Mitotic Recombination and Adaptive Genomic Changes in Human Pathogenic Fungi

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sources of Mitotic DSBs
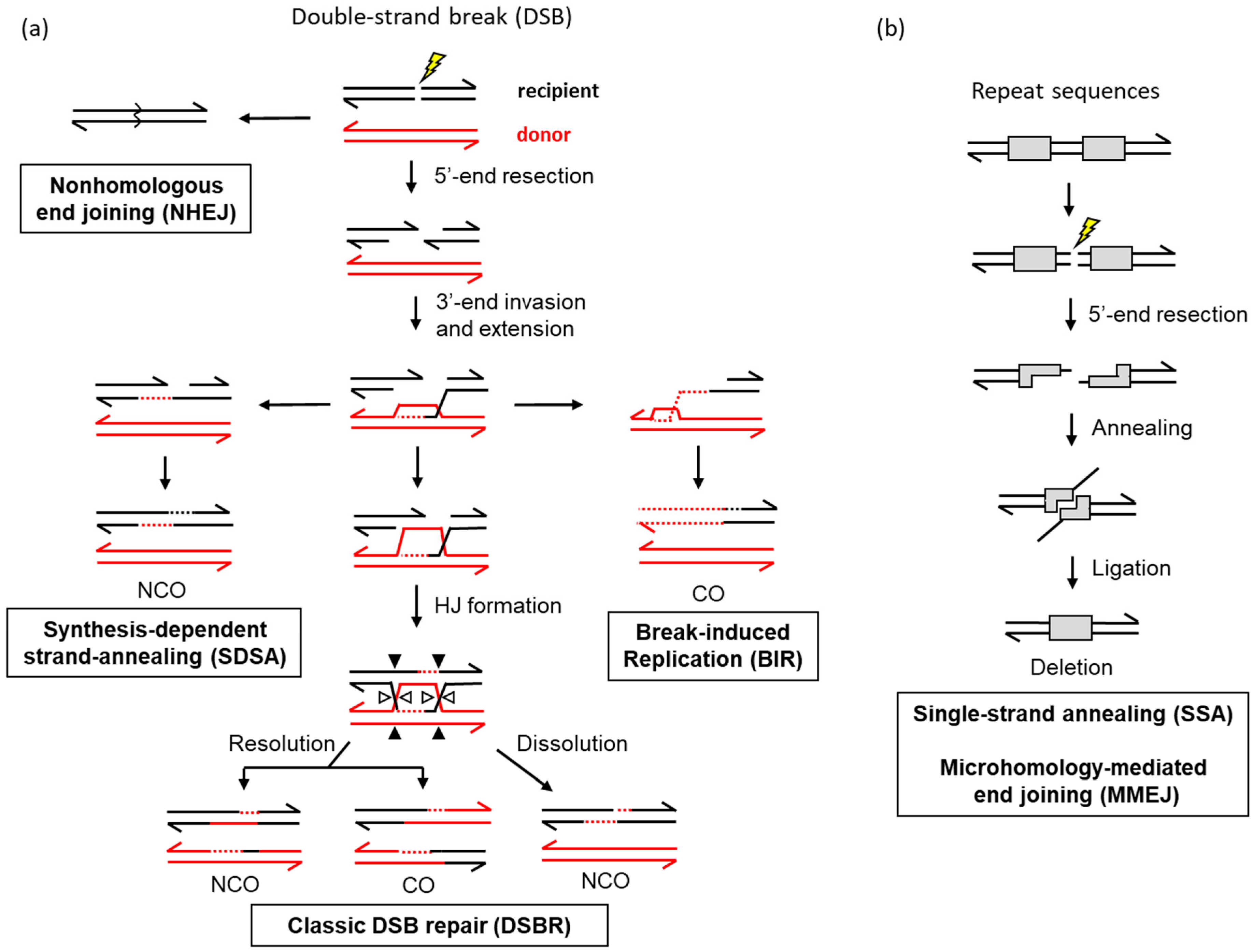
3. Mechanisms of Homologous Recombination in Yeast
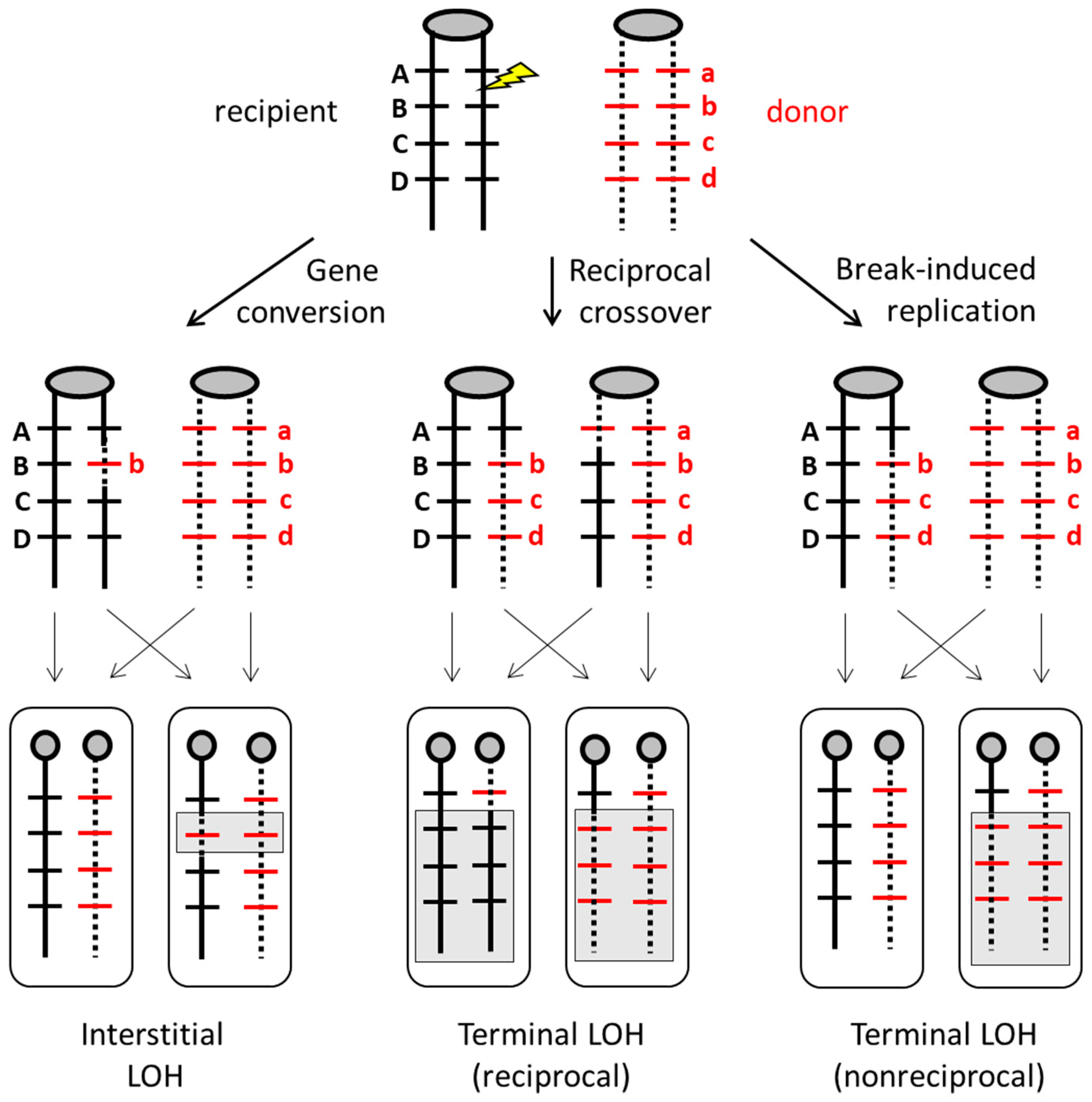
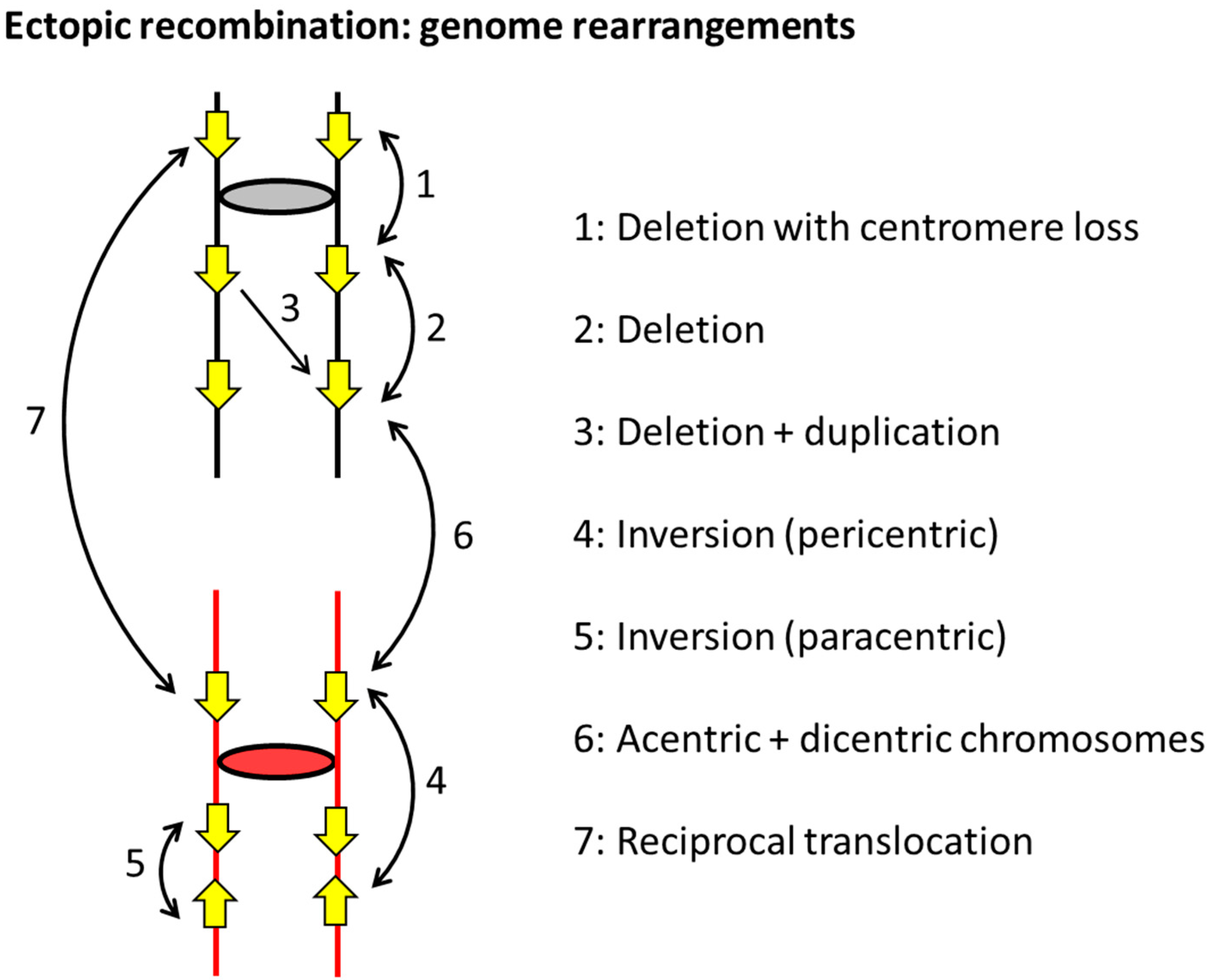
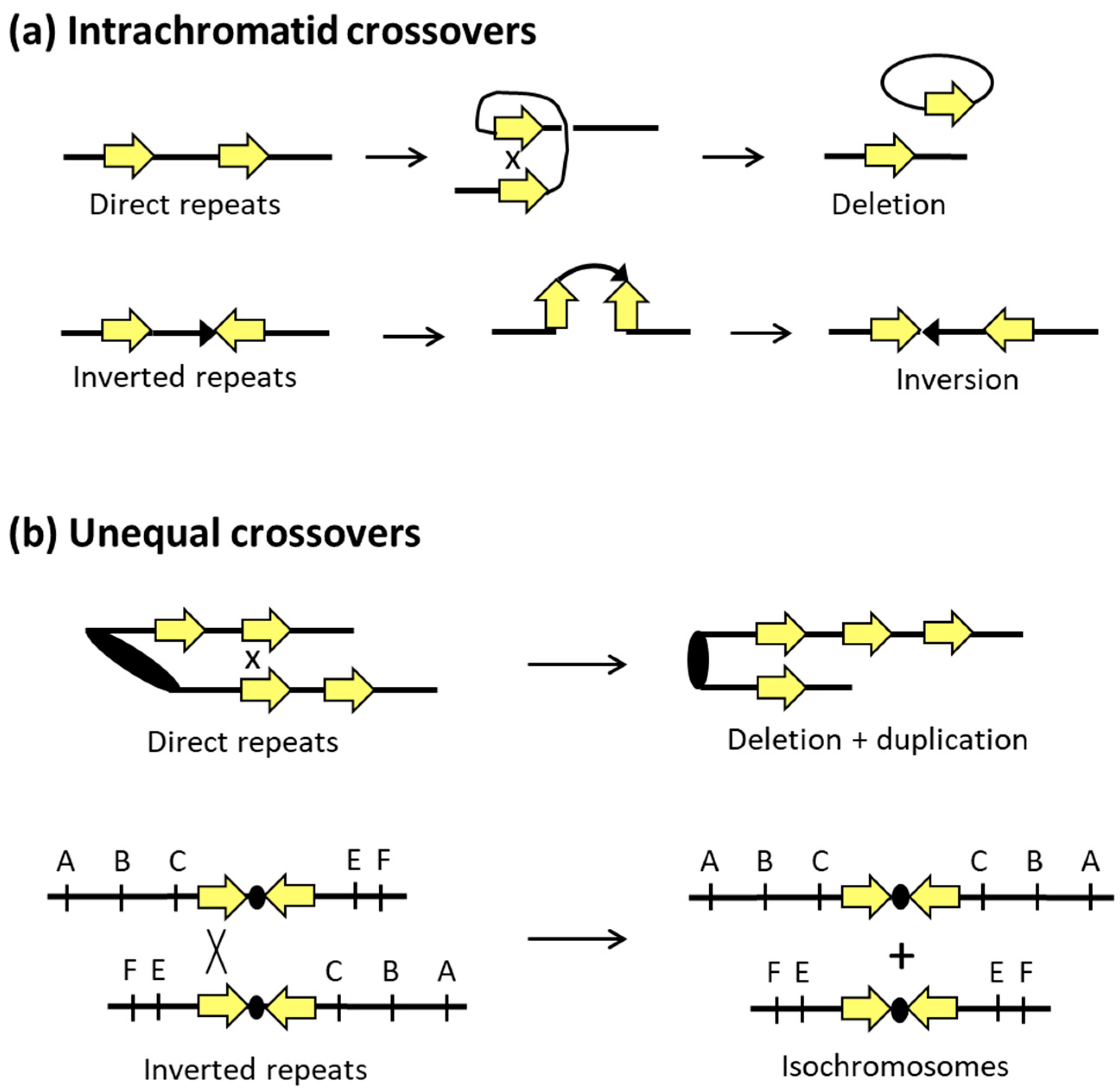
4. Mitotic Recombination and Genome Alterations in S. cerevisiae
5. Genetic Diversity and Stress Adaptation in Candida albicans
6. Genetic Diversity and Stress Adaptation in Cryptococcus neoformans
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forche, A. Large-scale chromosomal changes and associated fitness consequences in pathogenic fungi. Curr. Fungal Infect. Rep. 2014, 8, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, K.L.; Fraser, J.A. Balancing stability and flexibility within the genome of the pathogen Cryptococcus neoformans. PLoS Pathog. 2013, 9, e1003764. [Google Scholar] [CrossRef] [PubMed]
- Ene, I.V.; Farrer, R.A.; Hirakawa, M.P.; Agwamba, K.; Cuomo, C.A.; Bennett, R.J. Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc. Natl. Acad. Sci. USA 2018, 115, E8688–E8697. [Google Scholar] [CrossRef] [PubMed]
- Forche, A.; Cromie, G.; Gerstein, A.C.; Solis, N.V.; Pisithkul, T.; Srifa, W.; Jeffery, E.; Abbey, D.; Filler, S.G.; Dudley, A.M.; et al. Rapid phenotypic and genotypic diversification after exposure to the oral host niche in Candida albicans. Genetics 2018, 209, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Farrer, R.A.; Giamberardino, C.; Sakthikumar, S.; Jones, A.; Yang, T.; Tenor, J.L.; Wagih, O.; Van Wyk, M.; Govender, N.P.; et al. Microevolution of serial clinical isolates of Cryptococcus neoformans var. grubii and C. gattii. mBio 2017, 8, e00166-17. [Google Scholar] [CrossRef]
- Hu, G.; Wang, J.; Choi, J.; Jung, W.H.; Liu, I.; Litvintseva, A.P.; Bicanic, T.; Aurora, R.; Mitchell, T.G.; Perfect, J.R.; et al. Variation in chromosome copy number influences the virulence of Cryptococcus neoformans and occurs in isolates from AIDS patients. BMC Genom. 2011, 12, 526. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, B.J.; Arendrup, M.C. Invasive candidiasis. N. Engl. J. Med. 2015, 373, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Horn, D.L.; Neofytos, D.; Anaissie, E.J.; Fishman, J.A.; Steinbach, W.J.; Olyaei, A.J.; Marr, K.A.; Pfaller, M.A.; Chang, C.-H.; Webster, K.M. Epidemiology and outcomes of candidemia in 2019 patients: Data from the prospective antifungal therapy alliance registry. Clin. Infect. Dis. 2009, 48, 1695–1703. [Google Scholar] [CrossRef]
- Clancy, C.J.; Nguyen, M.H. Emergence of Candida auris: An international call to arms. Clin. Infect. Dis. 2016, 64, 141–143. [Google Scholar] [CrossRef]
- Lin, X.; Heitman, J. The biology of the Cryptococcus neoformans species complex. Annu. Rev. Microbiol. 2006, 60, 69–105. [Google Scholar] [CrossRef]
- Zhao, Y.; Lin, J.; Fan, Y.; Lin, X. Life cycle of Cryptococcus neoformans. Annu. Rev. Microbiol. 2019, 73, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef]
- Stone, N.R.H.; Rhodes, J.; Fisher, M.C.; Mfinanga, S.; Kivuyo, S.; Rugemalila, J.; Segal, E.S.; Needleman, L.; Molloy, S.F.; Kwon-Chung, J.; et al. Dynamic ploidy changes drive fluconazole resistance in human cryptococcal meningitis. J. Clin. Investig. 2019, 129, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Federspiel, N.A.; Chibana, H.; Dungan, J.; Kalman, S.; Magee, B.B.; Newport, G.; Thorstenson, Y.R.; Agabian, N.; Magee, P.T.; et al. The diploid genome sequence of Candida albicans. Proc. Natl. Acad. Sci. USA 2004, 101, 7329–7334. [Google Scholar] [CrossRef] [PubMed]
