Identification of Key Genes Involved in Embryo Development and Differential Oil Accumulation in Two Contrasting Maize Genotypes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sampling
2.2. Simple and Transmission Electron Microscopy
2.3. RNA Extraction, cDNA Library Construction, and High-Throughput Sequencing
2.4. Sequences Analysis and Assembly
2.5. Differential Expression of Unigenes
2.6. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analyses
2.7. Quantitative Real-Time Polymerase Chain Reaction (RT-PCR) Analysis
3. Results
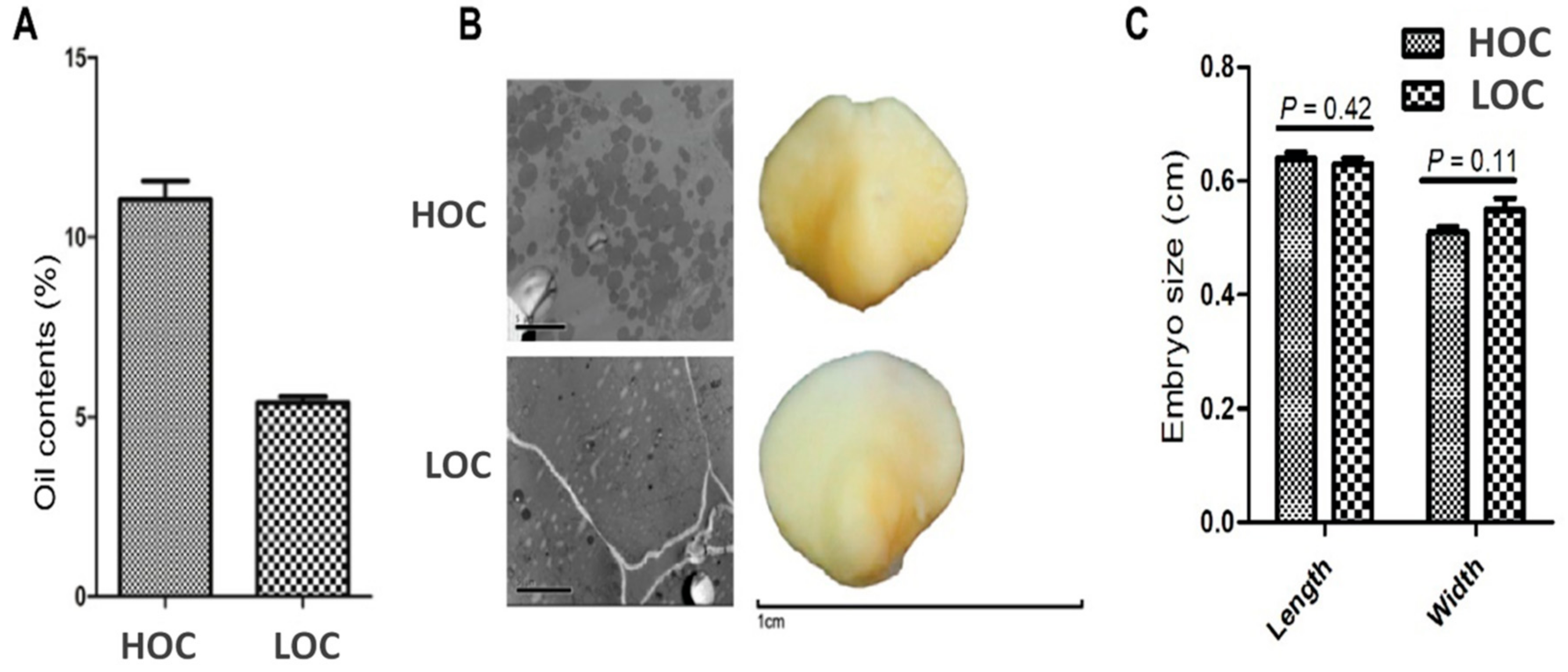
3.1. Characterization of Seed Oil Content and Embryo Phenotype in Contrasting Maize Cultivars
3.2. Generation and Analysis of Dynamic Transcriptome Profiles
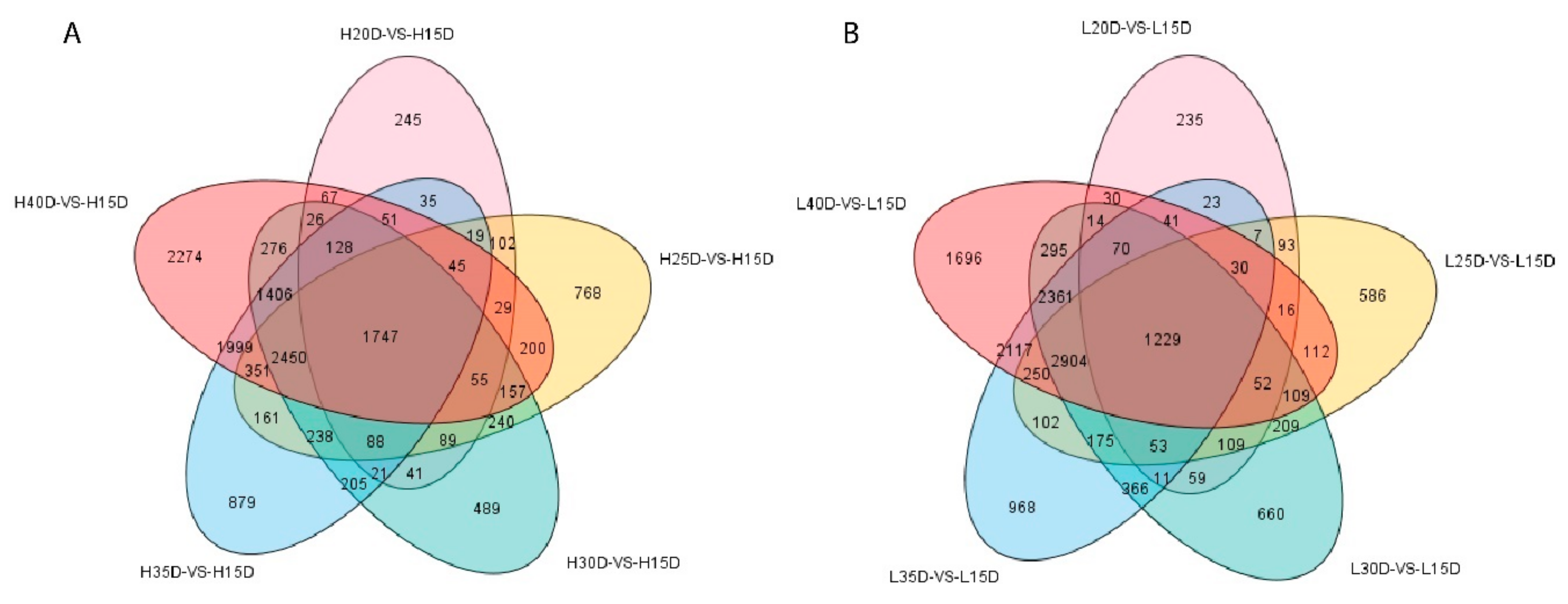
3.3. Differential Gene Expression Analysis Related to Embryo Development
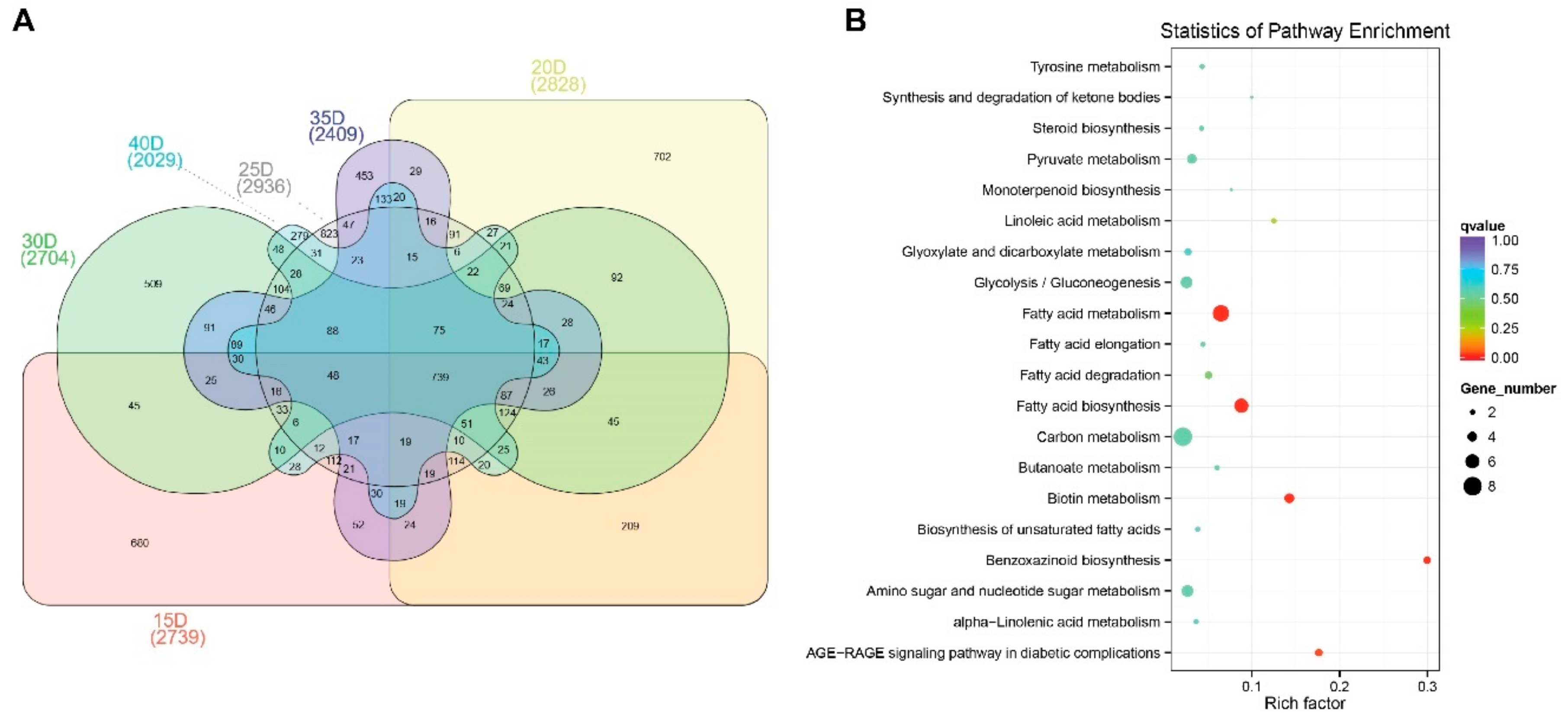
3.4. Differential Gene Expression Analysis Related to Oil Metabolism
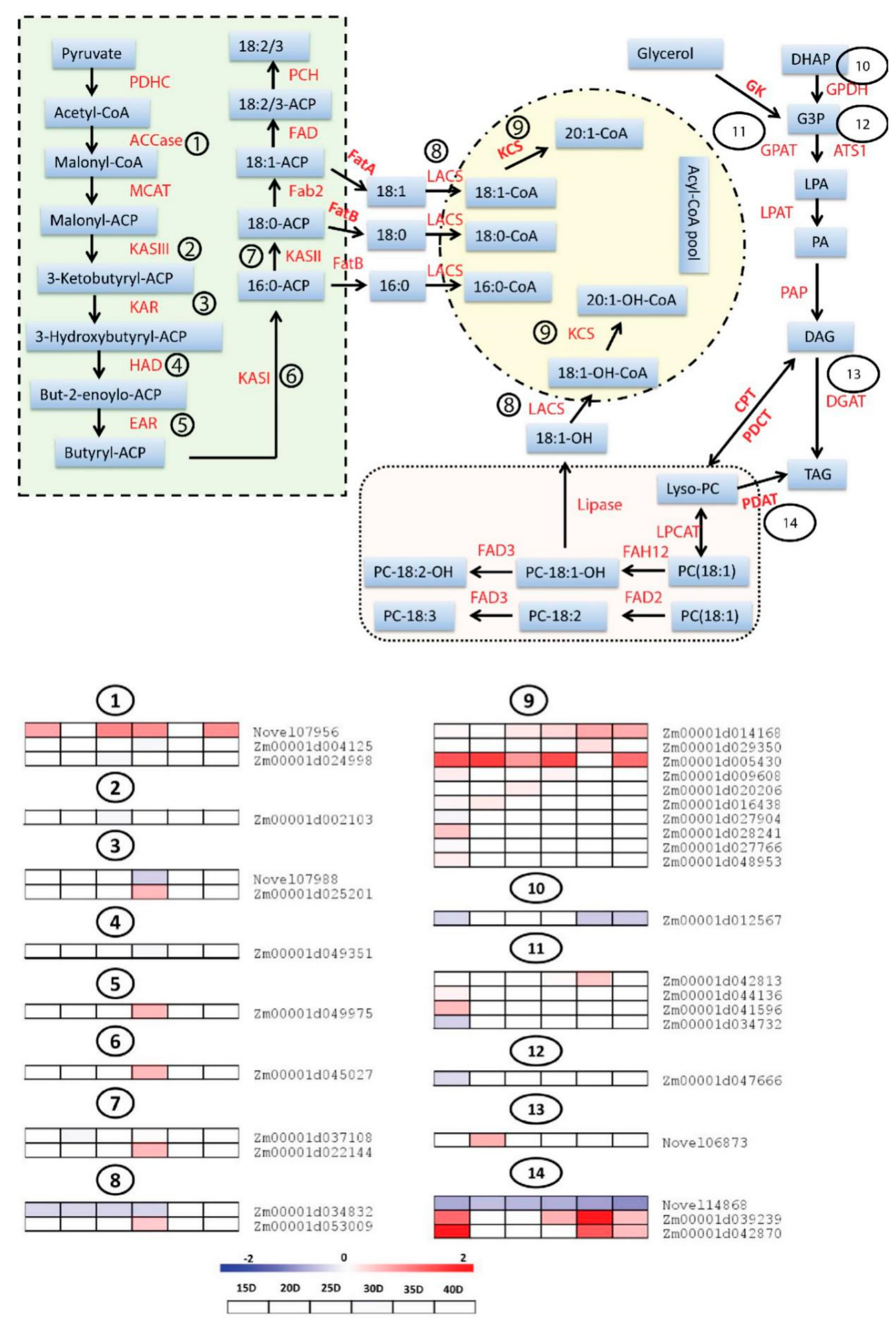
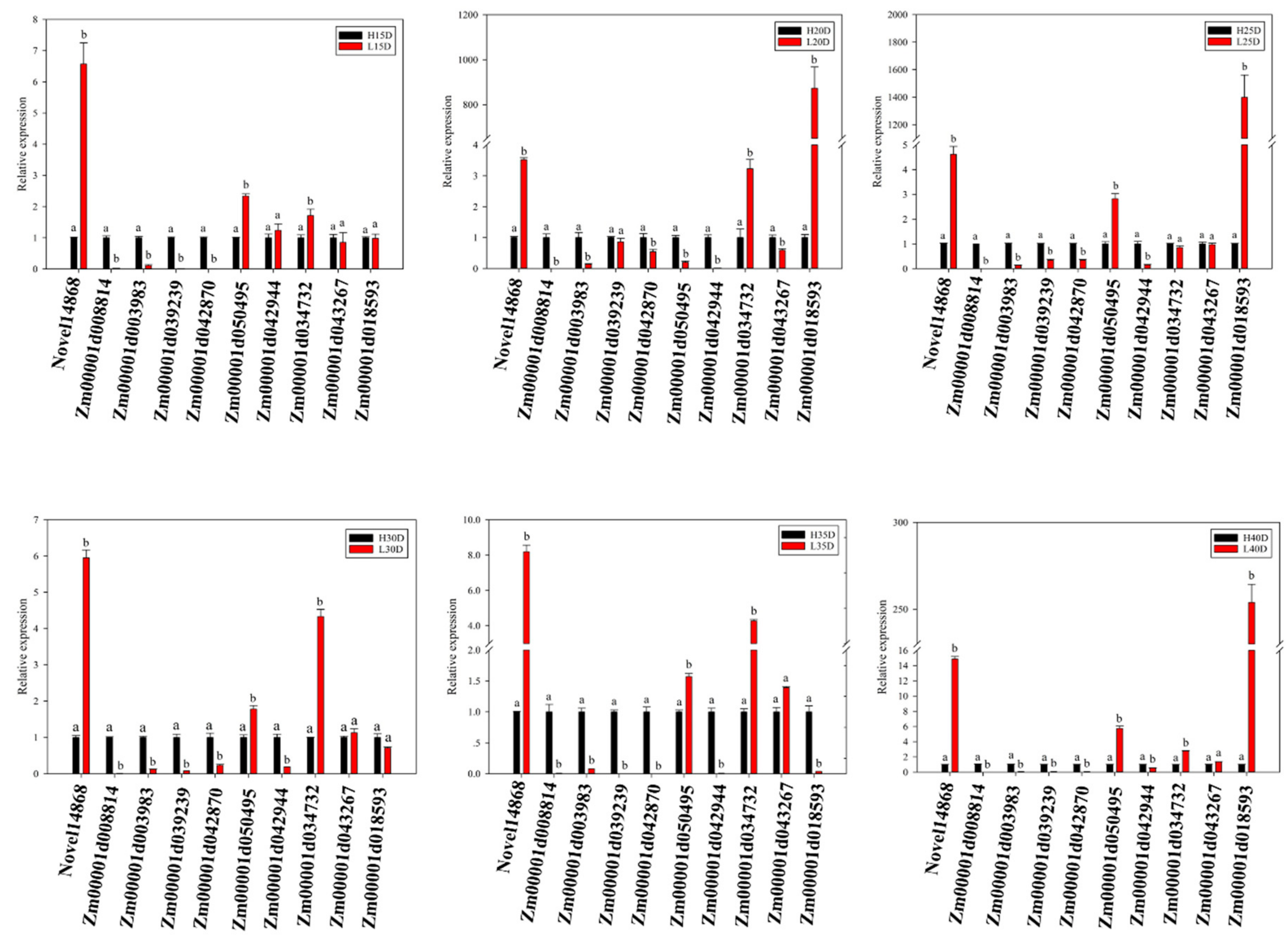
3.5. Differential Expression Analysis of Genes Involved in Fatty Acids (FA) and Triacylglycerols (TAG) Biosynthesis
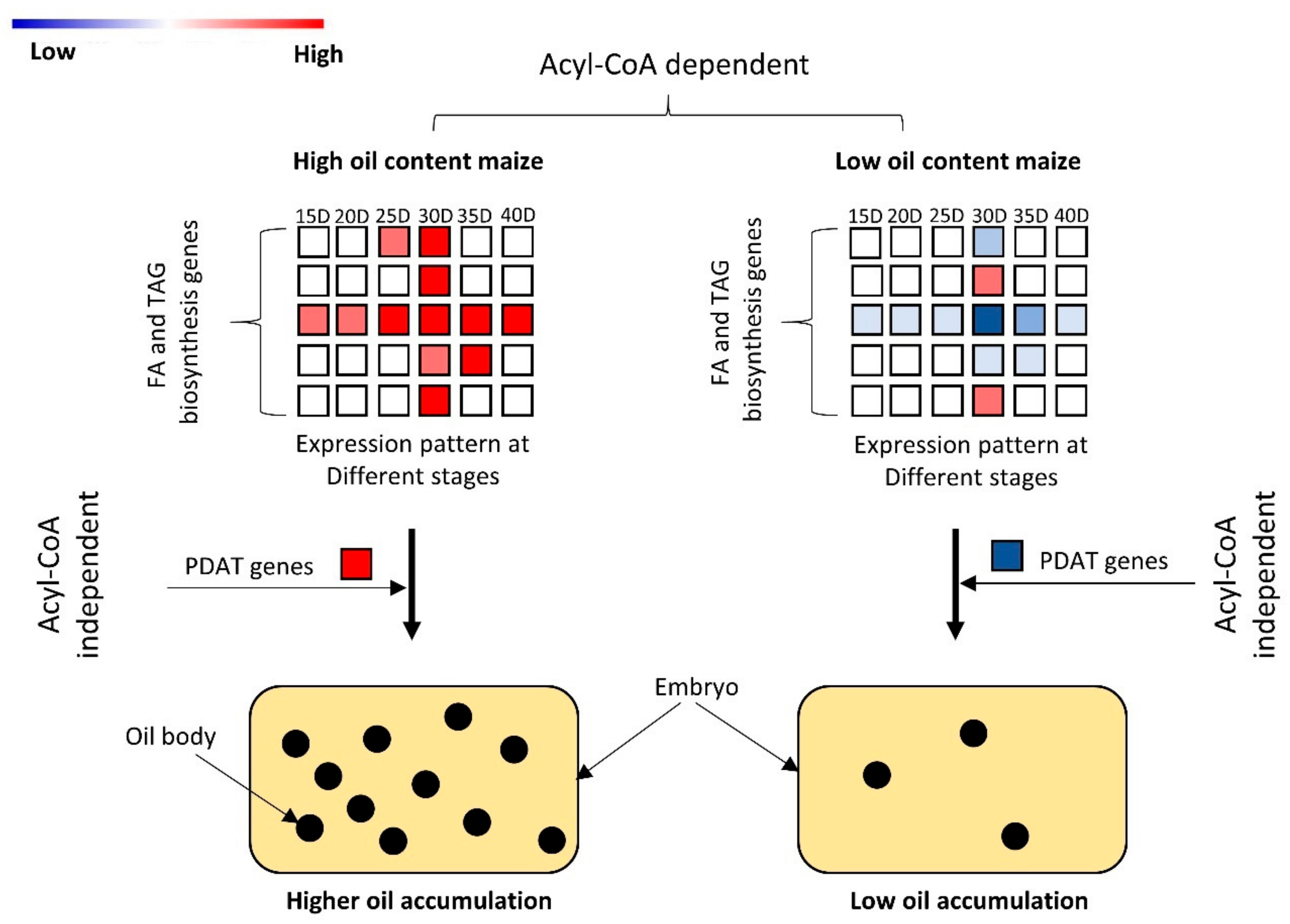
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Godfray, H.C.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Scince (N.Y.) 2010, 327, 812–818. [Google Scholar] [CrossRef]
