TILLING by Sequencing: A Successful Approach to Identify Rare Alleles in Soybean Populations


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Growth Conditions
2.2. DNA Extraction, Amplification, Sequencing, and Mutant Identification
2.3. Sequence Analysis
2.4. SNP Assays
2.5. Carbohydrate Measurement
3. Results
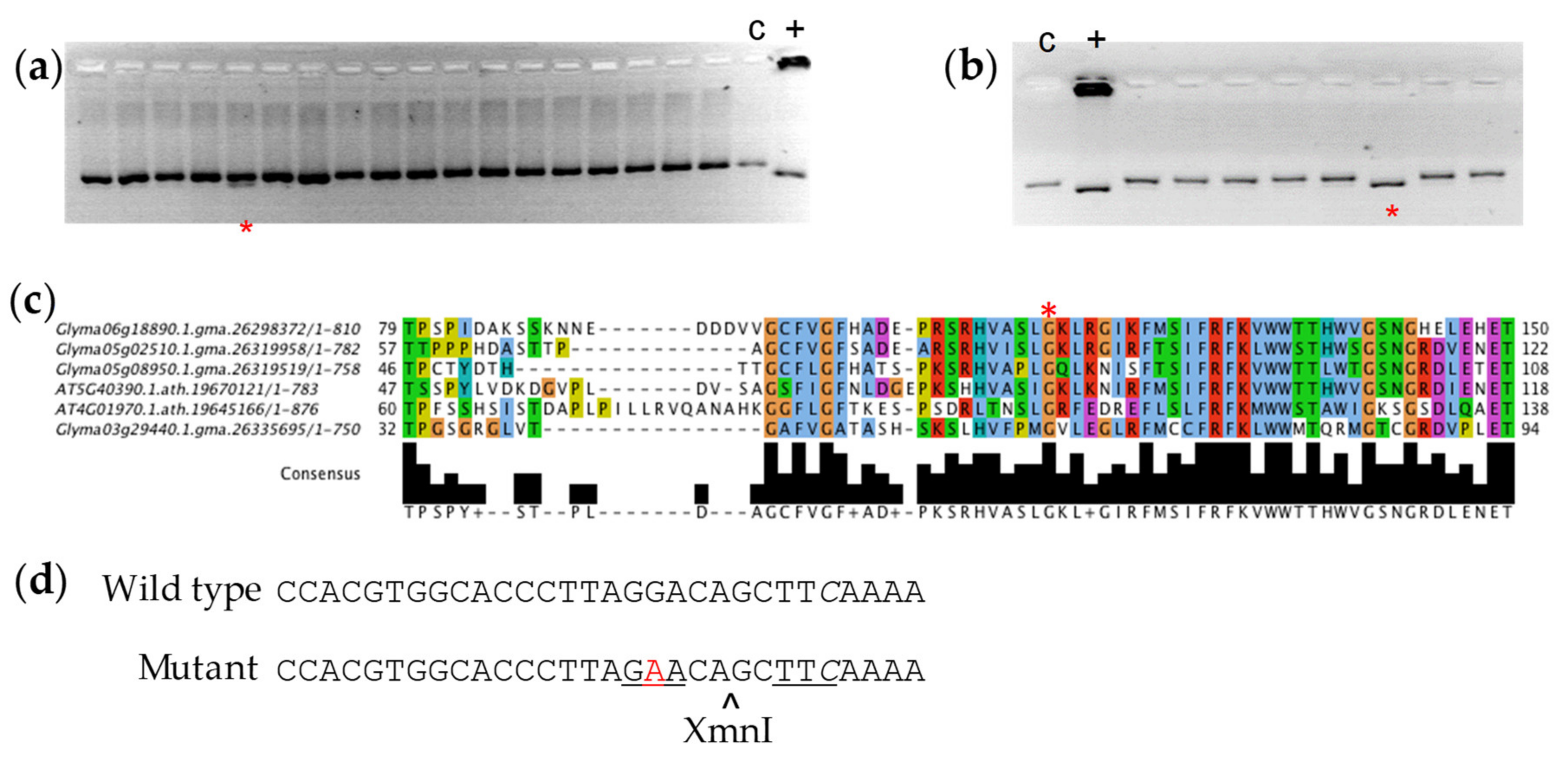