- Loftus, B.J.; Fung, E.; Roncaglia, P.; Rowley, D.; Amedeo, P.; Bruno, D.; Vamathevan, J.; Miranda, M.; Anderson, I.J.; Fraser, J.A.; et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science 2005, 307, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Janbon, G.; Ormerod, K.L.; Paulet, D.; Byrnes, E.J., 3rd; Yadav, V.; Chatterjee, G.; Mullapudi, N.; Hon, C.-C.; Billmyre, R.B.; Brunel, F.; et al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLoS Genet. 2014, 10, e1004261. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.P.; Martinez, D.A.; Sakthikumar, S.; Anderson, M.Z.; Berlin, A.; Gujja, S.; Zeng, Q.; Zisson, E.; Wang, J.M.; Greenberg, J.M.; et al. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res. 2015, 25, 413–425. [Google Scholar] [CrossRef]
- Ropars, J.; Maufrais, C.; Diogo, D.; Marcet-Houben, M.; Perin, A.; Sertour, N.; Mosca, K.; Permal, E.; Laval, G.; Bouchier, C.; et al. Gene flow contributes to diversification of the major fungal pathogen Candida albicans. Nat. Commun. 2018, 9, 2253. [Google Scholar] [CrossRef]
- Ene, I.V.; Bennett, R.J. The cryptic sexual strategies of human fungal pathogens. Nat. Rev. Microbiol. 2014, 12, 239. [Google Scholar] [CrossRef]
- Wang, J.M.; Bennett, R.J.; Anderson, M.Z. The genome of the human pathogen Candida albicans is shaped by mutation and cryptic sexual recombination. mBio 2018, 9, e01205-18. [Google Scholar] [CrossRef]
- Hickman, M.A.; Paulson, C.; Dudley, A.; Berman, J. Parasexual ploidy reduction drives population heterogeneity through random and transient aneuploidy in Candida albicans. Genetics 2015, 200, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.; Rasmussen, M.D.; Lin, M.F.; Santos, M.A.S.; Sakthikumar, S.; Munro, C.A.; Rheinbay, E.; Grabherr, M.; Forche, A.; Reedy, J.L.; et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 2009, 459, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.A.; Giamberardino, C.; Sykes, S.M.; Yu, C.-H.; Tenor, J.L.; Chen, Y.; Yang, T.; Jones, A.M.; Sun, S.; Haverkamp, M.R.; et al. Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans. Genome Res. 2017, 27, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Popp, C.; Ramírez-Zavala, B.; Schwanfelder, S.; Krüger, I.; Morschhäuser, J. Evolution of fluconazole-resistant Candida albicans; strains by drug-induced mating competence and parasexual recombination. mBio 2019, 10, e02740-18. [Google Scholar] [CrossRef]
- Selmecki, A.; Forche, A.; Berman, J. Genomic plasticity of the human fungal pathogen Candida albicans. Eukaryot. Cell 2010, 9, 991–1008. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Rodgers, K.; McVey, M. Error-prone repair of DNA double-strand breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef]
- Todd, R.T.; Wikoff, T.D.; Forche, A.; Selmecki, A. Genome plasticity in Candida albicans is driven by long repeat sequences. eLife 2019, 8. [Google Scholar] [CrossRef]
- Sun, S.; Xu, J. Chromosomal Rearrangements between Serotype A and D Strains in Cryptococcus neoformans. PLoS ONE 2009, 4, e5524. [Google Scholar] [CrossRef]
- Seidl, M.F.; Thomma, B.P.H.J. Sex or no sex: Evolutionary adaptation occurs regardless. BioEssays 2014, 36, 335–345. [Google Scholar] [CrossRef]
- Keeney, S. Spo11 and the formation of DNA double-strand breaks in meiosis. Genome Dyn. Stab. 2008, 2, 81–123. [Google Scholar] [CrossRef] [PubMed]
- Game, J.C.; Mortimer, R.K. A genetic study of x-ray sensitive mutants in yeast. Mutat. Res. 1974, 24, 281–292. [Google Scholar] [CrossRef]
- Daley, J.M.; Palmbos, P.L.; Wu, D.; Wilson, T.E. Nonhomologous end joining in yeast. Annu. Rev. Genet. 2005, 39, 431–451. [Google Scholar] [CrossRef] [PubMed]
- Deem, A.; Keszthelyi, A.; Blackgrove, T.; Vayl, A.; Coffey, B.; Mathur, R.; Chabes, A.; Malkova, A. Break-induced replication is highly inaccurate. PLoS Biol. 2011, 9, e1000594. [Google Scholar] [CrossRef] [PubMed]
- Strathern, J.N.; Shafer, B.; McGill, C.B. DNA synthesis errors associated with double-strand-break repair. Genetics 1995, 140, 965–972. [Google Scholar]