- Strable, J.; Scanlon, M.J. Maize (Zea mays): A model organism for basic and applied research in plant biology. Cold Spring Harb. Protoc. 2009, 2009, emo132. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Wang, G.; Hauck, A.; Lai, J. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Fareghi, S.; Mirlohi, A.F.; Saeidi, G.; Khamisabadi, H. Evaluation of SRAP marker efficiency in identifying the relationship between genetic diversities of corn inbred lines with seed quantity and quality in derived hybrids. Cell. Mol. Biol. 2019, 65, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, J. High-Oil Maize Genomics. In The Maize Genome; Bennetzen, J., Flint-Garcia, S., Hirsch, C., Tuberosa, R., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 305–317. [Google Scholar] [CrossRef]
- Ali, Q.; Anwar, F.; Ashraf, M.; Saari, N.; Perveen, R. Ameliorating effects of exogenously applied proline on seed composition, seed oil quality and oil antioxidant activity of maize (Zea mays L.) under drought stress. Int. J. Mol. Sci. 2013, 14, 818–835. [Google Scholar] [CrossRef] [PubMed]
- Wattoo, F.M.; Rana, R.M.; Fiaz, S.; Zafar, S.A.; Noor, M.A.; Hassan, H.M.; Bhatti, M.H.; Rehman, S.U.; Anis, G.B.; Amir, R.M. Identification of drought tolerant maize genotypes and seedling based morpho-physiological selection indices for crop improvement. Sains Malays 2018, 47, 295–302. [Google Scholar]
- Hussain, H.A.; Men, S.; Hussain, S.; Chen, Y.; Ali, S.; Zhang, S.; Zhang, K.; Li, Y.; Xu, Q.; Liao, C.; et al. Interactive effects of drought and heat stresses on morpho-physiological attributes, yield, nutrient uptake and oxidative status in maize hybrids. Sci. Rep. 2019, 9, 3890. