Effects of Substrate-Binding Site Residues on the Biochemical Properties of a Tau Class Glutathione S-Transferase from Oryza sativa

Abstract
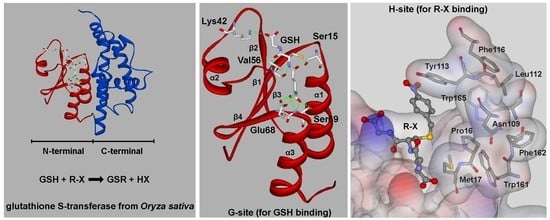
1. Introduction
2. Materials and Methods
2.1. Protein Sequence Alignments and Structural Modeling of the OsGSTU17 Protein
2.2. Site-Directed Mutagenesis of OsGSTU17
2.3. Prokaryotic Expression and Purification of the Recombinant Mutant Proteins
2.4. Substrate Activity
2.5. Kinetic Parameters
2.6. Thermal Stability
3. Results
3.1. Identification of Amino Acids Present at the G- and H-Sites of the OsGSTU17 Protein
3.2. Expression and Purification of the Mutant Proteins
3.3. Substrate Activity of Mutant Proteins
3.4. Kinetic Characterization of the Mutant Proteins
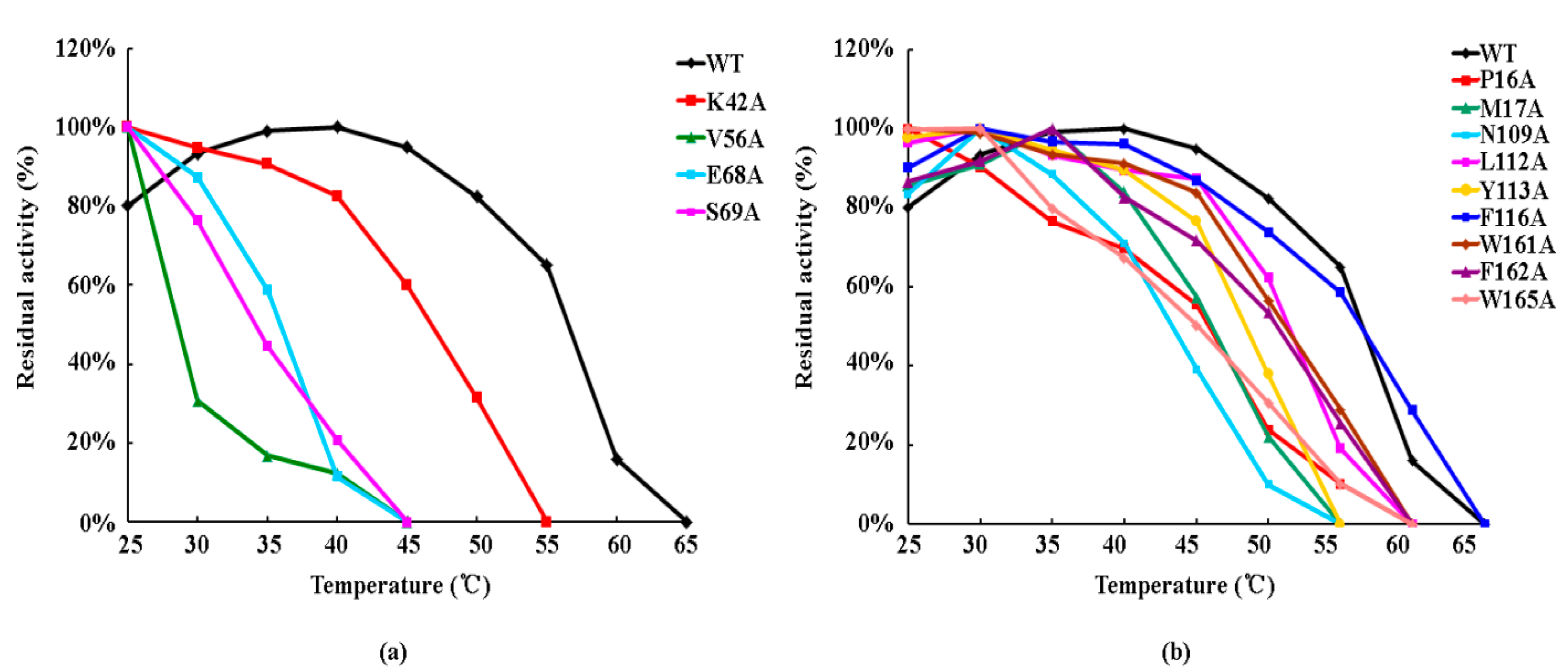
3.5. Effect of Side-Chain Groups on Thermal Stability of the Enzymes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Edwards, R.; Dixon, D.P.; Walbot, V. Plant glutathione S-transferases: Enzymes with multiple functions in sickness and in health. Trends Plant Sci. 2000, 5, 193–198. [Google Scholar] [CrossRef]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Moons, A. Regulatory and functional interactions of plant growth regulators and plant glutathione S-transferases (GSTs). Vitam. Horm. 2005, 72, 155–202. [Google Scholar] [PubMed]
- Cummins, I.; Dixon, D.P.; Freitag-Pohl, S.; Skipsey, M.; Edwards, R. Multiple roles for plant glutathione transferases in xenobiotic detoxification. Drug Metab. Rev. 2011, 43, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.P.; Lapthorn, A.; Edwards, R. Plant glutathione transferases. Genome Biol. 2002, 3, 3004. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frova, C. The plant glutathione transferase gene family: Genomic structure, functions, expression and evolution. Physiol. Plant. 2003, 119, 469–479. [Google Scholar] [CrossRef]
- Barozzi, F.; Di Sansebastiano, G.P.; Sabella, E.; Aprile, A.; Piro, G.; De Bellis, L.; Nutricati, E. Glutathione S-transferase related detoxification processes are correlated with receptor-mediated vacuolar sorting mechanisms. Plant Cell Rep. 2017, 36, 1361–1373. [Google Scholar] [CrossRef]
- Ding, N.; Wang, A.; Zhang, X.; Wu, Y.; Wang, R.; Cui, H.; Huang, R.; Luo, Y. Identification and analysis of glutathione S-transferase gene family in sweet potato reveal divergent GST-mediated networks in aboveground and underground tissues in response to abiotic stresses. BMC Plant Biol. 2017, 17, 225. [Google Scholar] [CrossRef]
- Ismaiel, A.A.; Papenbrock, J. Effect of Patulin from Penicillium vulpinum on the Activity of Glutathione-S-Transferase and Selected Antioxidative Enzymes in Maize. Int. J. Environ. Res. Public Health 2017, 14, 825. [Google Scholar] [CrossRef]
- Khan, N.; Hu, C.M.; Amjad Khan, W.; Hou, X. Genome-Wide Identification, Classification, and Expression Divergence of Glutathione-Transferase Family in Brassica rapa under Multiple Hormone Treatments. Biomed. Res. Int 2018, 2018, 6023457. [Google Scholar] [CrossRef]