- Symington, L.S.; Rothstein, R.; Lisby, M. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 2014, 198, 795–835. [Google Scholar] [CrossRef]
- Orr-Weaver, T.L.; Szostak, J.W.; Rothstein, R.J. Yeast transformation: A model system for the study of recombination. Proc. Natl. Acad. Sci. USA 1981, 78, 6354–6358. [Google Scholar] [CrossRef]
- Lichten, M.; Haber, J.E. Position effects in ectopic and allelic mitotic recombination in Saccharomyces cerevisiae. Genetics 1989, 123, 261–268. [Google Scholar]
- Jinks-Robertson, S.; Petes, T.D. Chromosomal translocations generated by high-frequency meiotic recombination between repeated yeast genes. Genetics 1986, 114, 731–752. [Google Scholar]
- Haber, J.E. In vivo biochemistry: Physical monitoring of recombination induced by site-specific endonucleases. BioEssays 1995, 17, 609–620. [Google Scholar] [CrossRef]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double-strand-break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef]
- Allers, T.; Lichten, M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 2001, 106, 47–57. [Google Scholar] [CrossRef]
- Bizard, A.H.; Hickson, I.D. The dissolution of double Holliday junctions. Cold Spring Harb. Perspect. Biol. 2014, 6, a016477. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.K.; Heyer, W.D. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma 2011, 120, 109–127. [Google Scholar] [CrossRef]
- Paques, F.; Leung, W.Y.; Haber, J.E. Expansions and contractions in a tandem repeat induced by double-strand break repair. Mol. Cell. Biol. 1998, 18, 2045–2054. [Google Scholar] [CrossRef]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef]
- Fishman-Lobell, J.; Rudin, N.; Haber, J.E. Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol. Cell. Biol. 1992, 1292, 1303. [Google Scholar] [CrossRef]
- Lyndaker, A.M.; Alani, E. A tale of tails: Insights into the coordination of 3′ end processing during homologous recombination. Bioessays 2009, 31, 315–321. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- Stern, C. Somatic crossing over and segregation in Drosophila melanogaster. Genetics 1936, 21, 625–730. [Google Scholar] [PubMed]
- Barbera, M.A.; Petes, T.D. Selection and analysis of spontaneous reciprocal mitotic cross-overs in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2006, 103, 12819–12824. [Google Scholar] [CrossRef] [PubMed]
- St. Charles, J.; Petes, T.D. High-resolution mapping of spontaneous mitotic recombination hotspots on the 1.1 Mb arm of yeast chromosome IV. PLoS Genet. 2013, 9, e1003434. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Petes, T.D. Genome-wide high-resolution mapping of UV-induced mitotic recombination events in Saccharomyces cerevisiae. PLoS Genet. 2013, 9, e1003894. [Google Scholar] [CrossRef] [PubMed]
- Jinks-Robertson, S.; Michelitch, M.; Ramcharan, S. Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 1993, 13, 3937–3950. [Google Scholar] [CrossRef]
- Munz, P.; Amstutz, H.; Kohli, J.; Leupold, U. Recombination between dispersed serine tRNA genes in Schizosaccharomyces pombe. Nature 1982, 300, 225–231. [Google Scholar] [CrossRef]
- Zhao, Y.; Dominska, M.; Petrova, A.; Bagshaw, H.; Kokoska, R.J.; Petes, T.D. Properties of mitotic and meiotic recombination in the tandemly-repeated CUP1 gene cluster in the yeast Saccharomyces cerevisiae. Genetics 2017, 206, 785–800. [Google Scholar] [CrossRef]
- Hull, R.M.; Cruz, C.; Jack, C.V.; Houseley, J. Environmental change drives accelerated adaptation through stimulated copy number variation. PLoS Biol. 2017, 15, e2001333. [Google Scholar] [CrossRef]
- Kobayashi, T.; Heck, D.J.; Nomura, M.; Horiuchi, T. Expansion and contraction of ribosomal DNA repeats in Sacchoromyces cerevisiae: Requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev. 1998, 12, 3821–3830. [Google Scholar] [CrossRef]
