthony, K.R.N.; Bakun, A.; Feldman, G.C.; Guldberg, O.H. Improved predictions of coral bleaching using seasonal baselines and higher spatial resolution. Limnol. Oceanogr. 2008, 53, 1369–1375. [Google Scholar] [CrossRef]
- Muller-Parker, G.; D’Elia, C.F.; Cook, C.B. Interactions between corals and their symbiotic algae. In Coral Reefs in The Anthropocene; Birkeland, C., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 99–116. [Google Scholar]
- Bourne, D.G.; Morrow, K.M.; Webster, N.S. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 2016, 70, 317–340. [Google Scholar] [CrossRef] [PubMed]
- Keshavmurthy, S.; Yang, S.-Y.; Alamaru, A.; Chuang, Y.-Y.; Pichon, M.; Obura, D.; Fontana, S.; De Palmas, S.; Stefani, F.; Benzoni, F.; et al. DNA barcoding reveals the coral “laboratory-rat”, Stylophora pistillata encompasses multiple identities. Sci. Rep. 2013, 3, 1520. [Google Scholar] [CrossRef] [PubMed]
- Maddamsetti, R.; Johnson, D.T.; Spielman, S.J.; Petrie, K.L.; Marks, D.S.; Meyer, J.R. Gain-of-function experiments with bacteriophage lambda uncover residues under diversifying selection in nature. Evolution 2018, 72, 2234–2243. [Google Scholar] [CrossRef]
- Lin, T.-Y. Simple sequence repeat variations expedite phage divergence: mechanisms of indels and gene mutations. Mutat. Res. Mol. Mech. Mutagen. 2016, 789, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P.C.B.A. Genomic repeats, genome plasticity and the dynamics of Mycoplasma evolution. Nucleic Acids Res. 2002, 30, 2031–2042. [Google Scholar] [CrossRef]
- Moxon, R.; Bayliss, C.; Hood, D. Bacterial contingency loci: the role of simple sequence DNA repeats in bacterial adaptation. Annu. Rev. Genet. 2006, 40, 307–333. [Google Scholar] [CrossRef]
- Zhou, K.; Aertsen, A.; Michiels, C.W. The role of variable DNA tandem repeats in bacterial adaptation. FEMS Microbiol. Rev. 2014, 38, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Schaper, E.; Anisimova, M. The evolution and function of protein tandem repeats in plants. New Phytol. 2015, 206, 397–410. [Google Scholar] [CrossRef]
- Pollak, Y.; Zelinger, E.; Raskina, O. Repetitive DNA in the architecture, repatterning, and diversification of the genome of Aegilops speltoides Tausch (Poaceae, Triticeae). Front. Plant Sci. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, L.; He, S.; Huang, M.; Tan, J.; Zhao, L.; Yan, S.; Li, H.; Zhou, K.; Liang, Y.; et al. Cold stress selectively unsilences tandem repeats in heterochromatin associated with accumulation of H3K9ac. Plant Cell Environ. 2012, 35, 2130–2142. [Google Scholar] [CrossRef]
- Droma, Y.; Hanaoka, M.; Basnyat, B.; Arjyal, A.; Neupane, P.; Pandit, A.; Sharma, D.; Ito, M.; Miwa, N.; Katsuyama, Y. Adaptation to high altitude in Sherpas: association with the insertion/deletion polymorphism in the angiotensin-converting enzyme gene. Wilderness Environ. Med. 2008, 19, 22–29. [Google Scholar] [CrossRef]
- Ren, J.; Hou, Z.; Wang, H.; Sun, M.; Liu, X.; Liu, B.; Guo, X. Intraspecific variation in mitogenomes of five Crassostrea species provides insight into oyster diversification and speciation. Mar. Biotechnol. 2016, 18, 242–254. [Google Scholar] [CrossRef]
- Schüler, A.; Bornberg-Bauer, E. Evolution of protein domain repeats in Metazoa. Mol. Biol. Evol. 2016, 33, 3170–3182. [Google Scholar] [CrossRef]