- Neuefeind, T.; Reinemer, P.; Bieseler, B. Plant glutathione S-transferases and herbicide detoxification. Biol. Chem. 1997, 378, 199–205. [Google Scholar] [PubMed]
- Pislewska-Bednarek, M.; Nakano, R.T.; Hiruma, K.; Pastorczyk, M.; Sanchez-Vallet, A.; Singkaravanit-Ogawa, S.; Ciesiolka, D.; Takano, Y.; Molina, A.; Schulze-Lefert, P.; et al. Glutathione Transferase U13 Functions in Pathogen-Triggered Glucosinolate Metabolism. Plant Physiol. 2018, 176, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xing, X.J.; Tian, Y.S.; Peng, R.H.; Xue, Y.; Zhao, W.; Yao, Q.H. Transgenic Arabidopsis Plants Expressing Tomato Glutathione S-Transferase Showed Enhanced Resistance to Salt and Drought Stress. PLoS ONE 2015, 10, e0136960. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Li, H.L.; Guo, D.; Wang, Y.; Dai, H.F.; Mei, W.L.; Peng, S.Q. Transcriptome-wide identification and expression analysis of glutathione S-transferase genes involved in flavonoids accumulation in Dracaena cambodiana. Plant Physiol. Biochem. 2016, 104, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Lo Cicero, L.; Madesis, P.; Tsaftaris, A.; Lo Piero, A.R. Tobacco plants over-expressing the sweet orange tau glutathione transferases (CsGSTUs) acquire tolerance to the diphenyl ether herbicide fluorodifen and to salt and drought stresses. Phytochemistry 2015, 116, 69–77. [Google Scholar] [CrossRef]
- Lo Cicero, L.; Catara, V.; Strano, C.P.; Bella, P.; Madesis, P.; Lo Piero, A.R. Over-expression of CsGSTU promotes tolerance to the herbicide alachlor and resistance to Pseudomonas syringae pv. tabaci in transgenic tobacco. Biol. Plant. 2017, 61, 169–177. [Google Scholar] [CrossRef]
- Dixon, D.P.; McEwen, A.G.; Lapthorn, A.J.; Edwards, R. Forced evolution of a herbicide detoxifying glutathione transferase. J. Biol. Chem. 2003, 278, 23930–23935. [Google Scholar] [CrossRef]
- Kapoli, P.; Axarli, I.A.; Platis, D.; Fragoulaki, M.; Paine, M.; Hemingway, J.; Vontas, J.; Labrou, N.E. Engineering sensitive glutathione transferase for the detection of xenobiotics. Biosens. Bioelectron. 2008, 24, 498–503. [Google Scholar] [CrossRef]
- Labrou, N.E.; Kotzia, G.A.; Clonis, Y.D. Engineering the xenobiotic substrate specificity of maize glutathione S-transferase I. Protein Eng. Des. Sel. 2004, 17, 741–748. [Google Scholar] [CrossRef]
- Lan, T.; Yang, Z.L.; Yang, X.; Liu, Y.J.; Wang, X.R.; Zeng, Q.Y. Extensive functional diversification of the Populus glutathione S-transferase supergene family. Plant Cell 2009, 21, 3749–3766. [Google Scholar] [CrossRef]
