The Role of 3′ to 5′ Reverse RNA Polymerization in tRNA Fidelity and Repair

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Thg1 Maintains tRNAHis Aminoacylation Fidelity
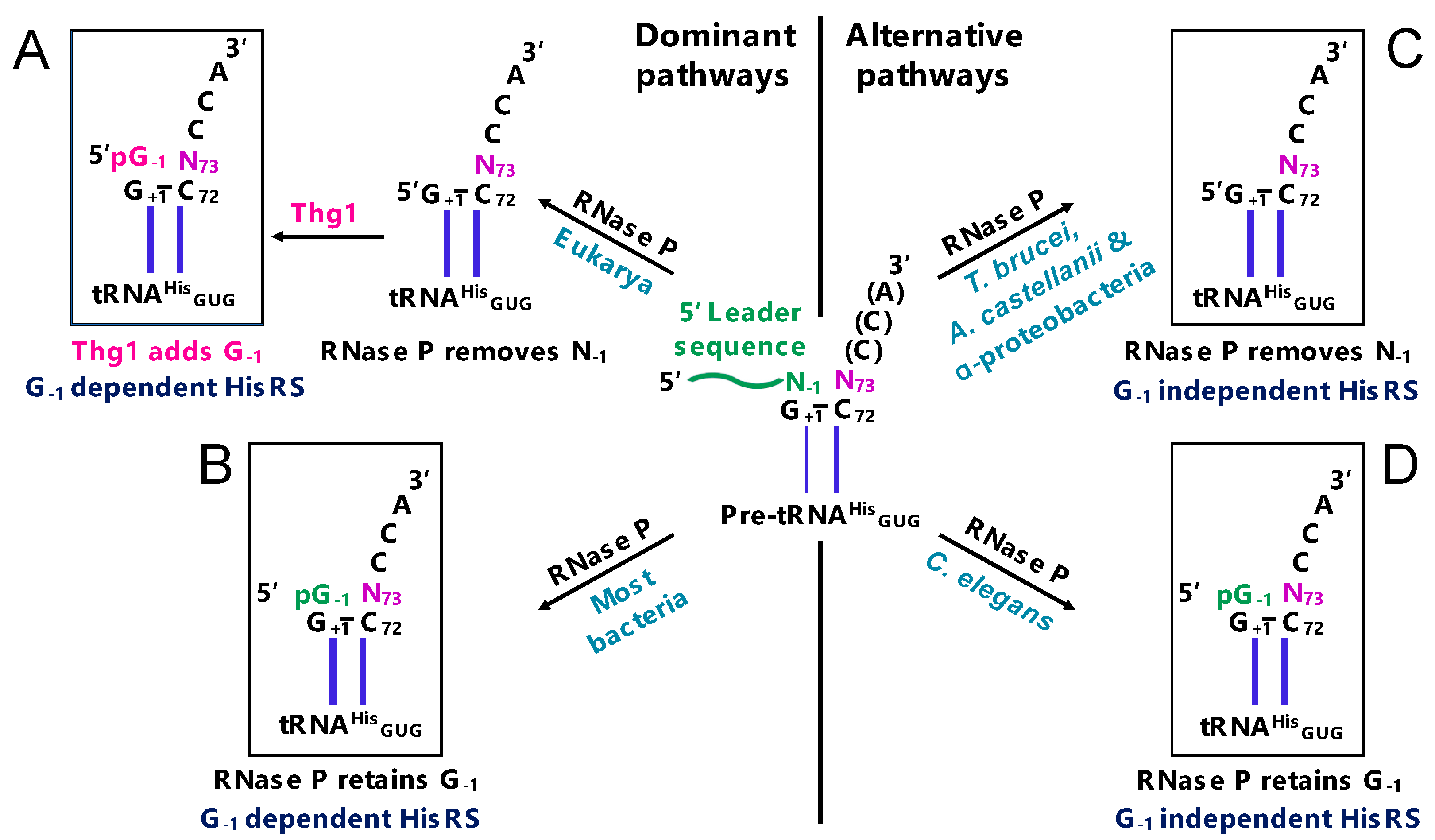
2.1. G-1 is a tRNAHis Identity Element
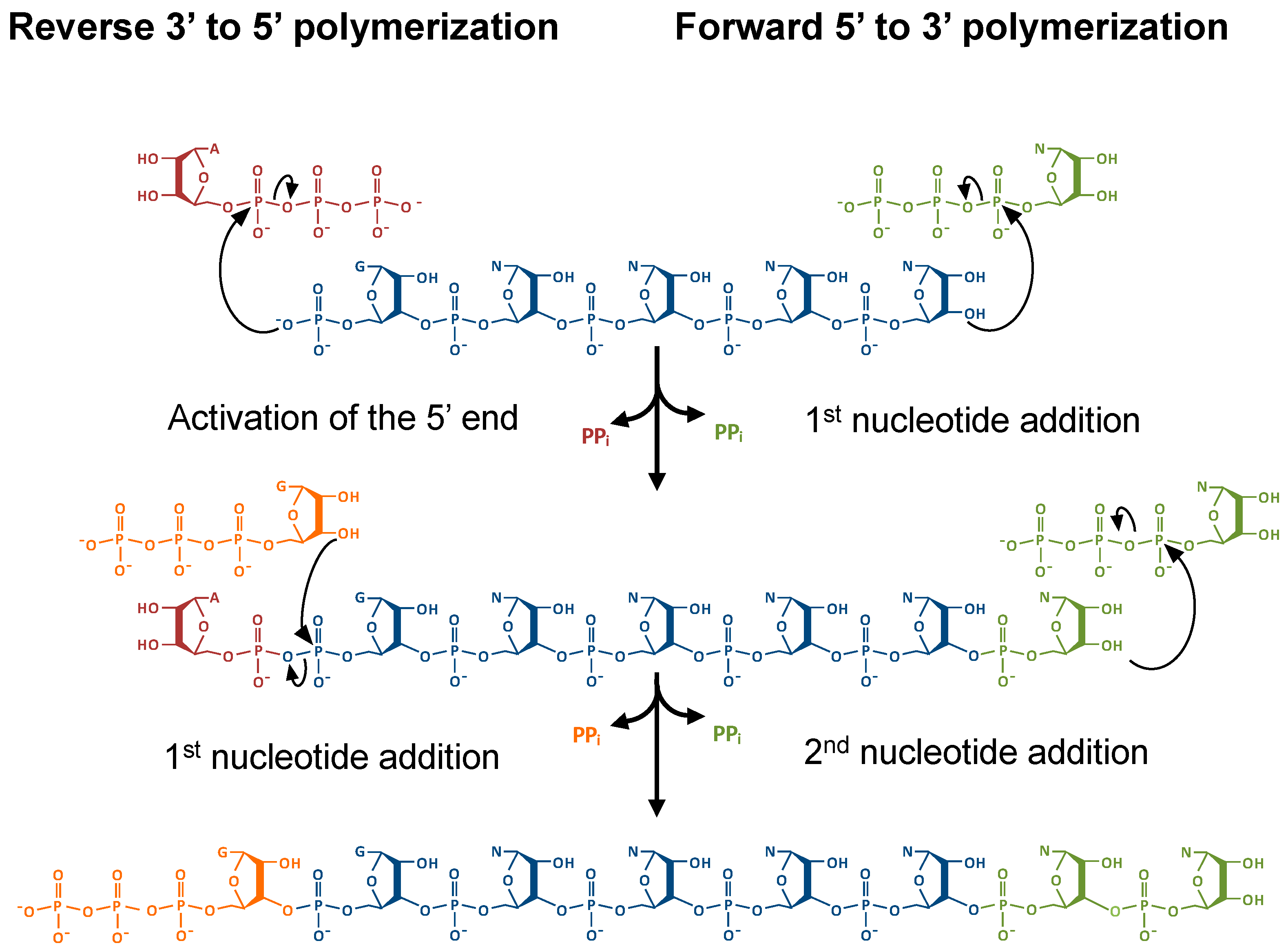
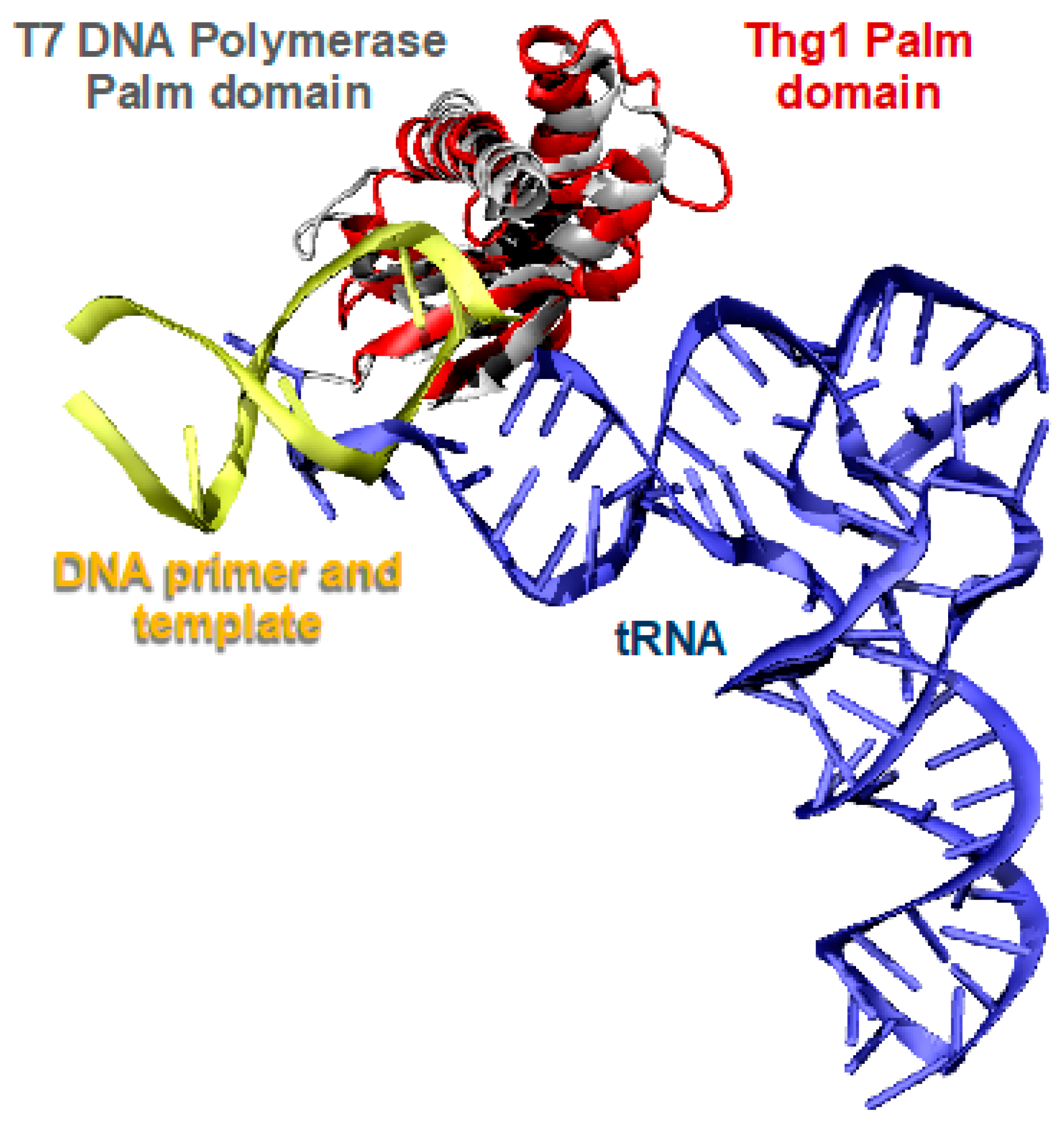
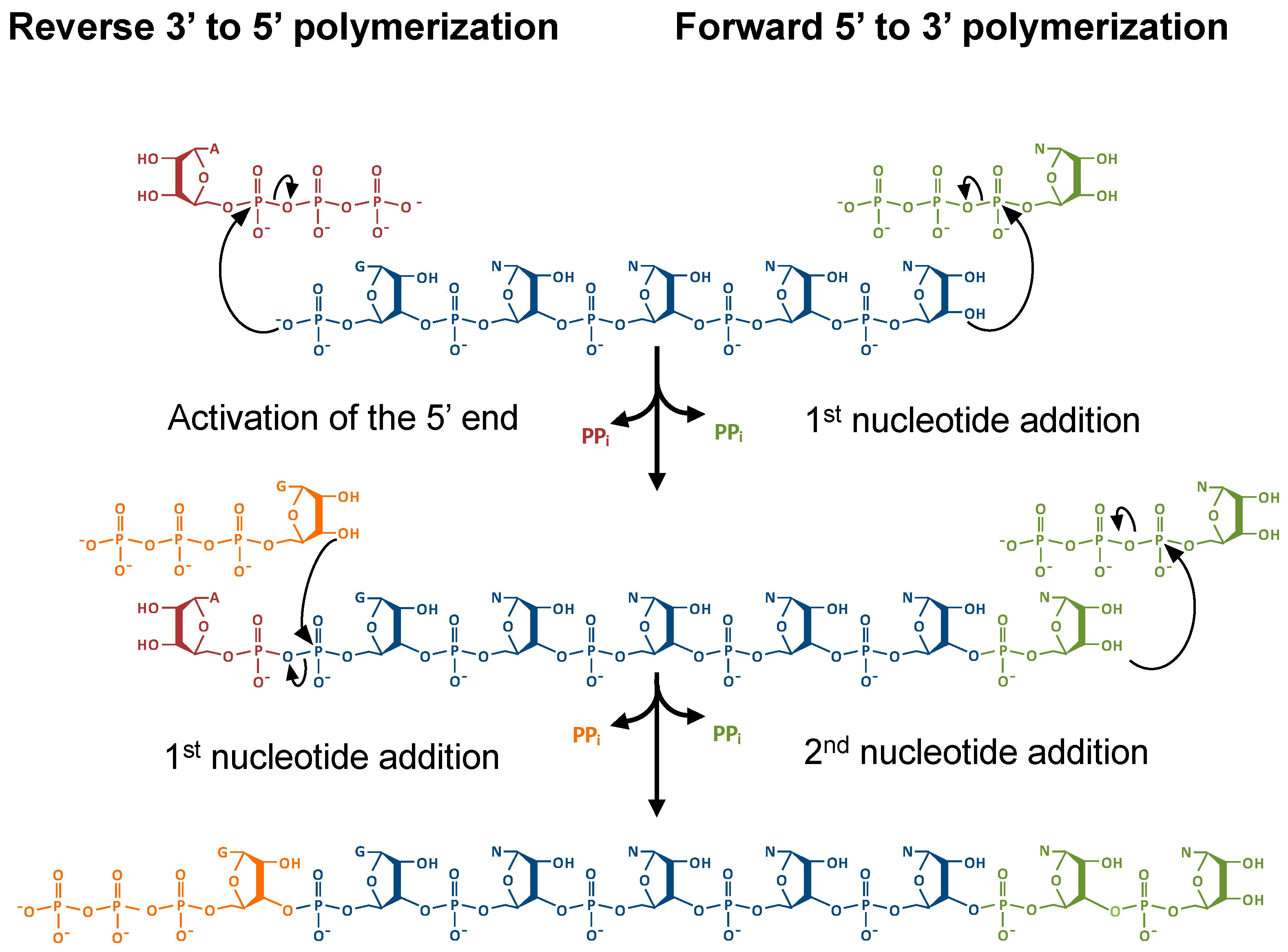
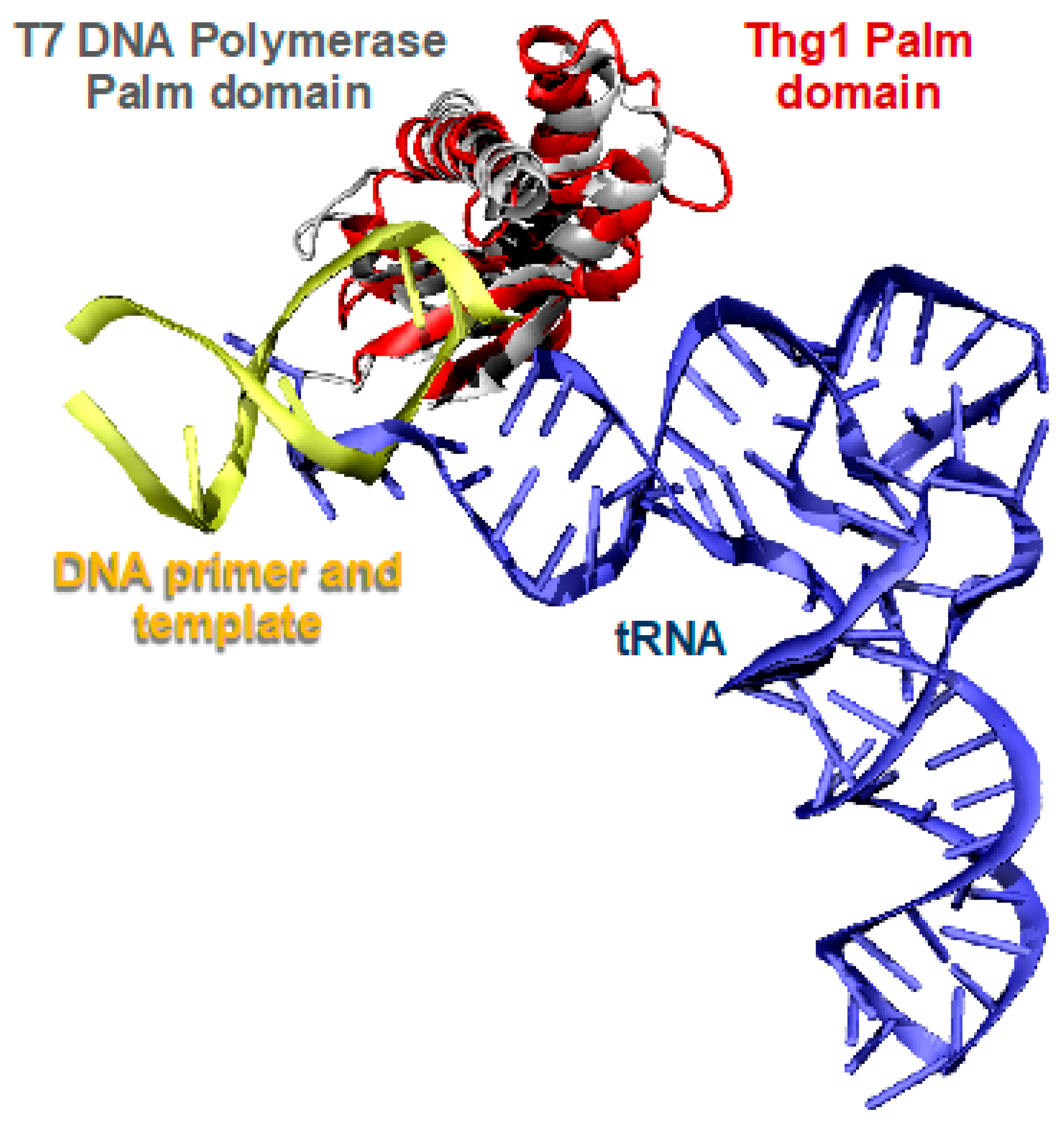
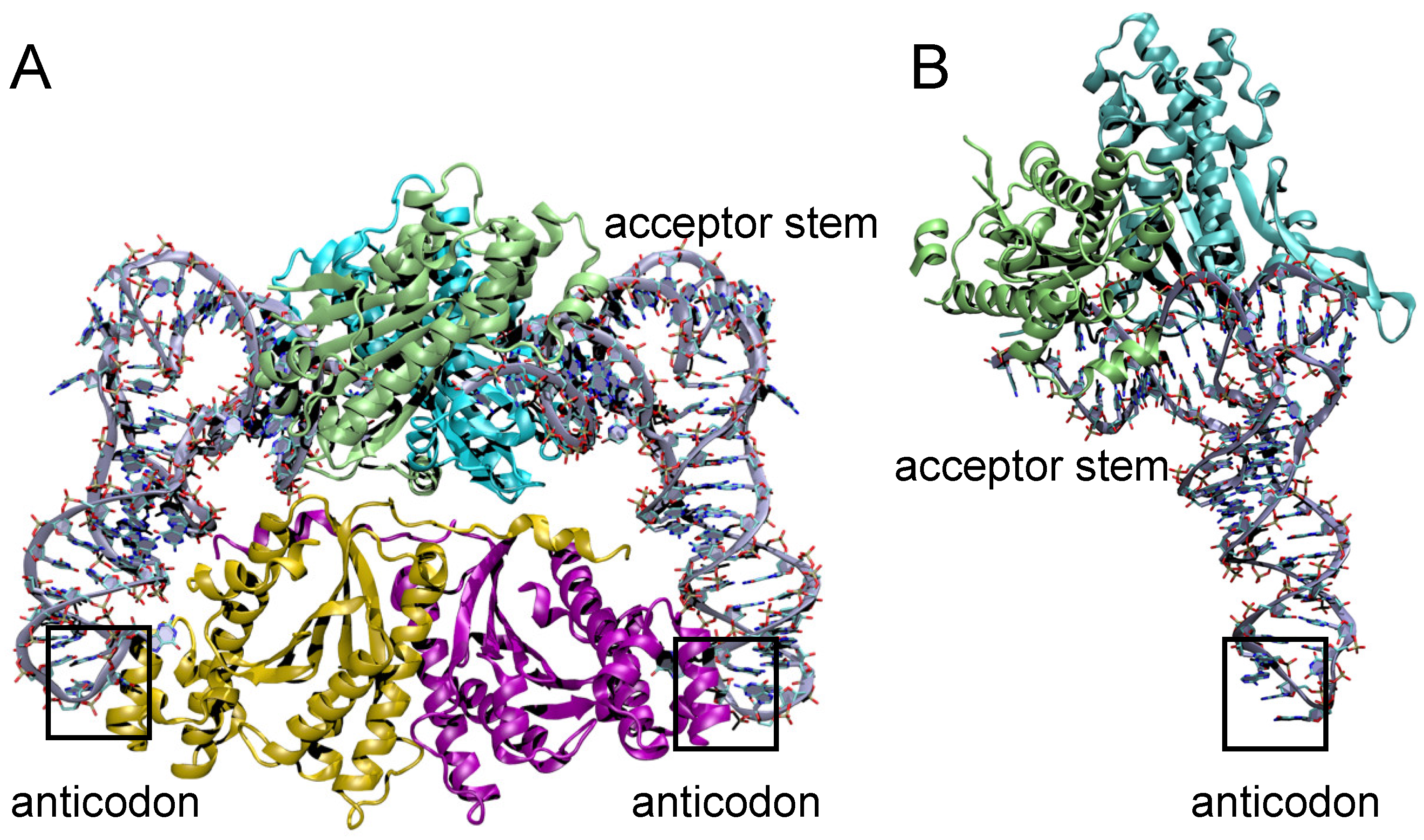
2.2. Thg1 Structurally Resembles Canonical 5′ to 3′ Polymerases
2.3. The Molecular Basis for tRNA Recognition
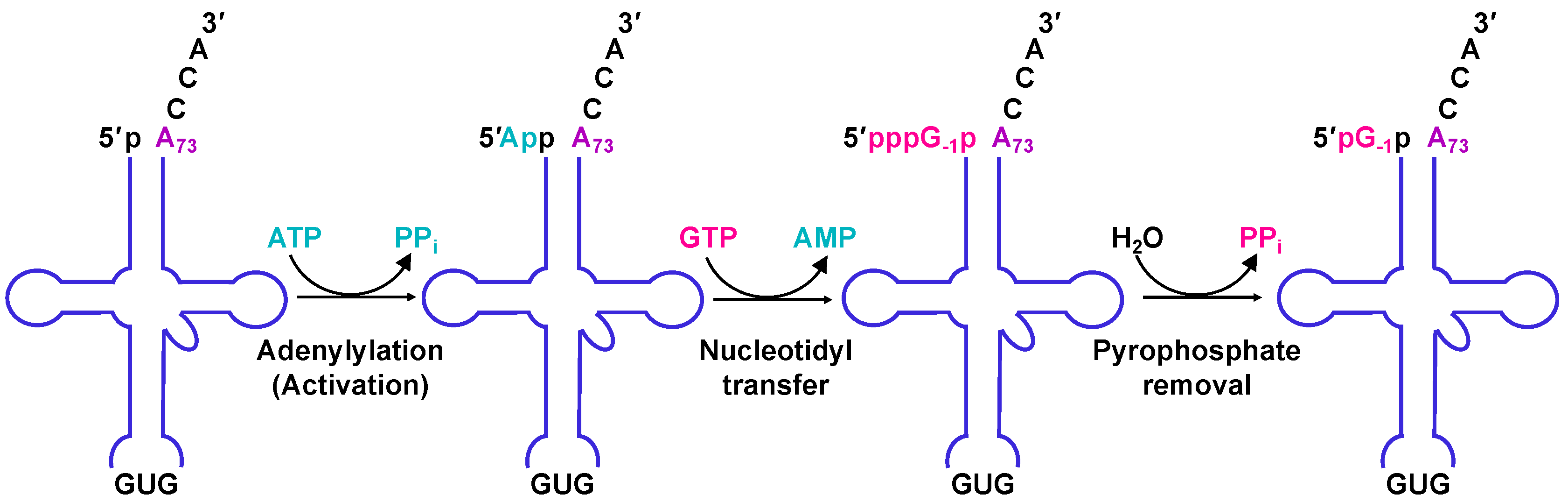
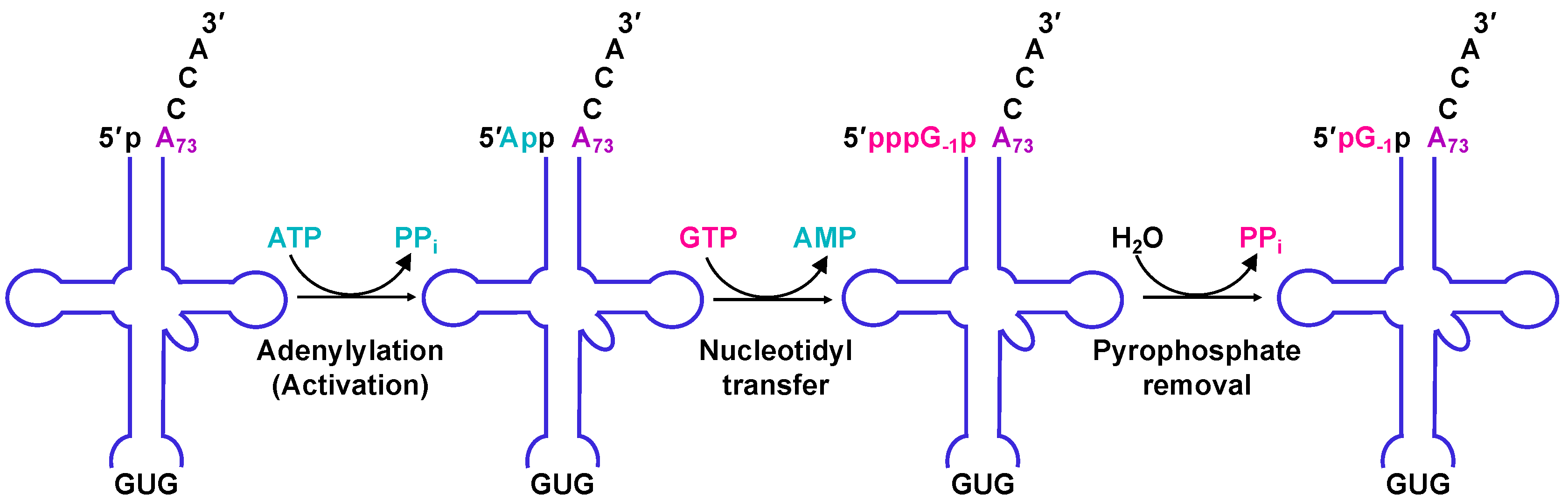
2.4. The Molecular Basis for Non-Watson–Crick G−1 Addition: tRNA Activation
2.5. The Molecular Basis for Non-Watson–Crick G−1 Addition: Nucleotidyl Transfer
2.6. The Molecular Basis for Non-Watson–Crick G-1 Addition: Pyrophosphate Removal
2.7. Maintenance of tRNAHis Fidelity
3. Thg1-Like Proteins Function in tRNA Repair
3.1. Thg1-Like Proteins Are Functionally and Phylogenetically Distinct from Thg1
3.2. The Discovery of TLPs in Bacteria and Archaea
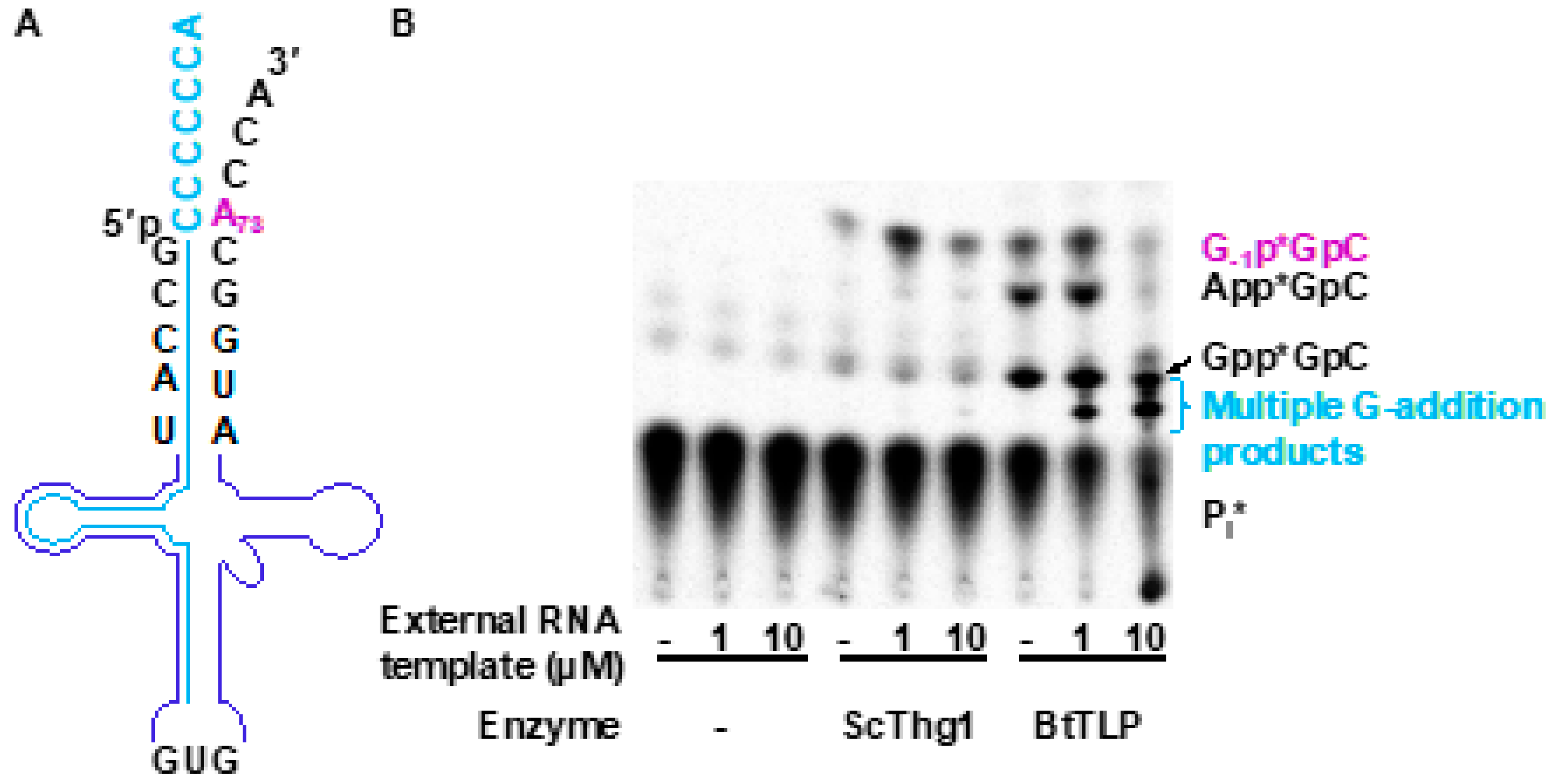
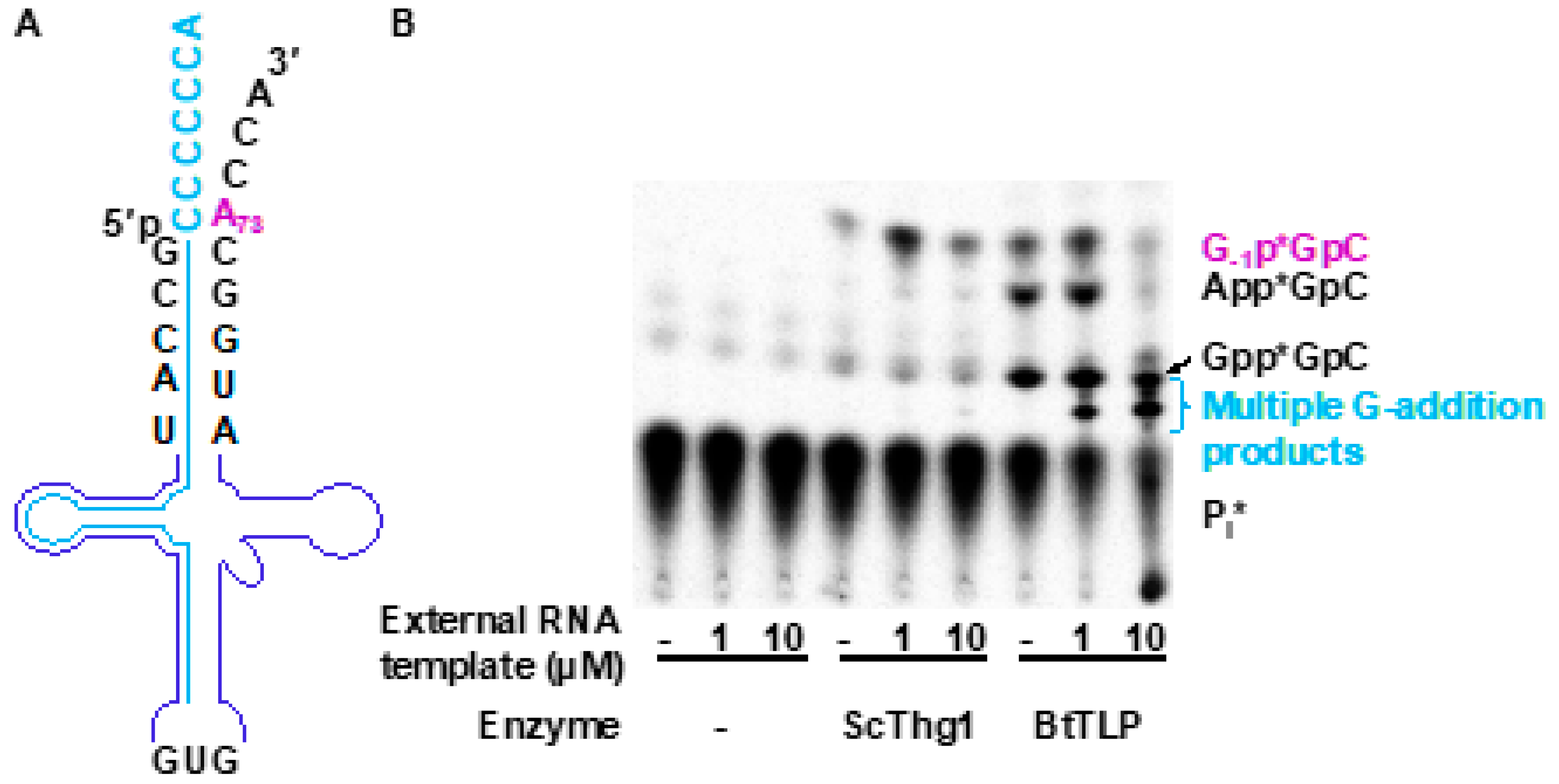
3.3. TLPs Catalyze Multiple Nucleotide Additions
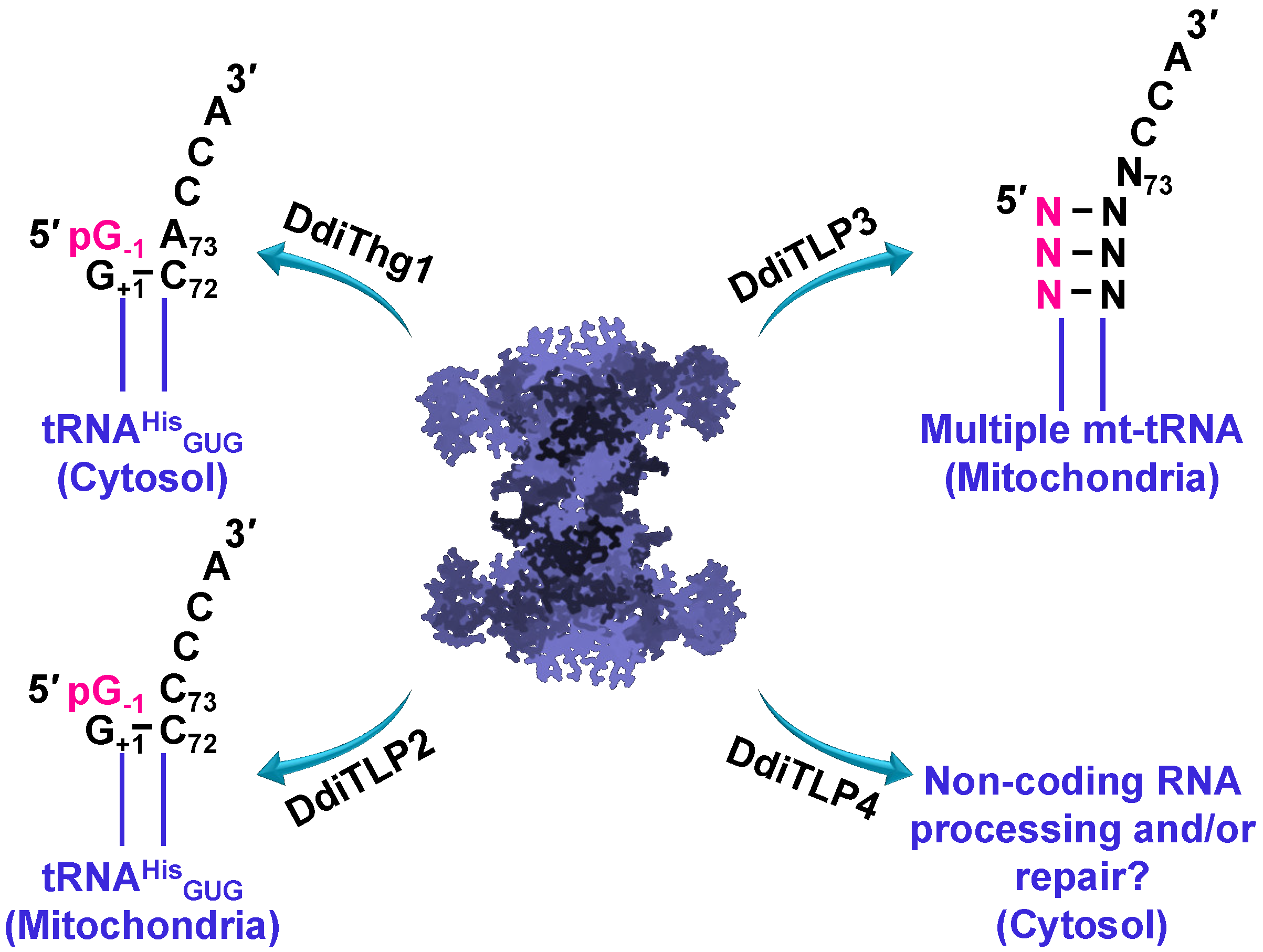
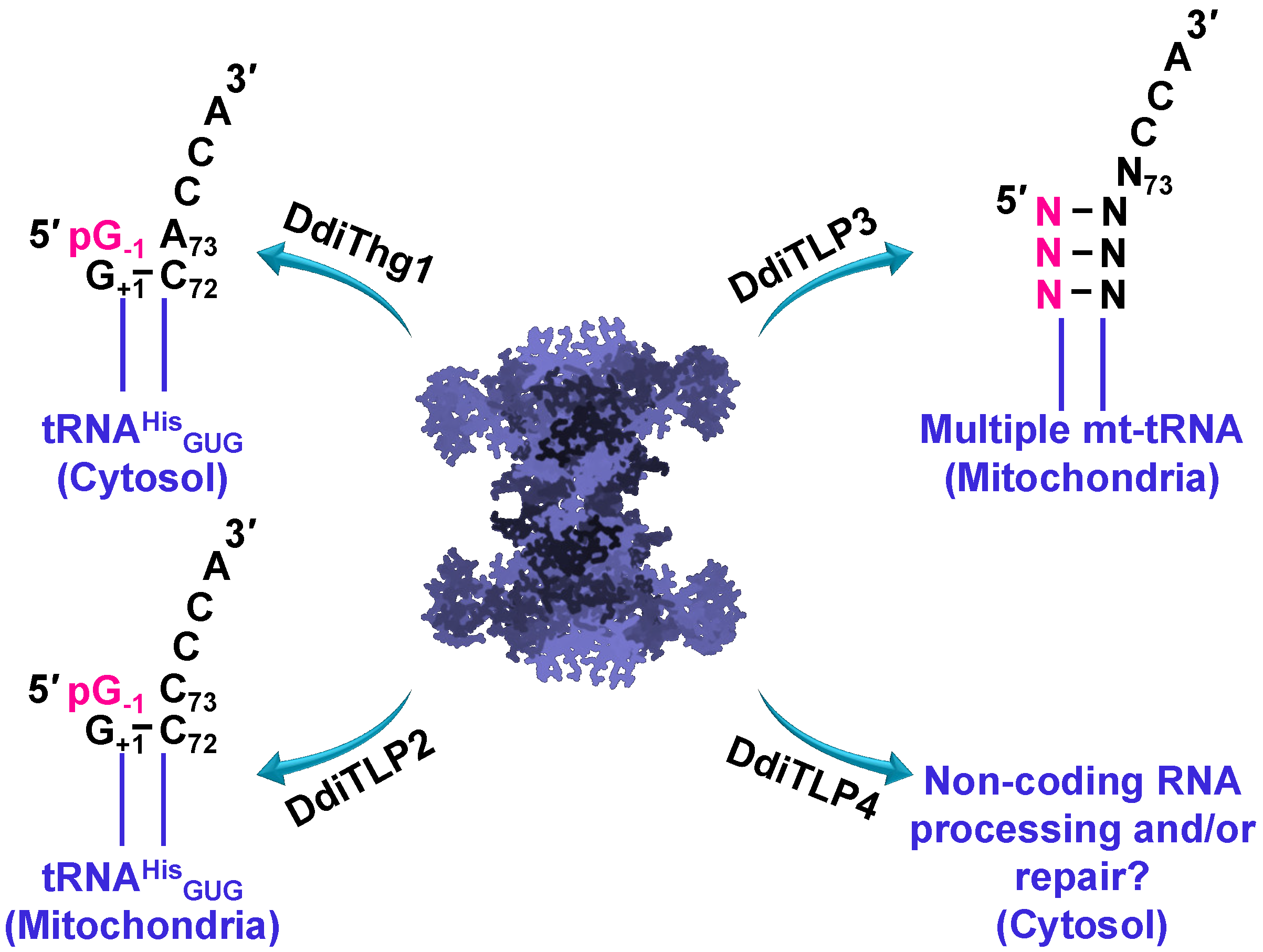
3.4. TLPs in Eukaryotes: Multiple TLPs Encoded by Dictyostelium Discoideum
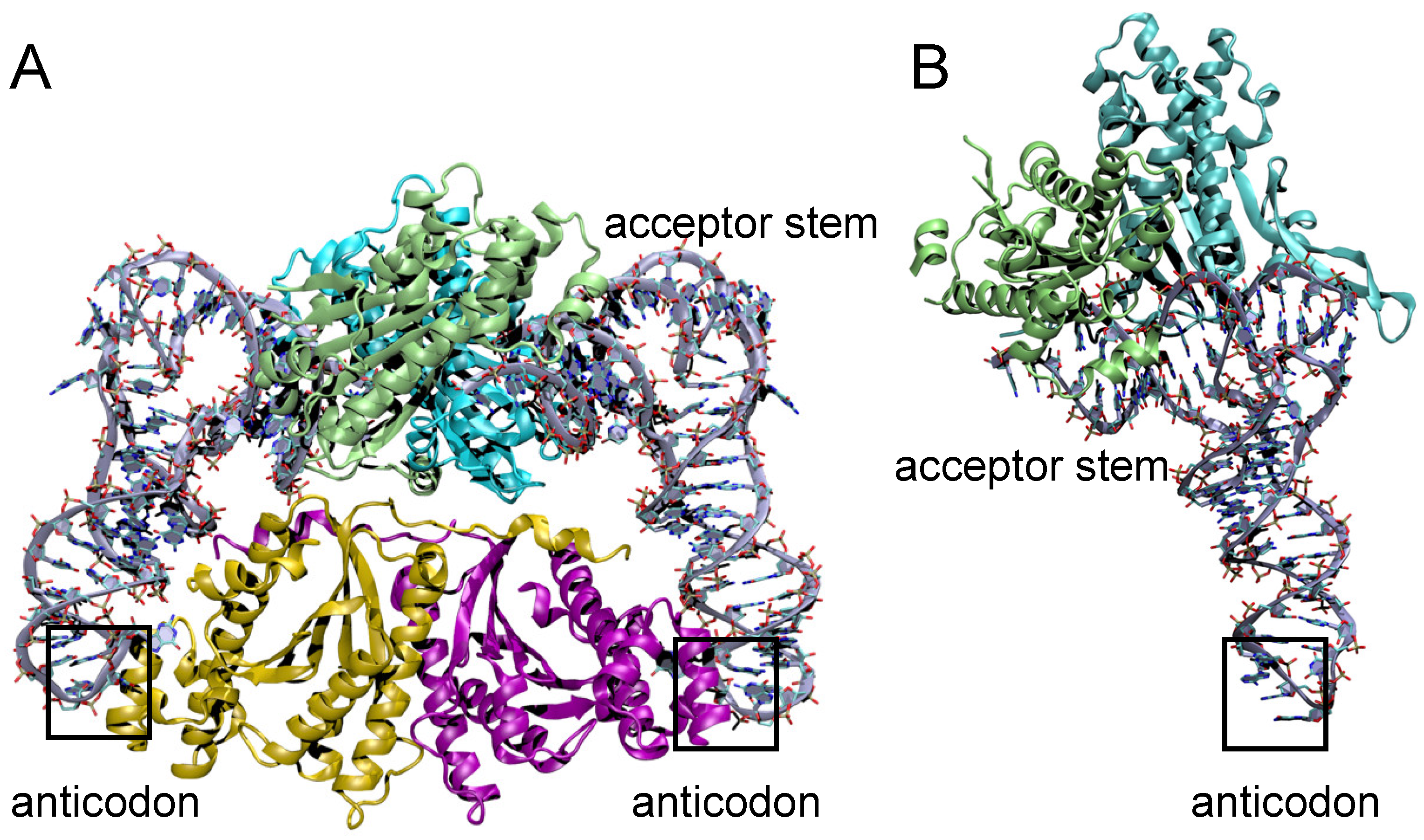
3.5. Structural Comparison of Thg1 and TLPs
3.6. 5′-End Activation and Nucleotidyl Transfer in TLPs
4. Synthetic Biology Applications and Thg1/TLP Engineering
4.1. TLPs Exhibit Broader RNA Recognition Properties Than Thg1 Homologs
4.2. Steps Toward Utilizing TLPs for Targeted 3′-5′ Addition Reactions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Joyce, C.M.; Steitz, T.A. Polymerase structures and function: Variations on a theme? J. Bacteriol. 1995, 177, 6321–6329. [Google Scholar] [CrossRef] [PubMed]