- Sharma, M.; Pandey, G.K. Expansion and function of repeat domain proteins during stress and development in plants. Front. Plant Sci. 2016, 6, 1218. [Google Scholar] [CrossRef] [PubMed]
- Kashi, Y.; King, D.G. Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 2006, 22, 253–259. [Google Scholar] [CrossRef]
- King, D.G.; Soller, M.; Kashi, Y. Evolutionary tuning knobs. Endeavour 1997, 21, 36–40. [Google Scholar] [CrossRef]
- Kashi, Y.; King, D.G. Has simple sequence repeat mutability been selected to facilitate evolution? Isr. J. Ecol. Evol. 2007, 52, 331–342. [Google Scholar] [CrossRef]
- Gemayel, R.; Vinces, M.D.; Legendre, M.; Verstrepen, K.J. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 2010, 44, 445–477. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.M.; Cruz, C.; Jack, C.V.; Houseley, J. Environmental change drives accelerated adaptation through stimulated copy number variation. PLoS Biol. 2017, 15, e2001333. [Google Scholar] [CrossRef]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S. Repetitive DNA in eukaryotic genomes. Chromosom. Res. 2015, 23, 415–420. [Google Scholar] [CrossRef]
- Verstrepen, K.J.; Jansen, A.; Lewitter, F.; Fink, G.R. Intragenic tandem repeats generate functional variability. Nat. Genet. 2005, 37, 986–990. [Google Scholar] [CrossRef]
- Sonay, T.B.; Carvalho, T.; Robinson, M.D.; Greminger, M.P.; Krützen, M.; Comas, D.; Highnam, G.; Mittelman, D.; Sharp, A.; Marques-Bonet, T.; et al. Tandem repeat variation in human and great ape populations and its impact on gene expression divergence. Genome Res. 2015, 25, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Lin, Y.; Santillan, B.A.; Yotnda, P.; Wilson, J.H. Environmental stress induces trinucleotide repeat mutagenesis in human cells. Proc. Natl. Acad. Sci. 2015, 112, 3764–3769. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Ahrens, J.B.; Nunez-Castilla, J.; Siltberg-Liberles, J. Evolution of intrinsic disorder in eukaryotic proteins. Cell. Mol. Life Sci. 2017, 74, 3163–3174. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins evolve by repeat expansion. Bioessays 2003, 25, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Light, S.; Sagit, R.; Sachenkova, O.; Ekman, D.; Elofsson, A. Protein expansion is primarily due to indels in intrinsically disordered regions. Mol. Biol. Evol. 2013, 30, 2645–2653. [Google Scholar] [CrossRef]
- James, L.C.; Tawfik, D.S. Conformational diversity and protein evolution – A 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003, 28, 361–368. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Protein dynamism and evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. 1996, 93, 11504–11509. [Google Scholar] [CrossRef] [PubMed]
- Vicedo, E.; Schlessinger, A.; Rost, B. Environmental pressure may change the composition protein disorder in prokaryotes. PloS One 2015, 10, e0133990. [Google Scholar] [CrossRef] [PubMed]
- Pietrosemoli, N.; García-Martín, J.A.; Solano, R.; Pazos, F. Genome-wide analysis of protein disorder in Arabidopsis thaliana: implications for plant environmental adaptation. PloS One 2013, 8, e55524. [Google Scholar] [CrossRef] [PubMed]
- Mahjoubi, H.; Ebel, C.; Hanin, M. Molecular and functional characterization of the durum wheat TdRL1, a member of the conserved Poaceae RSS1-like family that exhibits features of intrinsically disordered proteins and confers stress tolerance in yeast. Funct. Integr. Genomics 2015, 15, 717–728. [Google Scholar] [CrossRef]