- Liu, Y.J.; Han, X.M.; Ren, L.L.; Yang, H.L.; Zeng, Q.Y. Functional divergence of the glutathione S-transferase supergene family in Physcomitrella patens reveals complex patterns of large gene family evolution in land plants. Plant Physiol. 2013, 161, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, Y.J.; Zeng, Q.Y. Biochemical functions of the glutathione transferase supergene family of Larix kaempferi. Plant Physiol. Biochem. 2014, 77, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, Y.; Xia, D.; Gao, C.; Chao, W.; Yang, C. Overexpression of a GST gene ( ThGSTZ1 ) from Tamarix hispida improves drought and salinity tolerance by enhancing the ability to scavenge reactive oxygen species. Plant Cell Tissue Organ Cult. 2014, 117, 99–112. [Google Scholar] [CrossRef]
- Liu, H.J.; Tang, Z.X.; Han, X.M.; Yang, Z.L.; Zhang, F.M.; Yang, H.L.; Liu, Y.J.; Zeng, Q.Y. Divergence in Enzymatic Activities in the Soybean GST Supergene Family Provides New Insight into the Evolutionary Dynamics of Whole-Genome Duplicates. Mol. Biol. Evol. 2015, 32, 2844–2859. [Google Scholar] [CrossRef]
- Nianiou-Obeidat, I.; Madesis, P.; Kissoudis, C.; Voulgari, G.; Chronopoulou, E.; Tsaftaris, A.; Labrou, N.E. Plant glutathione transferase-mediated stress tolerance: Functions and biotechnological applications. Plant Cell Rep. 2017, 36, 791–805. [Google Scholar] [CrossRef]
- Axarli, I.; Dhavala, P.; Papageorgiou, A.C.; Labrou, N.E. Crystallographic and functional characterization of the fluorodifen-inducible glutathione transferase from Glycine max reveals an active site topography suited for diphenylether herbicides and a novel L-site. J. Mol. Biol. 2009, 385, 984–1002. [Google Scholar] [CrossRef]
- Dirr, H.; Reinemer, P.; Huber, R. X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function. Eur. J. Biochem. 1994; 220, 645–661. [Google Scholar]
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef]
- Armstrong, R.N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997, 10, 2–18. [Google Scholar] [CrossRef]
- Valenzuela-Chavira, I.; Contreras-Vergara, C.A.; Arvizu-Flores, A.A.; Serrano-Posada, H.; Lopez-Zavala, A.A.; Garcia-Orozco, K.D.; Hernandez-Paredes, J.; Rudino-Pinera, E.; Stojanoff, V.; Sotelo-Mundo, R.R.; et al. Insights into ligand binding to a glutathione S-transferase from mango: Structure, thermodynamics and kinetics. Biochimie 2017, 135, 35–45. [Google Scholar] [CrossRef]
- Yang, X.; Sun, W.; Liu, J.P.; Liu, Y.J.; Zeng, Q.Y. Biochemical and physiological characterization of a tau class glutathione transferase from rice (Oryza sativa). Plant Physiol. Biochem. 2009, 47, 1061–1068. [Google Scholar] [CrossRef]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [PubMed]
- Ricci, G.; Caccuri, A.M.; Lo Bello, M.; Pastore, A.; Piemonte, F.; Federici, G. Colorimetric and fluorometric assays of glutathione transferase based on 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole. Anal. Biochem. 1994, 218, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.; Dixon, D.P. Plant glutathione transferases. Methods Enzym. 2005, 401, 169–186. [Google Scholar]
- Zeng, Q.Y.; Wang, X.R. Catalytic properties of glutathione-binding residues in a tau class glutathione transferase (PtGSTU1) from Pinus tabulaeformis. FEBS Lett. 2005, 579, 2657–2662. [Google Scholar] [CrossRef]