- Lehman, I.R.; Richardson, C.C. The deoxyribonucleases of Escherichia coli. Iv. An exonuclease activity present in purified preparations of deoxyribonucleic acid polymerase. J. Biol. Chem. 1964, 239, 233–241. [Google Scholar] [PubMed]
- Uptain, S.M.; Kane, C.M.; Chamberlin, M.J. Basic mechanisms of transcript elongation and its regulation. Annu. Rev. Biochem. 1997, 66, 117–172. [Google Scholar] [CrossRef] [PubMed]
- Hyde, S.J.; Eckenroth, B.E.; Smith, B.A.; Eberley, W.A.; Heintz, N.H.; Jackman, J.E.; Doublie, S. tRNAHis guanylyltransferase (Thg1), a unique 3′-5′ nucleotidyl transferase, shares unexpected structural homology with canonical 5′-3′ DNA polymerases. Proc. Natl. Acad. Sci. USA 2010, 107, 20305–20310. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Nemoto, T.; Heinemann, I.U.; Yamashita, K.; Sonoda, T.; Komoda, K.; Tanaka, I.; Söll, D.; Yao, M. Structural basis of reverse nucleotide polymerization. Proc. Natl. Acad. Sci. USA 2013, 110, 20970–20975. [Google Scholar] [CrossRef]
- Jahn, D.; Pande, S. Histidine tRNA guanylyltransferase from Saccharomyces cerevisiae. Ii. Catalytic mechanism. J. Biol. Chem. 1991, 266, 22832–22836. [Google Scholar]
- Pande, S.; Jahn, D.; Söll, D. Histidine tRNA guanylyltransferase from Saccharomyces cerevisiae. I. Purification and physical properties. J. Biol. Chem. 1991, 266, 22826–22831. [Google Scholar] [PubMed]
- Gu, W.; Hurto, R.L.; Hopper, A.K.; Grayhack, E.J.; Phizicky, E.M. Depletion of Saccharomyces cerevisiae tRNAHis guanylyltransferase Thg1p leads to uncharged tRNAHis with additional m(5)c. Mol. Cell Biol. 2005, 25, 8191–8201. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Phizicky, E.M. tRNAHis guanylyltransferase catalyzes a 3′-5′ polymerization reaction that is distinct from G-1 addition. Proc. Natl. Acad. Sci. USA 2006, 103, 8640–8645. [Google Scholar] [CrossRef]
