New Insights into Long Terminal Repeat Retrotransposons in Mulberry Species


Abstract
1. Introduction
2. Results
2.1. Characterization of Long Terminal Repeat (LTR) Retrotransposons
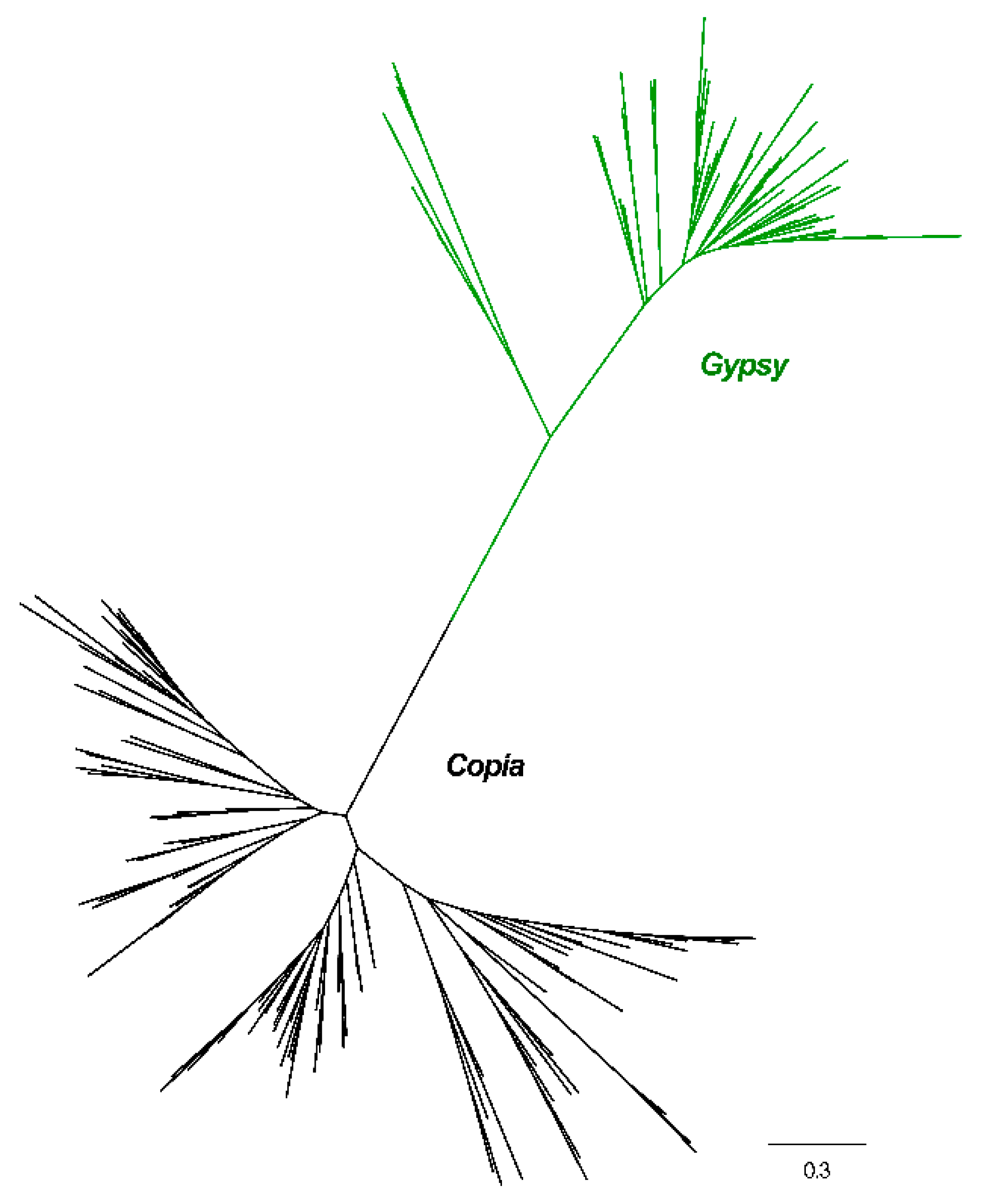
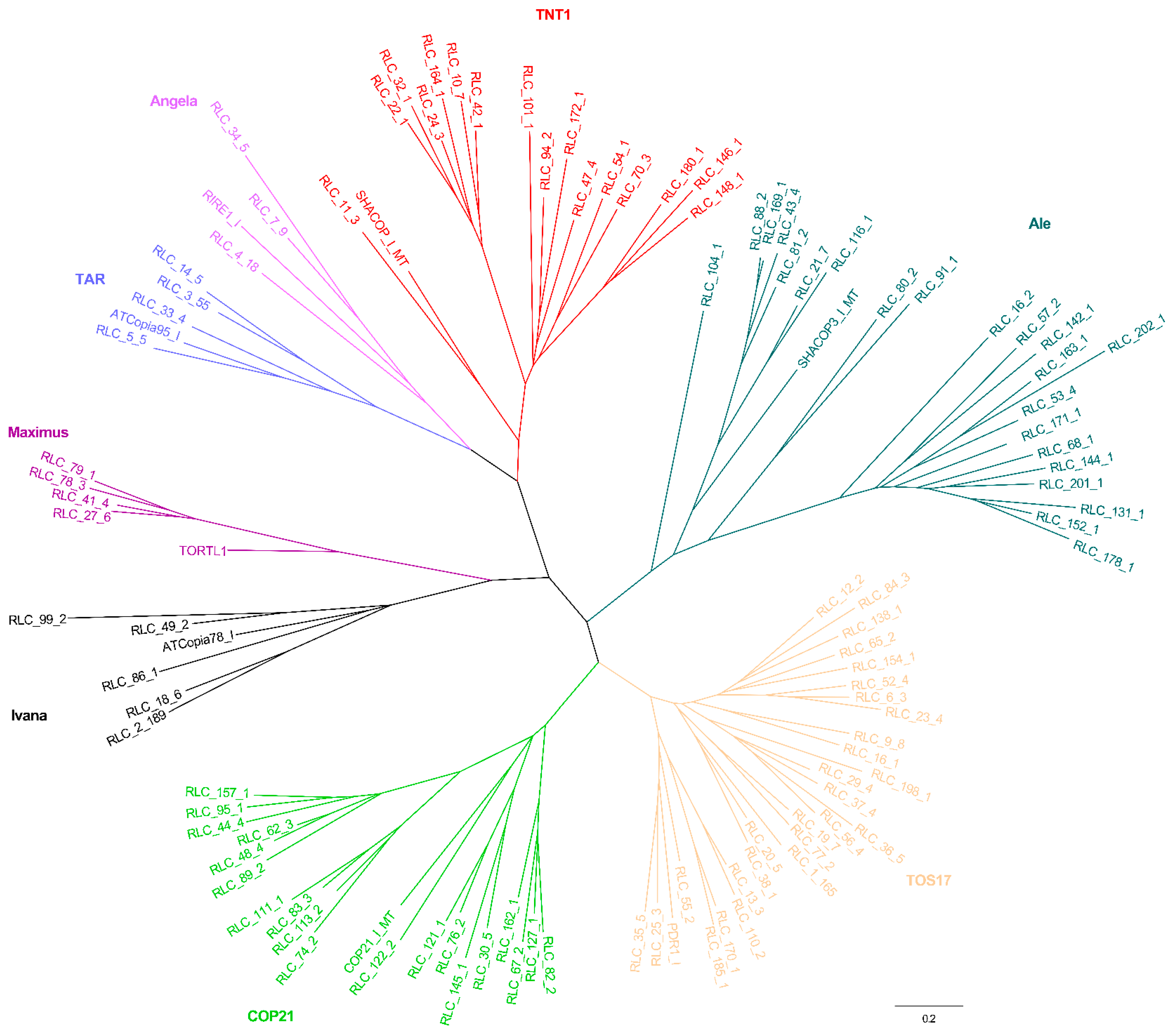
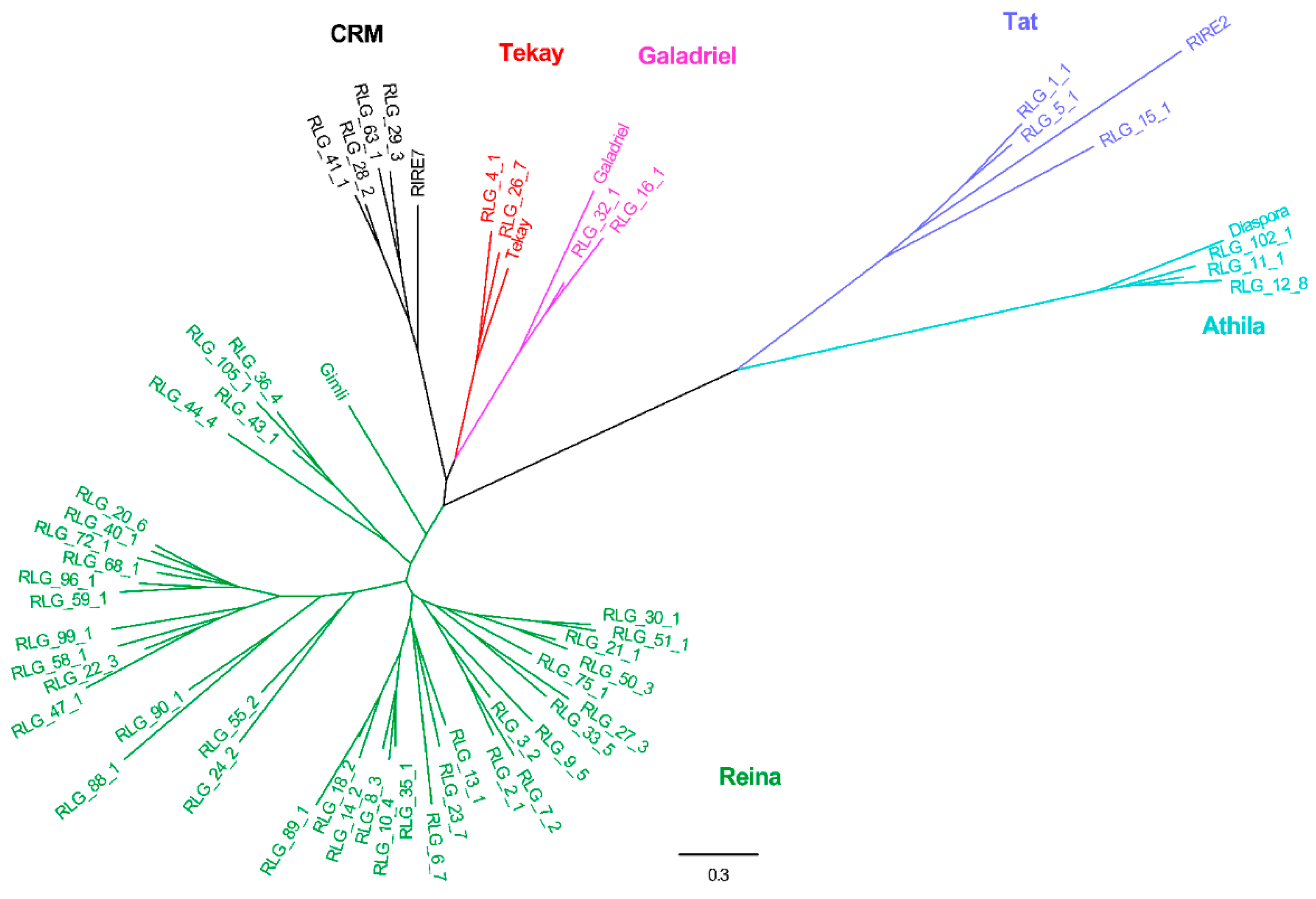
2.2. Phylogenetic Relationships
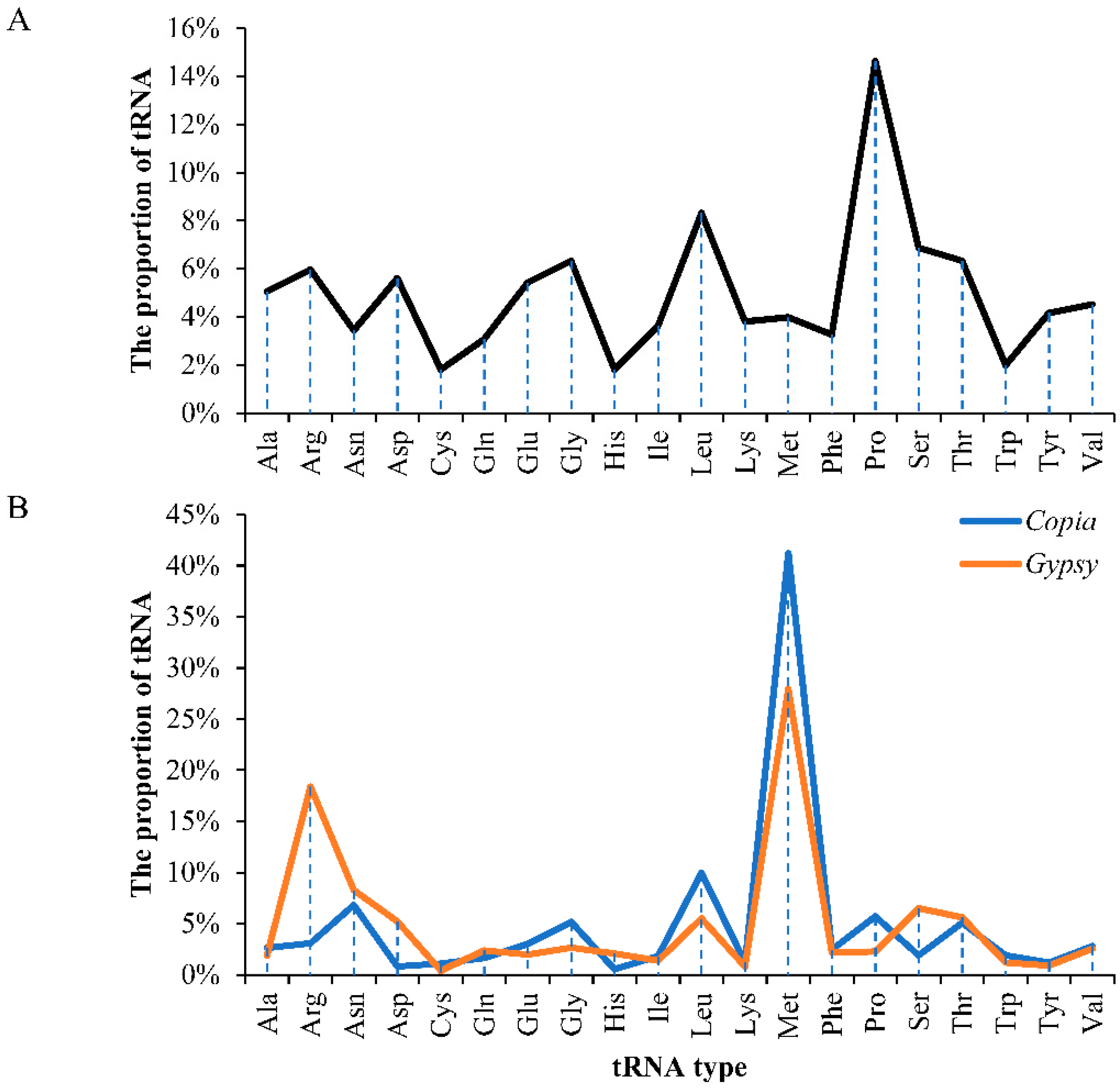
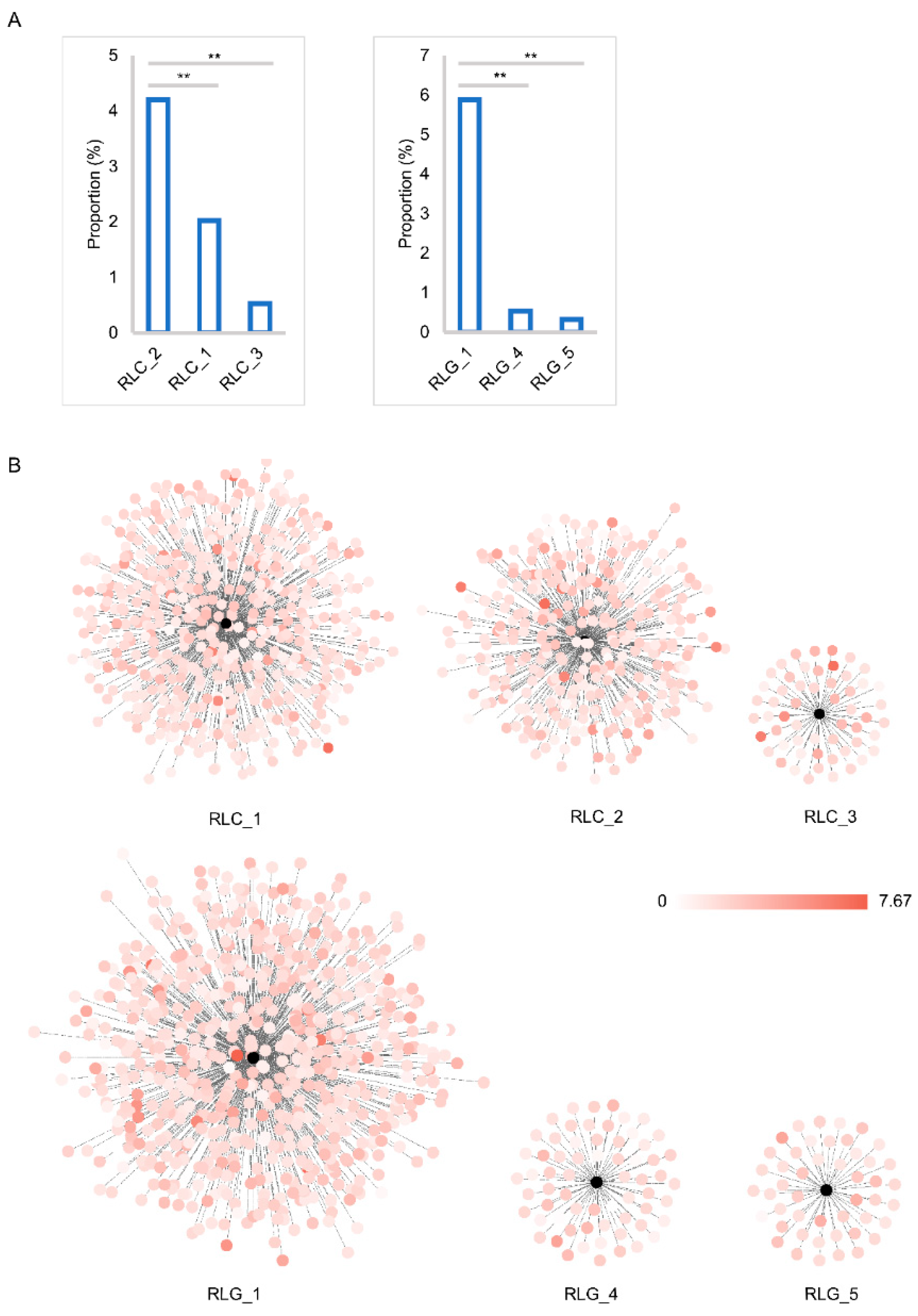
2.3. Insertion Time and Proportion Analysis
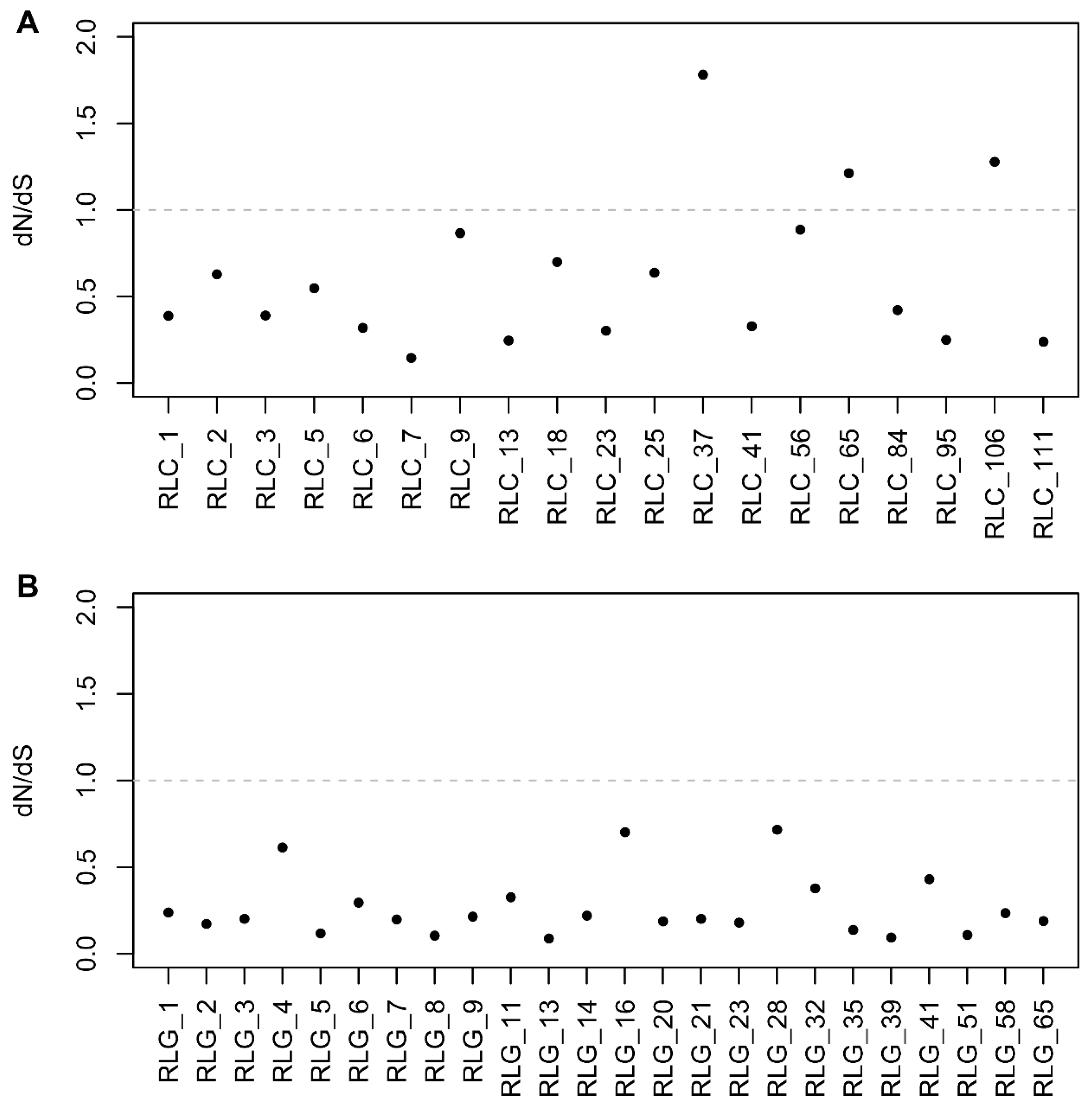
2.4. Selective Pressure Analysis
3. Discussion
3.1. Evolutionary Landscape of Copia and Gypsy Elements
3.2. Insertion and Deletion Dynamics of LTR Retrotransposons in the Mulberry Genome
4. Materials and Methods
4.1. Data Sources
4.2. Sequence and Phylogenetic Analysis
4.3. Estimation of Insertion Time
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LTR | Long terminal repeat |
| TE | transposable element |
| PBS | primer-binding site |
| PPT | polypurine tract |
| RT | reverse transcriptase |
| RH | RNase H |
| IN | integrase |
| PR | protease |
| MY | million years |
References