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, J.; He, L.; Zhang, Y.; Zhao, Y.; Xu, X.; Wei, Y.; Ge, S.; Ding, D.; Liu, M.; et al. Identification of Fatty Acid Desaturases in Maize and Their Differential Responses to Low and High Temperature. Genes 2019, 10, 455. [Google Scholar] [CrossRef]
- Yang, X.; Ma, H.; Zhang, P.; Yan, J.; Guo, Y.; Song, T.; Li, J. Characterization of QTL for oil content in maize kernel. TAG Theor. Appl. Genet. Theor. Und Angew. Genet. 2012, 125, 1169–1179. [Google Scholar] [CrossRef]
- Yang, X.; Guo, Y.; Yan, J.; Zhang, J.; Song, T.; Rocheford, T.; Li, J.S. Major and minor QTL and epistasis contribute to fatty acid compositions and oil concentration in high-oil maize. TAG Theor. Appl. Genet. Theor. Und Angew. Genet. 2010, 120, 665–678. [Google Scholar] [CrossRef]
- Sabelli, P.A.; Larkins, B.A. The development of endosperm in grasses. Plant Physiol. 2009, 149, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.H.C. Structure of plant seed oil bodies. Curr. Opin. Struct. Biol. 1994, 4, 493–498. [Google Scholar] [CrossRef]
- Du, H.; Huang, M.; Hu, J.; Li, J. Modification of the fatty acid composition in Arabidopsis and maize seeds using a stearoyl-acyl carrier protein desaturase-1 (ZmSAD1) gene. BMC Plant Biol. 2016, 16, 137. [Google Scholar] [CrossRef] [PubMed]
- Alrefai, R.; Berke, T.G.; Rocheford, T.R. Quantitative trait locus analysis of fatty acid concentrations in maize. Genome 1995, 38, 894–901. [Google Scholar] [CrossRef]