- Jackman, J.E.; Phizicky, E.M. tRNAHis guanylyltransferase adds G−1 to the 5′ end of trnahis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA 2006, 12, 1007–1014. [Google Scholar] [CrossRef]
- Ingle, R.A. Histidine biosynthesis. Arabidopsis Book 2011, 9, e0141. [Google Scholar] [CrossRef] [PubMed]
- Lant, J.T.; Berg, M.D.; Heinemann, I.U.; Brandl, C.J.; O’Donoghue, P. Pathways to disease from natural variations in human cytoplasmic tRNAs. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Liao, S.M.; Du, Q.S.; Meng, J.Z.; Pang, Z.W.; Huang, R.B. The multiple roles of histidine in protein interactions. Chem. Cent. J. 2013, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Sprinzl, M.; Vassilenko, K.S. Compilation of trna sequences and sequences of tRNA genes. Nucleic Acids Res. 2005, 33, D139–D140. [Google Scholar] [CrossRef]
- Connolly, S.A.; Rosen, A.E.; Musier-Forsyth, K.; Francklyn, C.S. G-1:C73 recognition by an arginine cluster in the active site of Escherichia coli histidyl-tRNA synthetase. Biochemistry 2004, 43, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Nameki, N.; Asahara, H.; Shimizu, M.; Okada, N.; Himeno, H. Identity elements of Saccharomyces cerevisiae tRNAHis. Nucleic Acids Res. 1995, 23, 389–394. [Google Scholar] [CrossRef]
- Rosen, A.E.; Musier-Forsyth, K. Recognition of G−1:C73 atomic groups by Escherichia coli histidyl-tRNA synthetase. J. Am. Chem. Soc. 2004, 126, 64–65. [Google Scholar] [CrossRef]
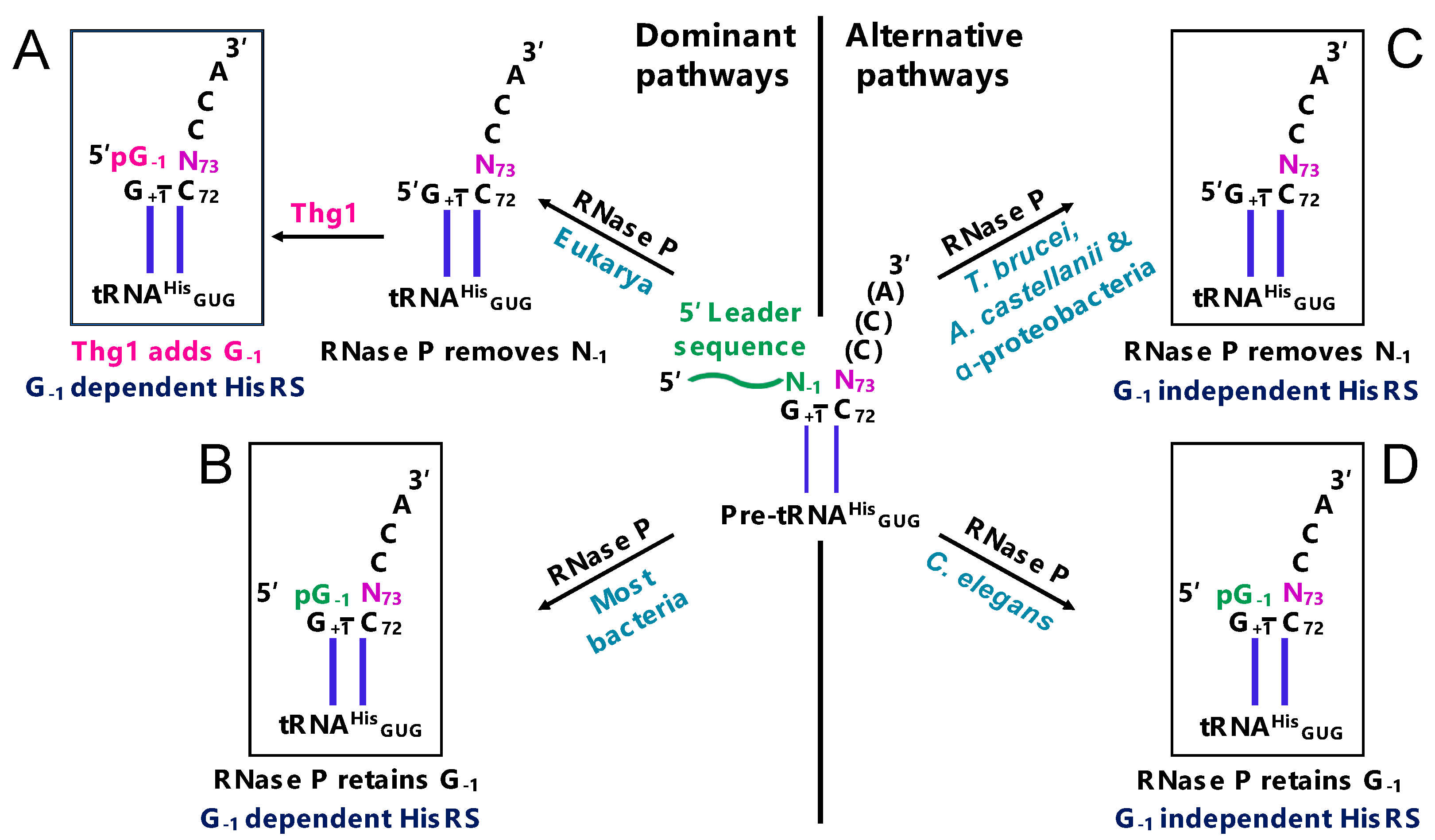
- Burkard, U.; Willis, I.; Söll, D. Processing of histidine transfer RNA precursors. Abnormal cleavage site for RNase P. J. Biol. Chem. 1988, 263, 2447–2451. [Google Scholar] [PubMed]