- Ganley, A.R.; Kobayashi, T. Ribosomal DNA and cellular senescence: New evidence supporting the connection between rDNA and aging. FEMS Yeast Res. 2014, 14, 49–59. [Google Scholar] [CrossRef]
- Ruiz, J.F.; Gomez-Gonzalez, B.; Aguilera, A. AID induces double-strand breaks at immunoglobulin switch regions and c-MYC causing chromosomal translocations in yeast THO mutants. PLoS Genet. 2011, 7, e1002009. [Google Scholar] [CrossRef] [PubMed]
- Piazza, A.; Wright, W.D.; Heyer, W.D. Multi-invasions are recombination byproducts that induce chromosomal rearrangements. Cell 2017, 170, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Hum, Y.F.; Jinks-Robertson, S. DNA strand-exchange patterns associated with double-strand break-induced and spontaneous mitotic crossovers in Saccharomyces cerevisiae. PLoS Genet. 2018, 14, e1007302. [Google Scholar] [CrossRef] [PubMed]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef] [PubMed]
- Sakofsky, C.J.; Ayyar, S.; Deem, A.K.; Chung, W.H.; Ira, G.; Malkova, A. Translesion polymerases drive microhomology-mediated break-induced replication leading to complex chromosomal rearrangements. Mol. Cell 2015, 60, 860–872. [Google Scholar] [CrossRef]
- Olaiya, A.F.; Sogin, S.J. Ploidy determination of Candida albicans. J. Bacteriol. 1979, 140, 1043–1049. [Google Scholar]
- Riggsby, W.S.; Torres-Bauza, L.J.; Wills, J.W.; Townes, T.M. DNA content, kinetic complexity, and the ploidy question in Candida albicans. Mol. Cell. Biol. 1982, 2, 853–862. [Google Scholar] [CrossRef]
- Whelan, W.L.; Magee, P.T. Natural heterozygosity in Candida albicans. J. Bacteriol. 1981, 145, 896–903. [Google Scholar]
- Hickman, M.A.; Zeng, G.; Forche, A.; Hirakawa, M.P.; Abbey, D.; Harrison, B.D.; Wang, Y.-M.; Su, C.-H.; Bennett, R.J.; Wang, Y.; et al. The ‘obligate diploid’ Candida albicans forms mating-competent haploids. Nature 2013, 494, 55. [Google Scholar] [CrossRef]
- Gräser, Y.; Volovsek, M.; Arrington, J.; Schönian, G.; Presber, W.; Mitchell, T.G.; Vilgalys, R. Molecular markers reveal that population structure of the human pathogen Candida albicans exhibits both clonality and recombination. Proc. Natl. Acad. Sci. USA 1996, 93, 12473–12477. [Google Scholar] [CrossRef]
- Pujol, C.; Reynes, J.; Renaud, F.; Raymond, M.; Tibayrenc, M.; Ayala, F.J.; Janbon, F.; Mallié, M.; Bastide, J.M. The yeast Candida albicans has a clonal mode of reproduction in a population of infected human immunodeficiency virus-positive patients. Proc. Natl. Acad. Sci. USA 1993, 90, 9456–9459. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.J.; Johnson, A.D. Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. EMBO J. 2003, 22, 2505–2515. [Google Scholar] [CrossRef] [PubMed]
- Alby, K.; Schaefer, D.; Bennett, R.J. Homothallic and heterothallic mating in the opportunistic pathogen Candida albicans. Nature 2009, 460, 890. [Google Scholar] [CrossRef] [PubMed]
- Forche, A.; Alby, K.; Schaefer, D.; Johnson, A.D.; Berman, J.; Bennett, R.J. The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS Biol. 2008, 6, e110. [Google Scholar] [CrossRef] [PubMed]
- Lephart, P.R.; Magee, P.T. Effect of the major repeat sequence on mitotic recombination in Candida Albicans. Genetics 2006, 174, 1737–1744. [Google Scholar] [CrossRef][Green Version]
- Forche, A.; Abbey, D.; Pisithkul, T.; Weinzierl, M.A.; Ringstrom, T.; Bruck, D.; Petersen, K.; Berman, J. Stress Alters Rates and Types of Loss of Heterozygosity in Candida albicans. mBio 2011, 2, e00129-11. [Google Scholar] [CrossRef]
- Berman, J.; Hadany, L. Does stress induce (para)sex? Implications for Candida albicans evolution. Trends Genet. 2012, 28, 197–203. [Google Scholar] [CrossRef]
- Chibana, H.; Iwaguchi, S.; Homma, M.; Chindamporn, A.; Nakagawa, Y.; Tanaka, K. Diversity of tandemly repetitive sequences due to short periodic repetitions in the chromosomes of Candida albicans. J. Bacteriol. 1994, 176, 3851–3858. [Google Scholar] [CrossRef][Green Version]