- Lan, T.; Wang, X.R.; Zeng, Q.Y. Structural and functional evolution of positively selected sites in pine glutathione S-transferase enzyme family. J. Biol. Chem. 2013, 288, 24441–24451. [Google Scholar] [CrossRef]
- Federici, L.; Lo Sterzo, C.; Pezzola, S.; Di Matteo, A.; Scaloni, F.; Federici, G.; Caccuri, A.M. Structural basis for the binding of the anticancer compound 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol to human glutathione s-transferases. Cancer Res. 2009, 69, 8025–8034. [Google Scholar] [CrossRef]
- Patskovsky, Y.; Patskovska, L.; Almo, S.C.; Listowsky, I. Transition state model and mechanism of nucleophilic aromatic substitution reactions catalyzed by human glutathione S-transferase M1a-1a. Biochemistry 2006, 45, 3852–3862. [Google Scholar] [CrossRef]
- Gutin, A.M.; Abkevich, V.I.; Shakhnovich, E.I. Is burst hydrophobic collapse necessary for protein folding? Biochemistry 1995, 34, 3066–3076. [Google Scholar] [CrossRef]
- Johnson, W.W.; Liu, S.; Ji, X.; Gilliland, G.L.; Armstrong, R.N. Tyrosine 115 participates both in chemical and physical steps of the catalytic mechanism of a glutathione S-transferase. J. Biol. Chem. 1993, 268, 11508–11511. [Google Scholar]
- Lo Piero, A.R.; Mercurio, V.; Puglisi, I.; Petrone, G. Different roles of functional residues in the hydrophobic binding site of two sweet orange tau glutathione S-transferases. FEBS J. 2010, 277, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Axarli, I.; Dhavala, P.; Papageorgiou, A.C.; Labrou, N.E. Crystal structure of Glycine max glutathione transferase in complex with glutathione: Investigation of the mechanism operating by the Tau class glutathione transferases. Biochem. J. 2009, 422, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Shokeer, A.; Mannervik, B. Minor modifications of the C-terminal helix reschedule the favored chemical reactions catalyzed by theta class glutathione transferase T1-1. J. Biol. Chem. 2010, 285, 5639–5645. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulou, E.G.; Labrou, N.E. Glutathione transferases: Emerging multidisciplinary tools in red and green biotechnology. Recent Pat. Biotechnol. 2009, 3, 211–223. [Google Scholar] [CrossRef]
- Perperopoulou, F.; Pouliou, F.; Labrou, N.E. Recent advances in protein engineering and biotechnological applications of glutathione transferases. Crit. Rev. Biotechnol. 2018, 38, 511–528. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NBD-Cl | CDNB | NBC | Cumene Hydroperoxide |
|---|---|---|---|
 |  |  |  |
| Substrate | Specific Activity (μmol min−1 mg−1) | |||||
|---|---|---|---|---|---|---|
| WT [31] | S15A [31] | K42A | V56A | E68A | S69A | |
| NBD-Cl (10 mM) | 0.203 ± 0.006 | ND | <0.01 | <0.01 | 0.016 ± 0.002 | <0.01 |
| CDNB (60 mM) | 0.113 ± 0.019 | ND | 0.011 ± 0.001 | 0.024 ± 0.002 | 0.029 ± 0.002 | 0.035 ± 0.002 |
| NBC (60 mM) | 1.153 ± 0.046 | ND | ND | 0.082 ± 0.014 | 0.063 ± 0.010 | 0.101 ± 0.016 |
| Cum-OOH (69 mM) | 0.013 ± 0.002 | ND | <0.01 | ND | <0.01 | ND |