- Orellana, O.; Cooley, L.; Söll, D. The additional guanylate at the 5′ terminus of Escherichia coli tRNAHis is the result of unusual processing by RNase P. Mol. Cell Biol. 1986, 6, 525–529. [Google Scholar] [CrossRef]
- Cooley, L.; Appel, B.; Söll, D. Post-transcriptional nucleotide addition is responsible for the formation of the 5′ terminus of histidine tRNA. Proc. Natl. Acad. Sci. USA 1982, 79, 6475–6479. [Google Scholar] [CrossRef]
- Gu, W.; Jackman, J.E.; Lohan, A.J.; Gray, M.W.; Phizicky, E.M. tRNAHis maturation: An essential yeast protein catalyzes addition of a guanine nucleotide to the 5′ end of tRNAHis. Genes Dev. 2003, 17, 2889–2901. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Hu, K.; Lei, Y.; Wang, Y.; Ma, T.; He, D. Identification and characterization of a novel cytoplasm protein Icf45 that is involved in cell cycle regulation. J. Biol. Chem. 2004, 279, 53498–53505. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.S.; Maris, E.L.; Jackman, J.E. tRNA 5′-end repair activities of tRNAHis guanylyltransferase (Thg1)-like proteins from bacteria and archaea. Nucleic Acids Res. 2011, 39, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Gogakos, T.; Babina, A.M.; Söll, D.; Randau, L. Change of tRNA identity leads to a divergent orthogonal histidyl-tRNA synthetase/ tRNAHis pair. Nucleic Acids Res. 2011, 39, 2286–2293. [Google Scholar] [CrossRef]
- Wang, C.; Sobral, B.W.; Williams, K.P. Loss of a universal tRNA feature. J. Bacteriol. 2007, 189, 1954–1962. [Google Scholar] [CrossRef]
- Rao, B.S.; Mohammad, F.; Gray, M.W.; Jackman, J.E. Absence of a universal element for tRNAHis identity in Acanthamoeba castellanii. Nucleic Acids Res. 2013, 41, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.S.; Jackman, J.E. Life without post-transcriptional addition of g-1: Two alternatives for trnahis identity in eukarya. RNA 2015, 21, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Hyde, S.J.; Rao, B.S.; Eckenroth, B.E.; Jackman, J.E.; Doublie, S. Structural studies of a bacterial tRNAHis guanylyltransferase (Thg1)-like protein, with nucleotide in the activation and nucleotidyl transfer sites. PLoS ONE 2013, 8, e67465. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Suzuki, T.; Chen, M.; Kato, K.; Yu, J.; Nakamura, A.; Tanaka, I.; Yao, M. Template-dependent nucleotide addition in the reverse (3′-5′) direction by Thg1-like protein. Sci. Adv. 2016, 2, e1501397. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, E.H.; Son, J.; Hwang, K.Y. Crystal structure of tRNAHis guanylyltransferase from Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2017, 490, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Phizicky, E.M. Identification of critical residues for G−1 addition and substrate recognition by tRNAHis guanylyltransferase. Biochemistry 2008, 47, 4817–4825. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Gott, J.M.; Gray, M.W. Doing it in reverse: 3′-to-5′ polymerization by the Thg1 superfamily. RNA 2012, 18, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Jackman, J.E. Kinetic analysis of 3′-5′ nucleotide addition catalyzed by eukaryotic tRNAHis guanylyltransferase. Biochemistry 2012, 51, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Betat, H.; Long, Y.; Jackman, J.E.; Morl, M. From end to end: tRNA editing at 5′- and 3′-terminal positions. Int. J. Mol. Sci. 2014, 15, 23975–23998. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Kim, K.; Buchsenschutz, H.C.; Chen, A.W.; Bi, Y.; Mann, M.R.; Turk, M.A.; Chung, C.Z.; Heinemann, I.U. Minimal requirements for reverse polymerization and tRNA repair by tRNAHis guanylyltransferase. RNA Biol. 2018, 15, 614–622. [Google Scholar] [CrossRef]