3.1. Sequencing of RS2 and RS3 Amplicons
3.2. Polymorphism Validation
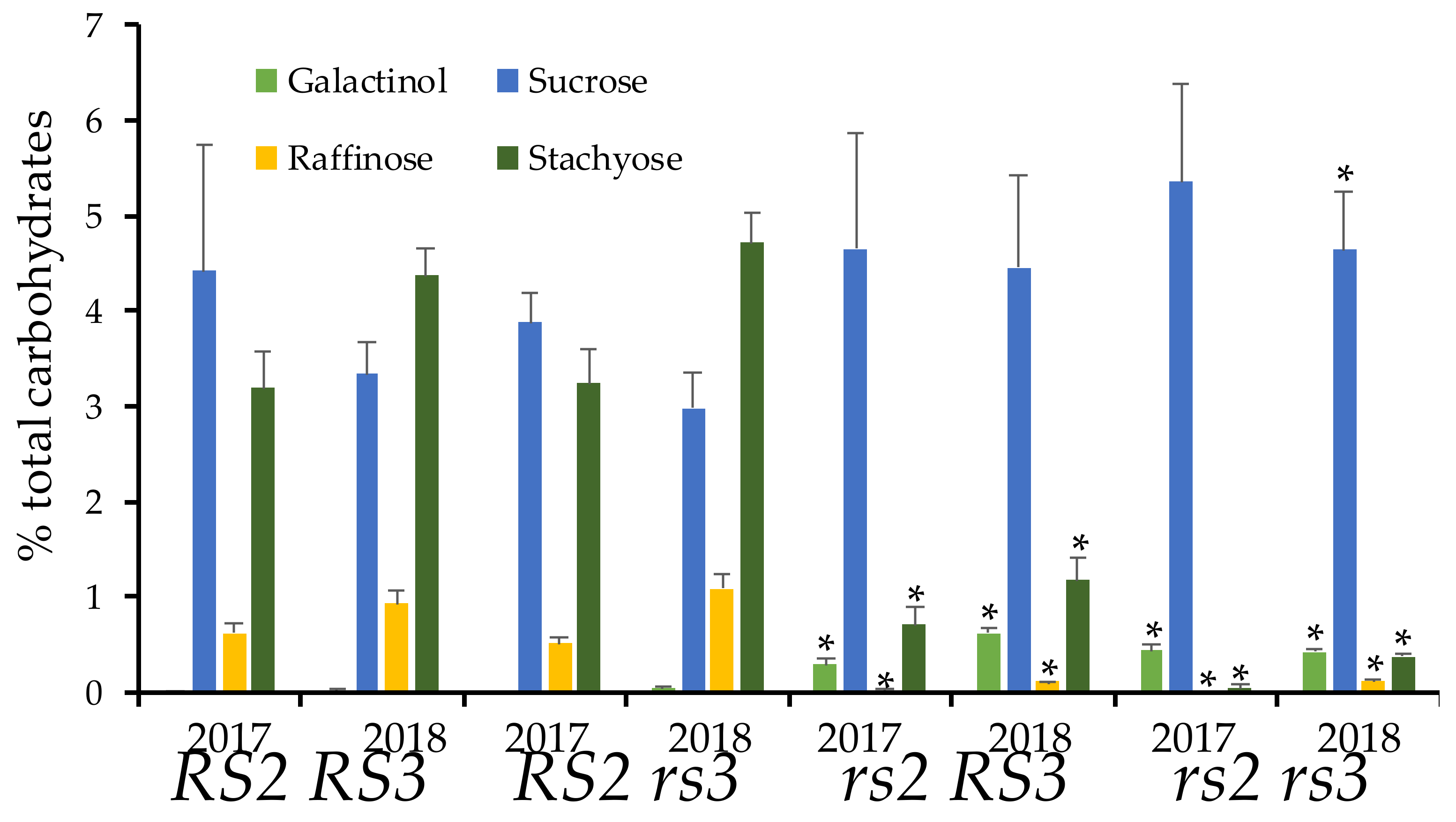
3.3. Low RFO Phenotype in rs2rs3 Double Mutants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- www.soystats.com. Soystats.com. Available online: http://www.soystats.com/2012/Default-frames.htm (accessed on 19 October 2019).