- Bennetzen, J.L. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr. Opin. Genet. Dev. 2005, 15, 621–627. [Google Scholar] [CrossRef]
- Bucher, E.; Reinders, J.; Mirouze, M. Epigenetic control of transposon transcription and mobility in Arabidopsis. Curr. Opin. Plant Biol. 2012, 15, 503–510. [Google Scholar] [CrossRef]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef]
- Laten, H.M.; Majumdar, A.; Gaucher, E.A. SIRE-1, a copia/Ty1-like retroelement from soybean, encodes a retroviral envelope-like protein. Proc. Natl. Acad. Sci. USA 1998, 95, 6897–6902. [Google Scholar] [CrossRef]
- Wright, D.A.; Voytas, D.F. Athila4 of Arabidopsis and Calypso of soybean define a lineage of endogenous plant retroviruses. Genome Res. 2002, 12, 122–131. [Google Scholar] [CrossRef]
- Xiong, Y.; Eickbush, T.H. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990, 9, 3353–3362. [Google Scholar] [CrossRef]
- Ragupathy, R.; You, F.M.; Cloutier, S. Arguments for standardizing transposable element annotation in plant genomes. Trends Plant Sci. 2013, 18, 367–376. [Google Scholar] [CrossRef]
- Pereira, V. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 2004, 5, R79. [Google Scholar] [CrossRef]