- Smith, B.A.; Jackman, J.E. Saccharomyces cerevisiae Thg1 uses 5′-pyrophosphate removal to control addition of nucleotides to tRNAHis. Biochemistry 2014, 53, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Lant, J.T.; Berg, M.D.; Sze, D.H.W.; Hoffman, K.S.; Akinpelu, I.C.; Turk, M.A.; Heinemann, I.U.; Duennwald, M.L.; Brandl, C.J.; O’Donoghue, P. Visualizing tRNA-dependent mistranslation in human cells. RNA Biol. 2018, 15, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Preston, M.A.; D’Silva, S.; Kon, Y.; Phizicky, E.M. tRNAHis 5-methylcytidine levels increase in response to several growth arrest conditions in Saccharomyces cerevisiae. RNA 2013, 19, 243–256. [Google Scholar] [CrossRef]
- Rice, T.S.; Ding, M.; Pederson, D.S.; Heintz, N.H. The highly conserved tRNAHis guanylyltransferase Thg1p interacts with the origin recognition complex and is required for the G2/M phase transition in the yeast Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 832–835. [Google Scholar] [CrossRef]
- Heinemann, I.U.; Randau, L.; Tomko, R.J., Jr.; Söll, D. 3′-5′ tRNAHis guanylyltransferase in bacteria. FEBS Lett. 2010, 584, 3567–3572. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, I.U.; Söll, D.; Randau, L. Transfer RNA processing in archaea: Unusual pathways and enzymes. FEBS Lett. 2010, 584, 303–309. [Google Scholar] [CrossRef]
- Abad, M.G.; Long, Y.; Willcox, A.; Gott, J.M.; Gray, M.W.; Jackman, J.E. A role for tRNAHis guanylyltransferase (Thg1)-like proteins from Dictyostelium discoideum in mitochondrial 5′-trna editing. RNA 2011, 17, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, I.U.; Nakamura, A.; O’Donoghue, P.; Eiler, D.; Söll, D. tRNAHis-guanylyltransferase establishes tRNAHis identity. Nucleic Acids Res. 2012, 40, 333–344. [Google Scholar] [CrossRef]
- Long, Y.; Abad, M.G.; Olson, E.D.; Carrillo, E.Y.; Jackman, J.E. Identification of distinct biological functions for four 3′-5′ RNA polymerases. Nucleic Acids Res. 2016, 44, 8395–8406. [Google Scholar] [CrossRef]