- Naczk, M.; Amarowicz, R.; Shahidi, F. α-Galactosides of Sucrose in Foods: Composition, Flatulence-Causing Effects, and Removal. In Antinutrients and Phytochemicals in Food; American Chemical Society: Washington, DC, USA, 1997; Volume 662, pp. 127–151. [Google Scholar]
- Hartwig, E.E.; Kuo, T.M.; Kenty, M.M. Seed Protein and its Relationship to Soluble Sugars in Soybean. Crop Sci. 1997, 37, 770–773. [Google Scholar] [CrossRef]
- Valentine, M.F.; De Tar, J.R.; Mookkan, M.; Firman, J.D.; Zhang, Z.J. Silencing of Soybean Raffinose Synthase Gene Reduced Raffinose Family Oligosaccharides and Increased True Metabolizable Energy of Poultry Feed. Front. Plant Sci. 2017, 8, 692. [Google Scholar] [CrossRef] [PubMed]
- Karr-Lilienthal, L.K.; Kadzere, C.T.; Grieshop, C.M.; Fahey, G.C. Chemical and nutritional properties of soybean carbohydrates as related to nonruminants: A review. Livest. Prod. Sci. 2005, 97, 1–12. [Google Scholar] [CrossRef]
- Dierking, E.C.; Bilyeu, K.D. Association of a Soybean Raffinose Synthase Gene with Low Raffinose and Stachyose Seed Phenotype. Plant Genome 2008, 1, 135–145. [Google Scholar] [CrossRef]
- Dierking, E.C.; Bilyeu, K.D. New sources of soybean seed meal and oil composition traits identified through TILLING. BMC Plant Biol. 2009, 9. [Google Scholar] [CrossRef]
- Jo, H.; Lorenz, A.J.; Rainey, K.M.; Shannon, J.G.; Chen, P.Y.; Bilyeu, K.D. Environmental Stability Study of Soybeans with Modified Carbohydrate Profiles in Maturity Groups 0 to V. Crop Sci. 2019, 59, 1531–1543. [Google Scholar] [CrossRef]
- Jo, H.; Lee, J.D.; Bilyeu, K.D. Environmental Stability of Carbohydrate Profiles in Different Soybean Genotypes. Crop Sci. 2018, 58, 773–782. [Google Scholar] [CrossRef]
- Bilyeu, K.D.; Wiebold, W.J. Environmental Stability of Seed Carbohydrate Profiles in Soybeans Containing Different Alleles of the Raffinose Synthase 2 (RS2) Gene. J. Agric. Food Chem. 2016, 64, 1071–1078. [Google Scholar] [CrossRef]
- Schillinger, J.A.; Dierking, E.C.; Bilyeu, K.D. Soybeans Having High Germination Rate and Ultra-Low Raffinose and Stachyose Content. U.S. Patent No 8471 107, 21 June 2013. [Google Scholar]
- Hagely, K.B.; Palmquist, D.; Bilyeu, K.D. Classification of Distinct Seed Carbohydrate Profiles in Soybean. J. Agr. Food Chem. 2013, 61, 1105–1111. [Google Scholar] [CrossRef]
- Leske, K.L.; Akavanichan, O.; Cheng, T.K.; Coon, C.N. Effect of Ethanol Extract on Nitrogen-Corrected True Metabolizable Energy for Soybean-Meal with Broilers and Roosters. Poult. Sci. 1991, 70, 892–895. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, Y.; Wang, L.L.; Liu, X.; Liu, Y.X.; Phillips, J.; Deng, X. A WRKY transcription factor participates in dehydration tolerance in Boea hygrometrica by binding to the W-box elements of the galactinol synthase (BhGolS1) promoter. Planta 2009, 230, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Koster, K.L.; Leopold, A.C. Sugars and Desiccation Tolerance in Seeds. Plant Physiol. 1988, 88, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Zuther, E.; Buchel, K.; Hundertmark, M.; Stitt, M.; Hincha, D.K.; Heyer, A.G. The role of raffinose in the cold acclimation response of Arabidopsis thaliana. FEBS Lett. 2004, 576, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Blochl, A.; Peterbauer, T.; Richter, A. Inhibition of raffinose oligosaccharide breakdown delays germination of pea seeds. J. Plant Physiol. 2007, 164, 1093–1096. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Y.M.; Wang, D.; Liu, Y.; Dirk, L.M.A.; Goodman, J.; Downie, A.B.; Wang, J.M.; Wang, G.Y.; Zhao, T.Y. Regulation of Seed Vigor by Manipulation of Raffinose Family Oligosaccharides in Maize and Arabidopsis thaliana. Mol. Plant 2017, 10, 1540–1555. [Google Scholar] [CrossRef]
- Gangl, R.; Tenhaken, R. Raffinose Family Oligosaccharides Act As Galactose Storesin Seeds and Are Required for Rapid Germination of Arabidopsisin the Dark. Front. Plant Sci. 2016, 7, 1115. [Google Scholar] [CrossRef]
- Dierking, E.C.; Bilyeu, K.D. Raffinose and stachyose metabolism are not required for efficient soybean seed germination. J. Plant Physiol. 2009, 166, 1329–1335. [Google Scholar] [CrossRef]