- Ma, J.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 2004, 304, 982. [Google Scholar] [CrossRef]
- Kashkush, K.; Feldman, M.; Levy, A.A. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat. Genet. 2003, 33, 102–106. [Google Scholar] [CrossRef]
- Neumann, P.; Navratilova, A.; Koblizkova, A.; Kejnovsky, E.; Hribova, E.; Hobza, R.; Widmer, A.; Dolezel, J.; Macas, J. Plant centromeric retrotransposons: A structural and cytogenetic perspective. Mob. DNA 2011, 2, 4. [Google Scholar] [CrossRef]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef]
- Nepal, M.P.; Ferguson, C.J. Phylogenetics of Morus (Moraceae) Inferred from ITS and trnL-trnF Sequence Data. Syst. Bot. 2012, 37, 442–450. [Google Scholar] [CrossRef]
- Berg, C.C. Moraceae Diversity in a Global Perspective; Friis, I., Balslev, H., Eds.; Royal Danish Academy of Science: Copenhagen, Denmark, 2005; pp. 423–440. [Google Scholar]
- He, N.; Zhang, C.; Qi, X.; Zhao, S.; Tao, Y.; Yang, G.; Lee, T.H.; Wang, X.; Cai, Q.; Li, D.; et al. Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 2013, 4, 2445. [Google Scholar] [CrossRef]