- Zheng, P.; Allen, W.B.; Roesler, K.; Williams, M.E.; Zhang, S.; Li, J.; Glassman, K.; Ranch, J.; Nubel, D.; Solawetz, W.; et al. A phenylalanine in DGAT is a key determinant of oil content and composition in maize. Nat. Genet. 2008, 40, 367–372. [Google Scholar] [CrossRef]
- Pouvreau, B.; Baud, S.; Vernoud, V.; Morin, V.; Py, C.; Gendrot, G.; Pichon, J.-P.; Rouster, J.; Paul, W.; Rogowsky, P.M. Duplicate Maize Wrinkled1 Transcription Factors Activate Target Genes Involved in Seed Oil Biosynthesis. Plant Physiol. 2011, 156, 674–686. [Google Scholar] [CrossRef]
- Baud, S.; Mendoza, M.S.; To, A.; Harscoët, E.; Lepiniec, L.; Dubreucq, B. WRINKLED1 specifies the regulatory action of LEAFY COTYLEDON2 towards fatty acid metabolism during seed maturation in Arabidopsis. Plant J. 2007, 50, 825–838. [Google Scholar] [CrossRef]
- Suzuki, M.; McCarty, D.R. Functional symmetry of the B3 network controlling seed development. Curr. Opin. Plant Biol. 2008, 11, 548–553. [Google Scholar] [CrossRef]
- Braybrook, S.A.; Harada, J.J. LECs go crazy in embryo development. Trends Plant Sci. 2008, 13, 624–630. [Google Scholar] [CrossRef]
- North, H.; Baud, S.; Debeaujon, I.; Dubos, C.; Dubreucq, B.; Grappin, P.; Jullien, M.; Lepiniec, L.; Marion-Poll, A.; Miquel, M. Arabidopsis seed secrets unravelled after a decade of genetic and omics-driven research. Plant J. 2010, 61, 971–981. [Google Scholar] [CrossRef]
- Shen, B.; Allen, W.B.; Zheng, P.; Li, C.; Glassman, K.; Ranch, J.; Nubel, D.; Tarczynski, M.C. Expression of ZmLEC1 and ZmWRI1 Increases Seed Oil Production in Maize. Plant Physiol. 