- La, T.; Large, E.; Taliercio, E.; Song, Q.; Gillman, J.D.; Xu, D.; Nguyen, H.T.; Shannon, G.; Scaboo, A. Characterization of Select Wild Soybean Accessions in the USDA Germplasm Collection for Seed Composition and Agronomic Traits. Crop Sci. 2019, 59, 233–251. [Google Scholar] [CrossRef]
- Cooper, J.L.; Till, B.J.; Laport, R.G.; Darlow, M.C.; Kleffner, J.M.; Jamai, A.; El-Mellouki, T.; Liu, S.; Ritchie, R.; Nielsen, N.; et al. TILLING to detect induced mutations in soybean. BMC Plant Biol. 2008, 8, 9. [Google Scholar] [CrossRef]
- Hoshino, T.; Watanabe, S.; Takagi, Y.; Anai, T. A novel GmFAD3-2a mutant allele developed through TILLING reduces alpha-linolenic acid content in soybean seed oil. Breed. Sci. 2014, 64, 371–377. [Google Scholar] [CrossRef]
- Lakhssassi, N.; Zhou, Z.; Liu, S.; Colantonio, V.; AbuGhazaleh, A.; Meksem, K. Characterization of the FAD2 Gene Family in Soybean Reveals the Limitations of Gel-Based TILLING in Genes with High Copy Number. Front. Plant Sci. 2017, 8, 324. [Google Scholar] [CrossRef]
- Hoshino, T.; Takagi, Y.; Anai, T. Novel GmFAD2-1b mutant alleles created by reverse genetics induce marked elevation of oleic acid content in soybean seeds in combination with GmFAD2-1a mutant alleles. Breed. Sci. 2010, 60, 419–425. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.; Howell, T.; Nitcher, R.; Missirian, V.; Watson, B.; Ngo, K.J.; Lieberman, M.; Fass, J.; Uauy, C.; Tran, R.K.; et al. Discovery of rare mutations in populations: TILLING by sequencing. Plant Physiol. 2011, 156, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.; Wiley, H.; Szymanski, D.; Nielsen, N. Targeting Induced Local Lesions in Genomes—TILLING. In Legume Crop Genomics; Wilson, R.F., Stalker, H.T., Brummer, E.C., Eds.; AOCS Press: Champaign, IL, USA, 2004; pp. 194–203. [Google Scholar]
- Thapa, R.; Carrero-Colón, M.; Addo-Quaye, C.; Held, J.; Dilkes, B.; Hudson, K.A. New Alleles of FAD3A Lower the Linolenic Acid Content of Soybean Seeds. Crop Sci. 2018, 58, 713–718. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Neff, M.M.; Neff, J.D.; Chory, J.; Pepper, A.E. dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: Experimental applications in Arabidopsis thaliana genetics. Plant J. 1998, 14, 387–392. [Google Scholar] [CrossRef]
- Hagely, K.B.; Jo, H.; Kim, J.-H.; Hudson, K.A.; Billyeu, K. Molecular assisted breeding for improved carbohydrate profiles in soybean seed. Theor. Appl. Genet. 2019, in press. [Google Scholar]
- Guo, Y.; Abernathy, B.; Zeng, Y.; Ozias-Akins, P. TILLING by sequencing to identify induced mutations in stress resistance genes of peanut (Arachis hypogaea). BMC Genom. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-I.; Tai, T.H. Identification of novel rice low phytic acid mutations via TILLING by sequencing. Mol. Breed. 2014, 34, 1717–1729. [Google Scholar] [CrossRef]
- Potapov, V.; Ong, J.L. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE 2017, 12, e0169774. [Google Scholar] [CrossRef] [PubMed]
- Shagin, D.A.; Shagina, I.A.; Zaretsky, A.R.; Barsova, E.V.; Kelmanson, I.V.; Lukyanov, S.; Chudakov, D.M.; Shugay, M. A high-throughput assay for quantitative measurement of PCR errors. Sci. Rep. 2017, 7, 2718. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapa, R.; Carrero-Colón, M.; Rainey, K.M.; Hudson, K. TILLING by Sequencing: A Successful Approach to Identify Rare Alleles in Soybean Populations. Genes 2019, 10, 1003. https://doi.org/10.3390/genes10121003
Thapa R, Carrero-Colón M, Rainey KM, Hudson K. TILLING by Sequencing: A Successful Approach to Identify Rare Alleles in Soybean Populations. Genes. 2019; 10(12):1003. https://doi.org/10.3390/genes10121003
Chicago/Turabian StyleThapa, Rima, Militza Carrero-Colón, Katy M. Rainey, and Karen Hudson. 2019. "TILLING by Sequencing: A Successful Approach to Identify Rare Alleles in Soybean Populations" Genes 10, no. 12: 1003. https://doi.org/10.3390/genes10121003
APA StyleThapa, R., Carrero-Colón, M., Rainey, K. M., & Hudson, K. (2019). TILLING by Sequencing: A Successful Approach to Identify Rare Alleles in Soybean Populations. Genes, 10(12), 1003. https://doi.org/10.3390/genes10121003