- Du, J.; Tian, Z.; Hans, C.S.; Laten, H.M.; Cannon, S.B.; Jackson, S.A.; Shoemaker, R.C.; Ma, J. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: Insights from genome-wide analysis and multi-specific comparison. Plant J. 2010, 63, 584–598. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.S. LTR retrotransposon landscape in Medicago truncatula: More rapid removal than in rice. BMC Genomics 2008, 9, 382. [Google Scholar] [CrossRef]
- Wicker, T.; Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007, 17, 1072–1081. [Google Scholar] [CrossRef]
- Hribova, E.; Neumann, P.; Matsumoto, T.; Roux, N.; Macas, J.; Dolezel, J. Repetitive part of the banana (Musa acuminata) genome investigated by low-depth 454 sequencing. BMC Plant Biol. 2010, 10, 204. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Gao, L.Z. Rapid and Recent Evolution of LTR Retrotransposons Drives Rice Genome Evolution During the Speciation of AA-Genome Oryza Species. G3 2017, 7, 1875–1885. [Google Scholar] [CrossRef]
- Neumann, P.; Pozarkova, D.; Macas, J. Highly abundant pea LTR retrotransposon Ogre is constitutively transcribed and partially spliced. Plant Mol. Biol. 2003, 53, 399–410. [Google Scholar] [CrossRef]