2010, 153, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Martin, J.M.; Beecher, B.; Lu, C.; Hannah, L.C.; Wall, M.L.; Altosaar, I.; Giroux, M.J. The ectopic expression of the wheat Puroindoline genes increase germ size and seed oil content in transgenic corn. Plant Mol. Biol. 2010, 74, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Val, L.D.; Schwartz, S.H.; Kerns, M.R.; Deikman, J. Development of a High Oil Trait for Maize. In Molecular Genetic Approaches to Maize Improvement; Kriz, A.L., Larkins, B.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 303–323. [Google Scholar] [CrossRef]
- Ting, J.T.; Lee, K.; Ratnayake, C.; Platt, K.A.; Balsamo, R.A.; Huang, A.H. Oleosin genes in maize kernels having diverse oil contents are constitutively expressed independent of oil contents. Size and shape of intracellular oil bodies are determined by the oleosins/oils ratio. Planta 1996, 199, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Wang, L.S.; Shu, Q.Y.; Wu, J.; Chen, L.G.; Shao, S.; Yin, D.D. Fatty acid composition of developing tree peony (Paeonia section Moutan DC.) seeds and transcriptome analysis during seed development. BMC Genom. 2015, 16, 208. [Google Scholar] [CrossRef]
- Lin, P.; Wang, K.; Zhou, C.; Xie, Y.; Yao, X.; Yin, H. Seed Transcriptomics Analysis in Camellia oleifera Uncovers Genes Associated with Oil Content and Fatty Acid Composition. Int. J. Mol. Sci. 2018, 19, 118. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Y.; Chen, J.; Li, P.; Li, C.; Niu, G.; Jiang, L. Transcriptome analysis revealed the dynamic oil accumulation in Symplocos paniculata fruit. BMC Genom. 2016, 17, 929. [Google Scholar] [CrossRef]