- Tantos, A.; Friedrich, P.; Tompa, P. Cold stability of intrinsically disordered proteins. FEBS Lett. 2009, 583, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Boothby, T.C.; Tapia, H.; Brozena, A.H.; Piszkiewicz, S.; Smith, A.E.; Giovannini, I.; Rebecchi, L.; Pielak, G.J.; Koshland, D.; Goldstein, B. Tardigrades use intrinsically disordered proteins to survive desiccation. Mol. Cell 2017, 65, 975–984.e5. [Google Scholar] [CrossRef]
- Fields, P.A. Review: protein function at thermal extremes: balancing stability and flexibility. Comp. Biochem. Physiol. A. Mol. Integr. Physiol. 2001, 129, 417–431. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Yu, X.; Luo, L.; Luo, Z.; Liu, G.; Liu, Y.; Xia, H.; Xiong, J.; Tao, T. RNA editing responses to oxidative stress between a wild abortive type male-sterile line and its maintainer line. Front. Plant Sci. 2017, 8, 1–12. [Google Scholar]
- Millar, A.H.; Whelan, J.; Soole, K.L.; Day, D.A. Organization and regulation of mitochondrial respiration in plants. Annu. Rev. Plant Biol. 2011, 62, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Green, R.R.; Pichersky, E. Hypothesis for the evolution of three-helix Chl a/b and Chl a/c light-harvesting antenna proteins from two-helix and four-helix ancestors. Photosynth. Res. 1994, 39, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Koziol, A.G.; Borza, T.; Ishida, K.-I.; Keeling, P.; Lee, R.W.; Durnford, D.G. Tracing the evolution of the light-harvesting antennae in chlorophyll a/b-containing organisms. Plant Physiol. 2007, 143, 1802–1816. [Google Scholar] [CrossRef]
- Heddad, M.; Engelken, J.; Adamska, I. Light stress proteins in viruses, cyanobacteria and photosynthetic eukaryota. In Photosynthesis. Advances in Photosynthesis and Respiration, vol 34; Eaton-Rye, J., Tripathy, B., Sharkey, T., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 299–317. [Google Scholar]
- Heddad, M.; Adamska, I. The evolution of light stress proteins in photosynthetic organisms. Comp. Funct. Genomics 2002, 3, 504–510. [Google Scholar] [CrossRef]
- Büchel, C. Evolution and function of light harvesting proteins. J. Plant Physiol. 2015, 172, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.E.; Sanchez-Puerta, M.V.; Delwiche, C.F. Evolution of light-harvesting complex proteins from Chl c-containing algae. BMC Evol. Biol. 2011, 11, 101. [Google Scholar] [CrossRef]
- Pascal, A.A.; Liu, Z.; Broess, K.; van Oort, B.; van Amerongen, H.; Wang, C.; Horton, P.; Robert, B.; Chang, W.; Ruban, A. Molecular basis of photoprotection and control of photosynthetic light-harvesting. Nature 2005, 436, 134–137. [Google Scholar] [CrossRef]
- Rochaix, J.-D.; Bassi, R. LHC-like proteins involved in stress responses and biogenesis/repair of the photosynthetic apparatus. Biochem. J. 2019, 476, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Birkeland, C. Coral Reefs in the Anthropocene; Springer: Dordrecht, The Netherlands, 2015. [Google Scholar]
- Dixon, G.B.; Davies, S.W.; Aglyamova, G.V.; Meyer, E.; Bay, L.K.; Matz, M.V. Genomic determinants of coral heat tolerance across latitudes. Science 2015, 348, 1460–1462. [Google Scholar] [CrossRef]
- Taviani, M. Post-Miocene reef faunas of the Red Sea: glacio-eustatic controls. In Sedimentation and Tectonics in Rift Basins Red Sea—Gulf of Aden; Springer: Dordrecht, The Netherlands, 1998; pp. 574–582. [Google Scholar]