- Chindamporn, A.; Nakagawa, Y.; Mizuguchi, I.; Chibana, H.; Doi, M.; Tanaka, K. Repetitive sequences (RPSs) in the chromosomes of Candida albicans are sandwiched between two novel stretches, HOK and RB2, common to each chromosome. Microbiology 1998, 144, 849–857. [Google Scholar] [CrossRef][Green Version]
- Freire-Benéitez, V.; Price, R.J.; Tarrant, D.; Berman, J.; Buscaino, A. Candida albicans repetitive elements display epigenetic diversity and plasticity. Sci. Rep. 2016, 6, 22989. [Google Scholar] [CrossRef]
- Rustchenko, E.P.; Curran, T.M.; Sherman, F. Variations in the number of ribosomal DNA units in morphological mutants and normal strains of Candida albicans and in normal strains of Saccharomyces cerevisiae. J. Bacteriol. 1993, 175, 7189–7199. [Google Scholar] [CrossRef] [PubMed]
- Wickes, B.; Staudinger, J.; Magee, B.B.; Kwon-Chung, K.J.; Magee, P.T.; Scherer, S. Physical and genetic mapping of Candida albicans: Several genes previously assigned to chromosome 1 map to chromosome R, the rDNA-containing linkage group. Infect. Immun. 1991, 59, 2480–2484. [Google Scholar] [PubMed]
- Christiaens, J.F.; Van Mulders, S.E.; Duitama, J.; Brown, C.A.; Ghequire, M.G.; De Meester, L.; Michiels, J.; Wenseleers, T.; Voordeckers, K.; Verstrepen, K.J. Functional divergence of gene duplicates through ectopic recombination. EMBO Rep. 2012, 13, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Cannon, R.D.; Holland, B.R.; Patchett, M.L.; Schmid, J. Impact of genetic background on allele selection in a highly mutable Candida albicans gene, PNG2. PLoS ONE 2010, 5, e9614. [Google Scholar] [CrossRef]
- Chu, W.S.; Rikkerink, E.H.; Magee, P.T. Genetics of the white-opaque transition in Candida albicans: Demonstration of switching recessivity and mapping of switching genes. J. Bacteriol. 1992, 174, 2951–2957. [Google Scholar] [CrossRef][Green Version]
- Selmecki, A.; Forche, A.; Berman, J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 2006, 313, 367–370. [Google Scholar] [CrossRef]
- Forche, A.; Magee, P.T.; Selmecki, A.; Berman, J.; May, G. Evolution in Candida albicans populations during a single passage through a mouse host. Genetics 2009, 182, 799–811. [Google Scholar] [CrossRef]
- Bouchonville, K.; Forche, A.; Tang, K.E.S.; Selmecki, A.; Berman, J. Aneuploid chromosomes are highly unstable during DNA transformation of Candida albicans. Eukaryot. Cell 2009, 8, 1554–1566. [Google Scholar] [CrossRef]
- Zhu, X.; Dunn, J.M.; Goddard, A.D.; Squire, J.A.; Becker, A.; Phillips, R.A.; Gallie, B.L. Mechanisms of loss of heterozygosity in retinoblastoma. Cytogenet. Cell Genet. 1992, 59, 248–252. [Google Scholar] [CrossRef]
- Feri, A.; Loll-Krippleber, R.; Commere, P.-H.; Maufrais, C.; Sertour, N.; Schwartz, K.; Sherlock, G.; Bougnoux, M.-E.; d’Enfert, C.; Legrand, M. Analysis of repair mechanisms following an induced double-strand break uncovers recessive deleterious alleles in the Candida albicans diploid genome. mBio 2016, 7, e01109-16. [Google Scholar] [CrossRef]
- Tso, G.H.W.; Reales-Calderon, J.A.; Tan, A.S.M.; Sem, X.; Le, G.T.T.; Tan, T.G.; Lai, G.C.; Srinivasan, K.G.; Yurieva, M.; Liao, W.; et al. Experimental evolution of a fungal pathogen into a gut symbiont. Science 2018, 362, 589. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.-H.; Anderson, M.Z.; Hirakawa, M.P.; Wang, J.M.; Frazer, C.; Alaalm, L.M.; Thomson, G.J.; Ene, I.V.; Bennett, R.J. Hemizygosity enables a mutational transition governing fungal virulence and commensalism. Cell Host Microbe 2019, 25, 418–431.e416. [Google Scholar] [CrossRef] [PubMed]
- Perepnikhatka, V.; Fischer, F.J.; Niimi, M.; Baker, R.A.; Cannon, R.D.; Wang, Y.K.; Sherman, F.; Rustchenko, E. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J. Bacteriol. 1999, 181, 4041–4049. [Google Scholar] [PubMed]