- Kim, H.U.; Lee, K.-R.; Shim, D.; Lee, J.H.; Chen, G.Q.; Hwang, S. Transcriptome analysis and identification of genes associated with ω-3 fatty acid biosynthesis in Perilla frutescens (L.) var. frutescens. BMC Genom. 2016, 17, 474. [Google Scholar] [CrossRef]
- Shahid, M.; Cai, G.; Zu, F.; Zhao, Q.; Qasim, M.U.; Hong, Y.; Fan, C.; Zhou, Y. Comparative Transcriptome Analysis of Developing Seeds and Silique Wall Reveals Dynamic Transcription Networks for Effective Oil Production in Brassica napus L. Int. J. Mol. Sci. 2019, 20, 1982. [Google Scholar] [CrossRef]
- Teoh, K.T.; Requesens, D.V.; Devaiah, S.P.; Johnson, D.; Huang, X.; Howard, J.A.; Hood, E.E. Transcriptome analysis of embryo maturation in maize. BMC Plant Biol. 2013, 13, 19. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Lee, S.I.; Muthusamy, M.; Nawaz, M.A.; Hong, J.K.; Lim, M.H.; Kim, J.A.; Jeong, M.J. Genome-wide analysis of spatiotemporal gene expression patterns during floral organ development in Brassica rapa. Mol. Genet. Genom. MGG 2019, 294, 1403–1420. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Wang, Y.; Wang, C.; Lyu, J.; Wang, Y.; Dong, H.; Long, W.; Wang, D.; Kong, W.; Wang, L.; et al. ALR encoding dCMP deaminase is critical for DNA damage repair, cell cycle progression and plant development in rice. J. Exp. Bot. 2017, 68, 5773–5786. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Dossa, K.; Mmadi, M.A.; Zhou, R.; Zhang, T.; Su, R.; Zhang, Y.; Wang, L.; You, J.; Zhang, X. Depicting the core transcriptome modulating multiple abiotic stresses responses in sesame (Sesamum indicum L.). Int. J. Mol. Sci. 2019, 20, 3930. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Tzen, J.T.C.; Cao, Y.; Laurent, P.; Ratnayake, C.; Huang, A.H.C. Lipids, Proteins, and Structure of Seed Oil Bodies from Diverse Species. Plant Physiol. 1993, 101, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Ji, C.; Han, X.; Zhao, X.; Li, X.; Mao, Y.; Wong, L.L.; Bartlam, M.; Rao, Z. Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1). J. Mol. Biol. 2006, 357, 858–869. [Google Scholar] [CrossRef]
- Andre, C.; Haslam, R.P.; Shanklin, J. Feedback regulation of plastidic acetyl-CoA carboxylase by 18:1-acyl carrier protein in Brassica napus. Proc. Natl. Acad. Sci. USA 2012, 109, 10107–10112. [Google Scholar] [CrossRef]
- Cahoon, E.B.; Ohlrogge, J.B. Apparent Role of Phosphatidylcholine in the Metabolism of Petroselinic Acid in Developing Umbelliferae Endosperm. Plant Physiol. 1994, 104, 845–855. [Google Scholar] [CrossRef]
- Ohlrogge, J.; Browse, J. Lipid biosynthesis. Plant Cell 1995, 7, 957–970. [Google Scholar] [CrossRef]
- Chai, Y.; Hao, X.; Yang, X.; Allen, W.B.; Li, J.; Yan, J.; Shen, B.; Li, J. Validation of DGAT1-2 polymorphisms associated with oil content and development of functional markers for molecular breeding of high-oil maize. Mol. Breed. 2012, 29, 939–949. [Google Scholar] [CrossRef]
- Roesler, K.; Shintani, D.; Savage, L.; Boddupalli, S.; Ohlrogge, J. Targeting of the Arabidopsis homomeric acetyl-coenzyme A carboxylase to plastids of rapeseeds. Plant Physiol. 1997, 113, 75–81. [Google Scholar] [CrossRef]