- Bruggemann, J.H.; Buffler, R.T.; Guillaume, M.M.; Walter, R.C.; Von Cosel, R.; Ghebretensae, B.N.; Berhe, S.M. Stratigraphy, palaeoenvironments and model for the deposition of the Abdur Reef Limestone: context for an important archaeological site from the last interglacial on the Red Sea coast of Eritrea. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2004, 203, 179–206. [Google Scholar] [CrossRef]
- Casazza, L.R. Pleistocene reefs of the Egyptian Red Sea: environmental change and community persistence. PeerJ 2017, 5, e3504. [Google Scholar] [CrossRef]
- Dullo, W.C. Facies, fossil record, and age of Pleistocene reefs from the Red Sea (Saudi Arabia). Facies 1990, 22, 1–45. [Google Scholar] [CrossRef]
- El-Sorogy, A.S. Contributions to the Pleistocene coral reefs of the Red Sea Coast, Egypt. Arab Gulf J. Sci. Res. 2008, 26, 63–85. [Google Scholar]
- Rohling, E.J.; Grant, K.; Hemleben, C.; Siddall, M.; Hoogakker, B.A.A.; Bolshaw, M.; Kucera, M. High rates of sea-level rise during the last interglacial period. Nat. Geosci. 2008, 1, 38–42. [Google Scholar] [CrossRef]
- Taviani, M.; López Correa, M.; Zibrowius, H.; Montagna, P.; McCulloch, M.; Ligi, M. Last glacial deep-water corals from the Red Sea. Bull. Mar. Sci. 2008, 81, 361–370. [Google Scholar]
- Trommer, G.; Siccha, M.; Rohling, E.J.; Grant, K.; van der Meer, M.T.J.; Schouten, S.; Hemleben, C.; Kucera, M. Millennial-scale variability in Red Sea circulation in response to Holocene insolation forcing. Paleoceanography 2010, 25, 25. [Google Scholar] [CrossRef]
- Fine, M.; Gildor, H.; Genin, A. A coral reef refuge in the Red Sea. Glob. Chang. Biol. 2013, 19, 3640–3647. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indices | Period Size | Copy Number | Consensus Size | Percent Indels | |
|---|---|---|---|---|---|
| RS_LinB | 315–741 | 27 | 16.9 | 27 | 11 |
| 318–741 | 54 | 8.4 | 54 | 13 | |
| 371–613 | 75 | 3.3 | 75 | 5 | |
| 501–634 | 21 | 6.1 | 21 | 10 | |
| 486–741 | 69 | 3.5 | 69 | 9 | |
| S. hystrix | 310–487 | 51 | 3.5 | 51 | 0 |
| 352–430 | 24 | 3.2 | 24 | 10 | |
| 310–528 | 51 | 4.3 | 51 | 1 | |
| 306–525 | 102 | 2.2 | 102 | 1 | |
| S. caliendrum | 265–436 | 51 | 3.4 | 51 | 0 |
| 265–477 | 102 | 2.1 | 102 | 1 | |
| 265–477 | 51 | 4.2 | 51 | 1 | |
| RS_LinA | 317–424 | 39 | 2.6 | 42 | 4 |
| 365–440 | 21 | 3.8 | 21 | 10 | |
| 350–444 | 39 | 2.4 | 39 | 7 | |
| 480–587 | 51 | 2.1 | 51 | 0 | |
| S. pistillata | 290–417 | 39 | 3.2 | 42 | 6 |
| 338–413 | 21 | 3.8 | 21 | 10 | |
| 453–611 | 51 | 3.1 | 51 | 0 |
| Posterior Probabilities | |||||||
|---|---|---|---|---|---|---|---|
| GO Term | Molecular Function | Madracis | Pocillopora | Seriatopora | Stylophora RS_LinB | Stylophora RS_LinA | Stylophora pistillata |
| GO:0005216 | ion channel activity | 0.913 | - | 0.628 | - | - | 0.581 |
| GO:0016817 | hydrolase activity, acting on acid anhydrides | 0.902 | 0.862 | 0.561 | 0.824 | 0.695 | 0.626 |
| GO:0015075 | ion transmembrane transporter activity | 0.88 | - | 0.654 | - | - | - |
| GO:0022890 | inorganic cation transmembrane transporter activity | 0.864 | - | - | - | - | - |
| GO:0008324 | cation transmembrane transporter activity | 0.858 | - | - | - | - | - |
| GO:0005524 | ATP binding | 0.833 | - | 0.832 | - | 0.739 | 0.502 |
| GO:0046873 | metal ion transmembrane transporter activity | 0.787 | - | - | - | - | - |