- Selmecki, A.; Gerami-Nejad, M.; Paulson, C.; Forche, A.; Berman, J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol. Microbiol. 2008, 68, 624–641. [Google Scholar] [CrossRef]
- Selmecki, A.M.; Dulmage, K.; Cowen, L.E.; Anderson, J.B.; Berman, J. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet. 2009, 5, e1000705. [Google Scholar] [CrossRef]
- Hull, C.M.; Heitman, J. Genetics of Cryptococcus neoformans. Ann. Rev. Genet. 2002, 36, 557–615. [Google Scholar] [CrossRef]
- Xu, J.; Vilgalys, R.; Mitchell, T.G. Multiple gene genealogies reveal recent dispersion and hybridization in the human pathogenic fungus Cryptococcus neoformans. Mol. Ecol. 2000, 9, 1471–1481. [Google Scholar] [CrossRef]
- Kwon-Chung, K.; Boekhout, T.; Wickes, B.L.; Fell, J. Systematics of the genus Cryptococcus and its type species C. neoformans. Cryptococcus Hum. Pathog. Model. Yeast 2011, 3–15. [Google Scholar] [CrossRef]
- Hagen, F.; Khayhan, K.; Theelen, B.; Kolecka, A.; Polacheck, I.; Sionov, E.; Falk, R.; Parnmen, S.; Lumbsch, H.T.; Boekhout, T. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet. Biol. 2015, 78, 16–48. [Google Scholar] [CrossRef]
- Casadevall, A.; Perfect, J.R. Cryptococcus neoformans; ASM Press: Washington, DC, USA, 1998. [Google Scholar] [CrossRef]
- Boekhout, T.; Theelen, B.; Diaz, M.; Fell, J.W.; Hop, W.C.J.; Abeln, E.C.A.; Dromer, F.; Meyer, W. Hybrid genotypes in the pathogenic yeast Cryptococcus neoformans. Microbiology 2001, 147, 891–907. [Google Scholar] [CrossRef]
- Fries, B.C.; Casadevall, A. Serial isolates of Cryptococcus neoformans from patients with AIDS differ in virulence for mice. J. Infect. Dis. 1998, 178, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Fries, B.C.; Chen, F.; Currie, B.P.; Casadevall, A. Karyotype instability in Cryptococcus neoformans infection. J. Clin. Microbiol. 1996, 34, 1531–1534. [Google Scholar] [PubMed]
- Sullivan, D.; Haynes, K.; Moran, G.; Shanley, D.; Coleman, D. Persistence, replacement, and microevolution of Cryptococcus neoformans strains in recurrent meningitis in AIDS patients. J. Clin. Microbiol. 1996, 34, 1739–1744. [Google Scholar] [PubMed]
- Blasi, E.; Brozzetti, A.; Francisci, D.; Neglia, R.; Cardinali, G.; Bistoni, F.; Vidotto, V.; Baldelli, F. Evidence of microevolution in a clinical case of recurrent Cryptococcus neoformans meningoencephalitis. Eur. J. Clin. Microbiol. Infect. Dis. 2001, 20, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Sukroongreung, S.; Lim, S.; Tantimavanich, S.; Eampokalap, B.; Carter, D.; Nilakul, C.; Kulkeratiyut, S.; Tansuphaswadikul, S. Phenotypic switching and genetic diversity of Cryptococcus neoformans. J. Clin. Microbiol. 2001, 39, 2060–2064. [Google Scholar] [CrossRef]
- Franzot, S.P.; Mukherjee, J.; Cherniak, R.; Chen, L.C.; Hamdan, J.S.; Casadevall, A. Microevolution of a standard strain of Cryptococcus neoformans resulting in differences in virulence and other phenotypes. Infect. Immun. 1998, 66, 89–97. [Google Scholar]
- Almeida, A.M.F.; Matsumoto, M.T.; Baeza, L.C.; De Oliveira e Silva, R.B.; Kleiner, A.A.P.; de Souza Carvalho Melhem, M.; Mendes Giannini, M.J.S.; Laboratory Group on Cryptococcosis. Molecular typing and antifungal susceptibility of clinical sequential isolates of Cryptococcus neoformans from Sao Paulo State, Brazil. FEMS Yeast Res. 2007, 7, 152–164. [Google Scholar] [CrossRef][Green Version]
- Garcia-Hermoso, D.; Dromer, F.; Janbon, G. Cryptococcus neoformans capsule structure evolution in vitro and during murine infection. Infect. Immun. 2004, 72, 3359–3365. [Google Scholar] [CrossRef]
- Steenwyk, J.L.; Soghigian, J.S.; Perfect, J.R.; Gibbons, J.G. Copy number variation contributes to cryptic genetic variation in outbreak lineages of Cryptococcus gattii from the North American Pacific Northwest. BMC Genom. 2016, 17, 700. [Google Scholar] [CrossRef]