- Mhaske, V.; Beldjilali, K.; Ohlrogge, J.; Pollard, M. Isolation and characterization of an Arabidopsis thaliana knockout line for phospholipid: Diacylglycerol transacylase gene (At5g13640). Plant Physiol. Biochem. 2005, 43, 413–417. [Google Scholar] [CrossRef]
- Zamora-Briseno, J.A.; Pereira-Santana, A.; Reyes-Hernandez, S.J.; Castano, E.; Rodriguez-Zapata, L.C. Global Dynamics in Protein Disorder during Maize Seed Development. Genes 2019, 10, 502. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Hong, M.; Wan, H.; Luo, L.; Yu, Z.; Guo, R. Identification of Key Genes Involved in Embryo Development and Differential Oil Accumulation in Two Contrasting Maize Genotypes. Genes 2019, 10, 993. https://doi.org/10.3390/genes10120993
Zhang X, Hong M, Wan H, Luo L, Yu Z, Guo R. Identification of Key Genes Involved in Embryo Development and Differential Oil Accumulation in Two Contrasting Maize Genotypes. Genes. 2019; 10(12):993. https://doi.org/10.3390/genes10120993
Chicago/Turabian StyleZhang, Xiangxiang, Meiyan Hong, Heping Wan, Lixia Luo, Zeen Yu, and Ruixing Guo. 2019. "Identification of Key Genes Involved in Embryo Development and Differential Oil Accumulation in Two Contrasting Maize Genotypes" Genes 10, no. 12: 993. https://doi.org/10.3390/genes10120993
APA StyleZhang, X., Hong, M., Wan, H., Luo, L., Yu, Z., & Guo, R. (2019). Identification of Key Genes Involved in Embryo Development and Differential Oil Accumulation in Two Contrasting Maize Genotypes. Genes, 10(12), 993. https://doi.org/10.3390/genes10120993