- Marin, I.; Llorens, C. Ty3/Gypsy retrotransposons: Description of new Arabidopsis thaliana elements and evolutionary perspectives derived from comparative genomic data. Mol. Biol. Evol. 2000, 17, 1040–1049. [Google Scholar] [CrossRef]
- Kordis, D. A genomic perspective on the chromodomain-containing retrotransposons: Chromoviruses. Gene 2005, 347, 161–173. [Google Scholar] [CrossRef]
- Llorens, C.; Munoz-Pomer, A.; Bernad, L.; Botella, H.; Moya, A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol. Direct. 2009, 4, 41. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef]
- Wright, S.I.; Agrawal, N.; Bureau, T.E. Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 2003, 13, 1897–1903. [Google Scholar]
- Baucom, R.S.; Estill, J.C.; Leebens-Mack, J.; Bennetzen, J.L. Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Res. 2009, 19, 243–254. [Google Scholar] [CrossRef]
- Staton, S.E.; Ungerer, M.C.; Moore, R.C. The genomic organization of Ty3/gypsy-like retrotransposons in Helianthus (Asteraceae) homoploid hybrid species. Am. J. Bot. 2009, 96, 1646–1655. [Google Scholar] [CrossRef]
- Lockton, S.; Gaut, B.S. The contribution of transposable elements to expressed coding sequence in Arabidopsis thaliana. J. Mol. Evol. 2009, 68, 80–89. [Google Scholar] [CrossRef]
- Nakashima, K.; Abe, J.; Kanazawa, A. Chromosomal distribution of soybean retrotransposon SORE-1 suggests its recent preferential insertion into euchromatic regions. Chromosome Res. 2018, 26, 199–210. [Google Scholar] [CrossRef]
- Gonzalez, L.G.; Deyholos, M.K. Identification, characterization and distribution of transposable elements in the flax (Linum usitatissimum L.) genome. BMC Genomics 2012, 13, 644. [Google Scholar] [CrossRef]
- Jiang, N.; Bao, Z.; Zhang, X.; Hirochika, H.; Eddy, S.R.; McCouch, S.R.; Wessler, S.R. An active DNA transposon family in rice. Nature 2003, 421, 163–167. [Google Scholar] [CrossRef]
- Nakazaki, T.; Okumoto, Y.; Horibata, A.; Yamahira, S.; Teraishi, M.; Nishida, H.; Inoue, H.; Tanisaka, T. Mobilization of a transposon in the rice genome. Nature 2003, 421, 170–172. [Google Scholar] [CrossRef]
- Momose, M.; Abe, Y.; Ozeki, Y. Miniature inverted-repeat transposable elements of Stowaway are active in potato. Genetics 2010, 186, 59–66. [Google Scholar] [CrossRef]