- Heinemann, I.U.; O’Donoghue, P.; Madinger, C.; Benner, J.; Randau, L.; Noren, C.J.; Söll, D. The appearance of pyrrolysine in trnahis guanylyltransferase by neutral evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 21103–21108. [Google Scholar] [CrossRef] [PubMed]
- Prat, L.; Heinemann, I.U.; Aerni, H.R.; Rinehart, J.; O’Donoghue, P.; Söll, D. Carbon source-dependent expansion of the genetic code in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 21070–21075. [Google Scholar] [CrossRef]
- Abad, M.G.; Rao, B.S.; Jackman, J.E. Template-dependent 3′-5′ nucleotide addition is a shared feature of tRNAHis guanylyltransferase enzymes from multiple domains of life. Proc. Natl. Acad. Sci. USA 2010, 107, 674–679. [Google Scholar] [CrossRef]
- Dodbele, S.; Moreland, B.; Gardner, S.M.; Bundschuh, R.; Jackman, J.E. 5′-end sequencing in Saccharomyces cerevisiae offers new insights into 5′-ends of tRNAHis and snoRNAs. FEBS Lett. 2019, in press. [Google Scholar] [CrossRef]
- Abad, M.G.; Long, Y.; Kinchen, R.D.; Schindel, E.T.; Gray, M.W.; Jackman, J.E. Mitochondrial tRNA 5′-editing in Dictyostelium discoideum and Polysphondylium pallidum. J. Biol. Chem. 2014, 289, 15155–15165. [Google Scholar] [CrossRef] [PubMed]
- Dodbele, S.; Jackman, J.E.; Gray, M.W. Mechanisms and evolution of tRNA 5′-editing in mitochondria. In RNA Metabolism in Mitochondria; Jorge, C.-R., Gray, M.W., Eds.; Springer: Cham, Switzerland, 2018; Volume 34. [Google Scholar]
- Gott, J.M.; Somerlot, B.H.; Gray, M.W. Two forms of RNA editing are required for tRNA maturation in Physarum mitochondria. RNA 2010, 16, 482–488. [Google Scholar] [CrossRef]