- Farrer, R.A.; Desjardins, C.A.; Sakthikumar, S.; Gujja, S.; Saif, S.; Zeng, Q.; Chen, Y.; Voelz, K.; Heitman, J.; May, R.C.; et al. Genome evolution and innovation across the four major lineages of Cryptococcus gattii. mBio 2015, 6, e00868-15. [Google Scholar] [CrossRef]
- Farrer, R.A.; Ford, C.B.; Rhodes, J.; Delorey, T.; May, R.C.; Fisher, M.C.; Cloutman-Green, E.; Balloux, F.; Cuomo, C.A. Transcriptional heterogeneity of Cryptococcus gattii VGII Compared with Non-VGII Lineages Underpins Key Pathogenicity Pathways. mSphere 2018, 3, e00445-18. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, C.A.; Kronstad, J.W.; Taylor, G.; Warren, R.; Yuen, M.; Hu, G.; Jung, W.H.; Sham, A.; Kidd, S.E.; Tangen, K.; et al. Genome variation in Cryptococcus gattii, an emerging pathogen of immunocompetent hosts. mBio 2011, 2, e00342-10. [Google Scholar] [CrossRef]
- Hu, G.; Chen, S.H.; Qiu, J.; Bennett, J.E.; Myers, T.G.; Williamson, P.R. Microevolution during serial mouse passage demonstrates FRE3 as a virulence adaptation gene in Cryptococcus neoformans. mBio 2014, 5, e00941-14. [Google Scholar] [CrossRef] [PubMed]
- Magditch, D.A.; Liu, T.-B.; Xue, C.; Idnurm, A. DNA mutations mediate microevolution between host-adapted forms of the pathogenic fungus Cryptococcus neoformans. PLoS Pathog. 2012, 8, e1002936. [Google Scholar] [CrossRef]
- Okagaki, L.H.; Strain, A.K.; Nielsen, J.N.; Charlier, C.; Baltes, N.J.; Chrétien, F.; Heitman, J.; Dromer, F.; Nielsen, K. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 2010, 6, e1000953. [Google Scholar] [CrossRef]
- Zaragoza, O.; García-Rodas, R.; Nosanchuk, J.D.; Cuenca-Estrella, M.; Rodríguez-Tudela, J.L.; Casadevall, A. Fungal cell gigantism during mammalian infection. PLoS Pathog. 2010, 6, e1000945. [Google Scholar] [CrossRef]
- Dambuza, I.M.; Drake, T.; Chapuis, A.; Zhou, X.; Correia, J.; Taylor-Smith, L.; LeGrave, N.; Rasmussen, T.; Fisher, M.C.; Bicanic, T.; et al. The Cryptococcus neoformans titan cell is an inducible and regulated morphotype underlying pathogenesis. PLoS Pathog. 2018, 14, e1006978. [Google Scholar] [CrossRef]
- Trevijano-Contador, N.; de Oliveira, H.C.; García-Rodas, R.; Rossi, S.A.; Llorente, I.; Zaballos, Á.; Janbon, G.; Ariño, J.; Zaragoza, Ó. Cryptococcus neoformans can form titan-like cells in vitro in response to multiple signals. PLoS Pathog. 2018, 14, e1007007. [Google Scholar] [CrossRef]
- Gerstein, A.C.; Fu, M.S.; Mukaremera, L.; Li, Z.; Ormerod, K.L.; Fraser, J.A.; Berman, J.; Nielsen, K. Polyploid titan cells produce haploid and aneuploid progeny to promote stress adaptation. mBio 2015, 6, e01340. [Google Scholar] [CrossRef]
- Hu, G.; Liu, I.; Sham, A.; Stajich, J.E.; Dietrich, F.S.; Kronstad, J.W. Comparative hybridization reveals extensive genome variation in the AIDS-associated pathogen Cryptococcus neoformans. Genome Biol. 2008, 9, R41. [Google Scholar] [CrossRef]
- Sionov, E.; Lee, H.; Chang, Y.C.; Kwon-Chung, K.J. Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog. 2010, 6, e1000848. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.W.L.; Morrow, C.A.; Djordjevic, J.T.; Wood, I.A.; Fraser, J.A. Microevolution of Cryptococcus neoformans driven by massive tandem gene amplification. Mol. Biol. Evol. 2012, 29, 1987–2000. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E.; Debatisse, M. Gene amplification: Yeast takes a turn. Cell 2006, 125, 1237–1240. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hastings, P.J. Adaptive amplification. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 271–283. [Google Scholar] [CrossRef]
- Green, B.M.; Finn, K.J.; Li, J.J. Loss of DNA replication control is a potent inducer of gene amplification. Science 2010, 329, 943–946. [Google Scholar] [CrossRef]
- Dunham, M.J.; Badrane, H.; Ferea, T.; Adams, J.; Brown, P.O.; Rosenzweig, F.; Botstein, D. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2002, 99, 16144–16149. [Google Scholar] [CrossRef]