- Dong, H.T.; Zhang, L.; Zheng, K.L.; Yao, H.G.; Chen, J.; Yu, F.C.; Yu, X.X.; Mao, B.Z.; Zhao, D.; Yao, J.; et al. A Gaijin-like miniature inverted repeat transposable element is mobilized in rice during cell differentiation. BMC Genomics 2012, 13, 135. [Google Scholar] [CrossRef]
- Patel, M.; Jung, S.; Moore, K.; Powell, G.; Ainsworth, C.; Abbott, A. High-oleate peanut mutants result from a MITE insertion into the FAD2 gene. Theor. Appl. Genet. 2004, 108, 1492–1502. [Google Scholar] [CrossRef]
- Li, T.; Qi, X.; Zeng, Q.; Xiang, Z.; He, N. MorusDB: A resource for mulberry genomics and genome biology. Database 2014, 2014, bau054. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Ma, B.; Li, T.; Xiang, Z.; He, N. MnTEdb, a collective resource for mulberry transposable elements. Database 2015, 2015, bav004. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing. 2017. Available online: https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing (accessed on 22 February 2019).
- Vitte, C.; Panaud, O.; Quesneville, H. LTR retrotransposons in rice (Oryza sativa, L.): Recent burst amplifications followed by rapid DNA loss. BMC Genomics 2007, 8, 218. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Copia | Gypsy |