- Lonergan, K.M.; Gray, M.W. Editing of transfer RNAs in Acanthamoeba castellanii mitochondria. Science 1993, 259, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, K.M.; Gray, M.W. Predicted editing of additional transfer RNAs in Acanthamoeba castellanii mitochondria. Nucleic Acids Res. 1993, 21, 4402. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Wang, D.; Komatsu, Y. Molecular mechanism of substrate recognition and specificity of tRNAHis guanylyltransferase during nucleotide addition in the 3′-5′ direction. RNA 2018, 24, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, W. Capture of a third Mg2+ is essential for catalyzing DNA synthesis. Science 2016, 352, 1334–1337. [Google Scholar] [CrossRef]
- Yang, W.; Weng, P.J.; Gao, Y. A new paradigm of DNA synthesis: Three-metal-ion catalysis. Cell Bioscience 2016, 6, 51. [Google Scholar] [CrossRef]
- Yuan, Y.; Altman, S. Selection of guide sequences that direct efficient cleavage of mRNA by human ribonuclease P. Science 1994, 263, 1269–1273. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, A.W.; Jayasinghe, M.I.; Chung, C.Z.; Rao, B.S.; Kenana, R.; Heinemann, I.U.; Jackman, J.E. The Role of 3′ to 5′ Reverse RNA Polymerization in tRNA Fidelity and Repair. Genes 2019, 10, 250. https://doi.org/10.3390/genes10030250
Chen AW, Jayasinghe MI, Chung CZ, Rao BS, Kenana R, Heinemann IU, Jackman JE. The Role of 3′ to 5′ Reverse RNA Polymerization in tRNA Fidelity and Repair. Genes. 2019; 10(3):250. https://doi.org/10.3390/genes10030250
Chicago/Turabian StyleChen, Allan W., Malithi I. Jayasinghe, Christina Z. Chung, Bhalchandra S. Rao, Rosan Kenana, Ilka U. Heinemann, and Jane E. Jackman. 2019. "The Role of 3′ to 5′ Reverse RNA Polymerization in tRNA Fidelity and Repair" Genes 10, no. 3: 250. https://doi.org/10.3390/genes10030250
APA StyleChen, A. W., Jayasinghe, M. I., Chung, C. Z., Rao, B. S., Kenana, R., Heinemann, I. U., & Jackman, J. E. (2019). The Role of 3′ to 5′ Reverse RNA Polymerization in tRNA Fidelity and Repair. Genes, 10(3), 250. https://doi.org/10.3390/genes10030250