| Substrate | Specific Activity (μmol∙min−1∙mg−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WT [31] | P16A | M17A | N109A | L112A | Y113A | F116A | W161A | F162A | W165A | |
| NBD-Cl (10 mM) | 0.203 ± 0.006 | 0.041 ± 0.002 | 0.183 ± 0.001 | 0.209 ± 0.002 | 0.380 ± 0.015 | 0.224 ± 0.005 | 0.246 ± 0.005 | 0.267 ± 0.003 | 0.192 ± 0.001 | 0.049 ± 0.009 |
| CDNB (60 mM) | 0.113 ± 0.019 | 0.349 ± 0.008 | 0.159 ± 0.003 | 0.189 ± 0.014 | 0.245 ± 0.001 | 0.129 ± 0.005 | 0.138 ± 0.001 | 0.347 ± 0.001 | 0.129 ± 0.001 | 0.080 ± 0.003 |
| NBC (60 mM) | 1.153 ± 0.046 | 0.017 ± 0.015 | 0.081 ± 0.012 | 0.083 ± 0.004 | 0.237± 0.040 | 0.069 ± 0.024 | 0.088 ± 0.003 | 0.164 ± 0.092 | 0.081 ± 0.001 | 0.189 ± 0.049 |
| Cum-OOH (69 mM) | 0.013 ± 0.002 | 0.081 ± 0.008 | 0.034 ± 0.011 | 0.032 ± 0.002 | 0.066 ± 0.004 | 0.032 ± 0.001 | 0.033 ± 0.003 | 0.037 ± 0.002 | 0.053 ± 0.002 | 0.028 ± 0.003 |
| GSH | NBD-Cl | |||||||
|---|---|---|---|---|---|---|---|---|
| Km | Vmax | kcat | kcat/Km | Km | Vmax | kcat | kcat/Km | |
| (mM) | (μM∙min−1∙mg−1) | (S−1) | (mM−1∙S−1) | (mM) | (μM∙min−1∙mg−1) | (S−1) | (mM−1∙S−1) | |
| WT [31] | 0.058 ± 0.006 | 0.225 ± 0.011 | 0.264 | 4.552 | 0.324 ± 0.016 | 0.219 ± 0.006 | 0.257 | 0.793 |
| M17A | 0.743 ± 0.049 | 0.397 ± 0.018 | 0.387 | 0.521 | 3.661 ± 0.843 | 0.830 ± 0.168 | 0.809 | 0.221 |
| N109A | 0.192 ± 0.003 | 0.268 ± 0.003 | 0.196 | 1.019 | 0.781 ± 0.034 | 0.364 ± 0.008 | 0.266 | 0.341 |
| L112A | 0.307 ± 0.010 | 0.485 ± 0.001 | 0.946 | 1.579 | 1.487 ± 0.079 | 0.896 ± 0.019 | 1.750 | 1.177 |
| Y113A | 0.120 ± 0.004 | 0.260 ± 0.003 | 0.220 | 1.842 | 0.471 ± 0.012 | 0.327 ± 0.008 | 0.277 | 0.589 |
| F116A | 0.166 ± 0.007 | 0.286 ± 0.005 | 0.322 | 1.942 | 0.882 ± 0.028 | 0.455 ± 0.014 | 0.512 | 0.581 |
| W161A | 0.221 ± 0.005 | 0.341 ± 0.006 | 0.573 | 2.588 | 1.117 ± 0.022 | 0.594 ± 0.011 | 0.999 | 0.894 |
| F162A | 0.360 ± 0.013 | 0.233 ± 0.003 | 0.421 | 1.170 | 3.866 ± 0.871 | 0.869 ± 0.198 | 1.573 | 0.407 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Wei, J.; Wu, Z.; Gao, J. Effects of Substrate-Binding Site Residues on the Biochemical Properties of a Tau Class Glutathione S-Transferase from Oryza sativa. Genes 2020, 11, 25. https://doi.org/10.3390/genes11010025
Yang X, Wei J, Wu Z, Gao J. Effects of Substrate-Binding Site Residues on the Biochemical Properties of a Tau Class Glutathione S-Transferase from Oryza sativa. Genes. 2020; 11(1):25. https://doi.org/10.3390/genes11010025
Chicago/Turabian StyleYang, Xue, Jinchi Wei, Zhihai Wu, and Jie Gao. 2020. "Effects of Substrate-Binding Site Residues on the Biochemical Properties of a Tau Class Glutathione S-Transferase from Oryza sativa" Genes 11, no. 1: 25. https://doi.org/10.3390/genes11010025
APA StyleYang, X., Wei, J., Wu, Z., & Gao, J. (2020). Effects of Substrate-Binding Site Residues on the Biochemical Properties of a Tau Class Glutathione S-Transferase from Oryza sativa. Genes, 11(1), 25. https://doi.org/10.3390/genes11010025