|---|---|---|
| Full-length number | 1532 | 1384 |
| Family number | 202 | 114 |
| Full length (bp) a | 7829 (1303–24,944) | 9526 (1468–23,704) |
| GC content | 0.41 | 0.41 |
| 5′LTR length (bp) a | 404 (97–2853) | 653 (102–3338) |
| 3′LTR length (bp) a | 404 (97–2834) | 653 (100–3352) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, B.; Kuang, L.; Xin, Y.; He, N. New Insights into Long Terminal Repeat Retrotransposons in Mulberry Species. Genes 2019, 10, 285. https://doi.org/10.3390/genes10040285
Ma B, Kuang L, Xin Y, He N. New Insights into Long Terminal Repeat Retrotransposons in Mulberry Species. Genes. 2019; 10(4):285. https://doi.org/10.3390/genes10040285
Chicago/Turabian StyleMa, Bi, Lulu Kuang, Youchao Xin, and Ningjia He. 2019. "New Insights into Long Terminal Repeat Retrotransposons in Mulberry Species" Genes 10, no. 4: 285. https://doi.org/10.3390/genes10040285
APA StyleMa, B., Kuang, L., Xin, Y., & He, N. (2019). New Insights into Long Terminal Repeat Retrotransposons in Mulberry Species. Genes, 10(4), 285. https://doi.org/10.3390/genes10040285