- Sun, S.; Yadav, V.; Billmyre, R.B.; Cuomo, C.A.; Nowrousian, M.; Wang, L.; Souciet, J.-L.; Boekhout, T.; Porcel, B.; Wincker, P.; et al. Fungal genome and mating system transitions facilitated by chromosomal translocations involving intercentromeric recombination. PLoS Biol. 2017, 15, e2002527. [Google Scholar] [CrossRef]
- Yadav, V.; Sun, S.; Billmyre, R.B.; Thimmappa, B.C.; Shea, T.; Lintner, R.; Bakkeren, G.; Cuomo, C.A.; Heitman, J.; Sanyal, K. RNAi is a critical determinant of centromere evolution in closely related fungi. Proc. Natl. Acad. Sci. USA 2018, 115, 3108–3113. [Google Scholar] [CrossRef]
- Kavanaugh, L.A.; Fraser, J.A.; Dietrich, F.S. Recent evolution of the human pathogen Cryptococcus neoformans by intervarietal transfer of a 14-gene fragment. Mol. Biol. Evol. 2006, 23, 1879–1890. [Google Scholar] [CrossRef]
- Morrow, C.A.; Lee, I.R.; Chow, E.W.L.; Ormerod, K.L.; Goldinger, A.; Byrnes, E.J.; Nielsen, K.; Heitman, J.; Schirra, H.J.; Fraser, J.A. A unique chromosomal rearrangement in the Cryptococcus neoformans type strain enhances key phenotypes associated with virulence. mBio 2012, 3, e00310-11. [Google Scholar] [CrossRef]
- Fraser, J.A.; Huang, J.C.; Pukkila-Worley, R.; Alspaugh, J.A.; Mitchell, T.G.; Heitman, J. Chromosomal translocation and segmental duplication in Cryptococcus neoformans. Eukaryot. Cell 2005, 4, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Arras, S.D.M.; Chitty, J.L.; Blake, K.L.; Schulz, B.L.; Fraser, J.A. A genomic safe haven for mutant complementation in Cryptococcus neoformans. PLoS ONE 2015, 10, e0122916. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chacko, N.; Wang, L.; Pavuluri, Y. Generation of stable mutants and targeted gene deletion strains in Cryptococcus neoformans through electroporation. Med. Mycol. 2014, 53, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Baudin, A.; Ozier-Kalogeropoulos, O.; Denouel, A.; Lacroute, F.; Cullin, C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993, 21, 3329–3330. [Google Scholar] [CrossRef] [PubMed]
- Goins, C.L.; Gerik, K.J.; Lodge, J.K. Improvements to gene deletion in the fungal pathogen Cryptococcus neoformans: Absence of Ku proteins increases homologous recombination, and co-transformation of independent DNA molecules allows rapid complementation of deletion phenotypes. Fungal Genet. Biol. 2006, 43, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Arras, S.D.M.; Fraser, J.A. Chemical inhibitors of non-homologous end joining increase targeted construct integration in Cryptococcus neoformans. PLoS ONE 2016, 11, e0163049. [Google Scholar] [CrossRef] [PubMed]
- Gasior, S.L.; Wakeman, T.P.; Xu, B.; Deininger, P.L. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 2006, 357, 1383–1393. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Gusa, A.; Averette, A.; Sun, S.; Heitman, J.; Jinks-Robertson, S. Transposon-mediated mutagenesis and drug resistance in Cryptococcus deneoformans in a host model of infection. In Proceedings of the HFP2019 Advanced Lecture Course, La Colle sur Loup, France, 18–24 May 2019. Abstract P4A. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gusa, A.; Jinks-Robertson, S. Mitotic Recombination and Adaptive Genomic Changes in Human Pathogenic Fungi. Genes 2019, 10, 901. https://doi.org/10.3390/genes10110901
Gusa A, Jinks-Robertson S. Mitotic Recombination and Adaptive Genomic Changes in Human Pathogenic Fungi. Genes. 2019; 10(11):901. https://doi.org/10.3390/genes10110901
Chicago/Turabian StyleGusa, Asiya, and Sue Jinks-Robertson. 2019. "Mitotic Recombination and Adaptive Genomic Changes in Human Pathogenic Fungi" Genes 10, no. 11: 901. https://doi.org/10.3390/genes10110901
APA StyleGusa, A., & Jinks-Robertson, S. (2019). Mitotic Recombination and Adaptive Genomic Changes in Human Pathogenic Fungi. Genes, 10(11), 901. https://doi.org/10.3390/genes10110901