| GO:0015077 | monovalent inorganic cation transmembrane transporter activity | 0.749 | - | - | - | - | - |
| GO:0003824 | catalytic activity | 0.748 | 0.762 | 0.771 | 0.528 | 0.739 | 0.681 |
| GO:0016818 | hydrolase activity, acting on acid anhydrides, in phosphorus-containing anhydrides | 0.707 | 0.602 | 0.602 | 0.666 | 0.695 | 0.619 |
| GO:0022857 | transmembrane transporter activity | 0.706 | 0.56 | 0.696 | - | - | 0.502 |
| GO:0005261 | cation channel activity | 0.688 | - | - | - | - | - |
| GO:0005215 | transporter activity | 0.67 | 0.548 | 0.684 | - | 0.561 | 0.515 |
| GO:0035639 | purine ribonucleoside triphosphate binding | 0.639 | 0.538 | 0.706 | - | 0.733 | 0.538 |
| GO:0008092 | cytoskeletal protein binding | 0.588 | 0.592 | 0.598 | 0.682 | 0.62 | 0.676 |
| GO:0000166 | nucleotide binding | - | 0.52 | 0.71 | - | 0.72 | - |
| GO:0001882 | nucleoside binding | - | - | 0.69 | - | 0.752 | - |
| GO:0032549 | ribonucleoside binding | - | - | 0.682 | - | 0.779 | - |
| GO:0017076 | purine nucleotide binding | - | - | 0.604 | - | 0.779 | 0.557 |
| GO:0022891 | substrate-specific transmembrane transporter activity | - | - | 0.68 | - | - | - |
| GO:0030554 | adenyl nucleotide binding | - | - | 0.648 | - | 0.547 | - |
| GO:0016301 | kinase activity | - | 0.53 | 0.549 | 0.617 | 0.857 | 0.655 |
| Posterior Probabilities | |||||||
|---|---|---|---|---|---|---|---|
| GO Term | Biological Process | Madracis | Pocillopora | Seriatopora | Stylophora RS_LinB | Stylophora RS_LinA | Stylophora pistillata |
| GO:0006810 | transport | 0.862 | 0.847 | 0.874 | 0.803 | 0.824 | 0.82 |
| GO:0034220 | ion transmembrane transport | 0.845 | - | 0.689 | - | - | - |
| GO:0019222 | regulation of metabolic process | 0.813 | 0.853 | 0.795 | 0.711 | 0.849 | 0.791 |
| GO:0007166 | cell surface receptor signaling pathway | 0.804 | 0.63 | 0.564 | 0.79 | 0.662 | 0.717 |
| GO:0009117 | nucleotide metabolic process | 0.728 | - | 0.697 | - | - | - |
| GO:0051649 | establishment of localization in cell | 0.694 | 0.692 | 0.721 | 0.538 | 0.698 | 0.659 |
| GO:0051641 | cellular localization | 0.647 | 0.631 | 0.64 | 0.632 | 0.643 | 0.636 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banguera-Hinestroza, E.; Ferrada, E.; Sawall, Y.; Flot, J.-F. Computational Characterization of the mtORF of Pocilloporid Corals: Insights into Protein Structure and Function in Stylophora Lineages from Contrasting Environments. Genes 2019, 10, 324. https://doi.org/10.3390/genes10050324
Banguera-Hinestroza E, Ferrada E, Sawall Y, Flot J-F. Computational Characterization of the mtORF of Pocilloporid Corals: Insights into Protein Structure and Function in Stylophora Lineages from Contrasting Environments. Genes. 2019; 10(5):324. https://doi.org/10.3390/genes10050324
Chicago/Turabian StyleBanguera-Hinestroza, Eulalia, Evandro Ferrada, Yvonne Sawall, and Jean-François Flot. 2019. "Computational Characterization of the mtORF of Pocilloporid Corals: Insights into Protein Structure and Function in Stylophora Lineages from Contrasting Environments" Genes 10, no. 5: 324. https://doi.org/10.3390/genes10050324
APA StyleBanguera-Hinestroza, E., Ferrada, E., Sawall, Y., & Flot, J.-F. (2019). Computational Characterization of the mtORF of Pocilloporid Corals: Insights into Protein Structure and Function in Stylophora Lineages from Contrasting Environments. Genes, 10(5), 324. https://doi.org/10.3390/genes10050324