Metabolic Constrains Rule Metastasis Progression


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
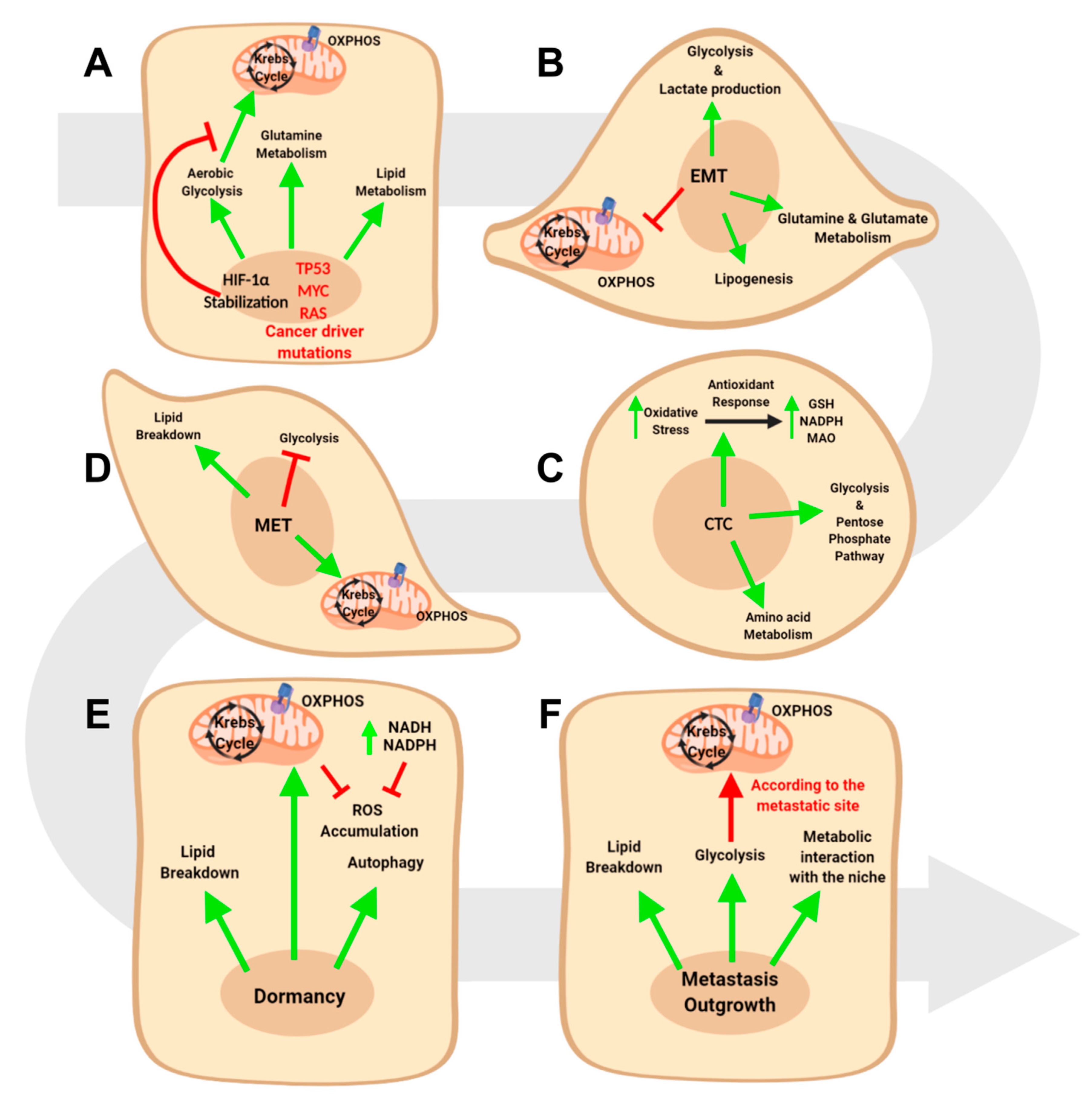
2. The Genetic Roots of Tumor Metabolism Reprogramming
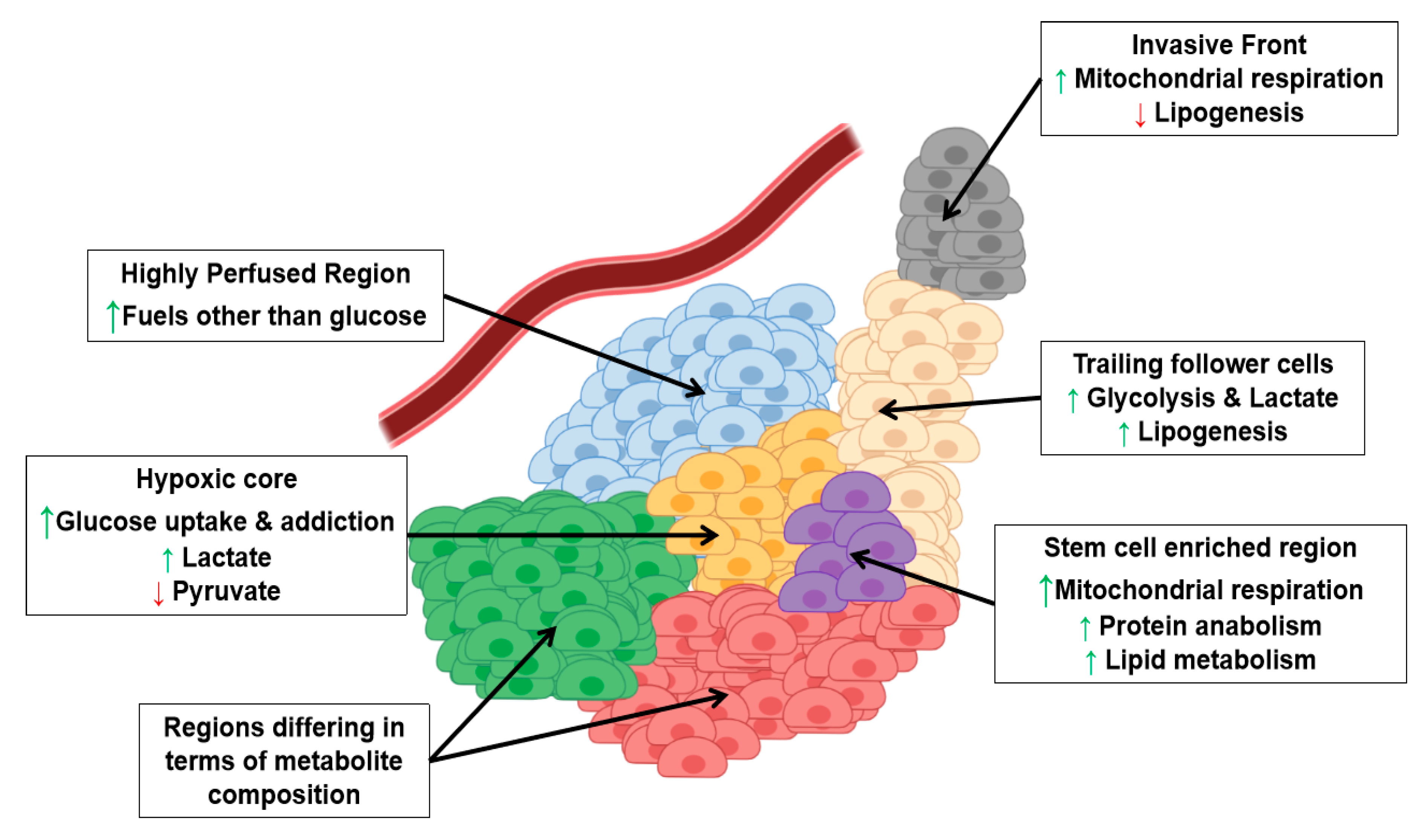
3. Metabolic Intra-Tumor Heterogeneity
4. Metabolic Reprogramming in Epithelial to Mesenchymal Transition
4.1. Glucose Metabolism and EMT
4.2. Lipid Metabolism and EMT
4.3. Amino Acid Metabolism and EMT
5. The Metabolic Reprogramming in Circulating Tumor Cells and Mesenchymal to Epithelial Transition
5.1. Metabolic Requirements of Circulating Tumor Cells
5.2. Metabolic Requirements of the Mesenchymal to Epithelial Transition
6. The Metabolic Evolution of Metastasis Outgrowth
6.1. Metabolic Features of In Situ and Metastatic Dormant Cancer Cells
6.2. Metabolic Requirements in Metastatic Outgrowth
7. Metabolism and Cancer Therapy
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Corcoran, S.E.; O’Neill, L.A. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Cliff, T.; Dalton, S.; Sartorelli, V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell 2015, 17, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Montes, I.; Escobar, B.; Palacios, B.; Gómez, M.J.; Izquierdo-Garcia, J.L.; Flores, L.; Jimenez-Borreguero, L.J.; Aragones, J.; Ruíz-Cabello, J.; Torres, M.; et al. Myocardial VHL-HIF Signaling Controls an Embryonic Metabolic Switch Essential for Cardiac Maturation. Dev. Cell 2016, 39, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhou, J.; Li, Y.; Zhao, Y.; Yuan, J.; Cao, Y.; Wang, L.; Zhang, Z.; Zhang, B.; Wang, C.C.; et al. YY1 regulates skeletal muscle regeneration through controlling metabolic reprogramming of satellite cells. EMBO J. 2019, 38, e99727. [Google Scholar] [CrossRef]
- Polyzos, A.A.; Lee, D.Y.; Datta, R.; Hauser, M.; Budworth, H.; Holt, A.; Mihalik, S.; Goldschmidt, P.; Frankel, K.; Trego, K.; et al. Metabolic Reprogramming in Astrocytes Distinguishes Region-Specific Neuronal Susceptibility in Huntington Mice. Cell Metab. 2019, 29, 1258–1273.e11. [Google Scholar] [CrossRef]
- Mogilenko, D.A.; Haas, J.T.; L’Homme, L.; Fleury, S.; Quemener, S.; Levavasseur, M.; Becquart, C.; Wartelle, J.; Bogomolova, A.; Pineau, L.; et al. Metabolic and Innate Immune Cues Merge into a Specific Inflammatory Response via the UPR. Cell 2019, 177, 1201–1216.e19. [Google Scholar] [CrossRef]
- Baardman, J.; Verberk, S.G.; Prange, K.H.; Van Weeghel, M.; Van Der Velden, S.; Ryan, D.G.; Wüst, R.C.I.; Neele, A.E.; Speijer, D.; Denis, S.W.; et al. A Defective Pentose Phosphate Pathway Reduces Inflammatory Macrophage Responses during Hypercholesterolemia. Cell Rep. 2018, 25, 2044–2052.e5. [Google Scholar] [CrossRef]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.-J.; Sakamoto, N.; Fearon, E.R.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef]
- Li, T.Y.; Sun, Y.; Liang, Y.; Liu, Q.; Shi, Y.; Zhang, C.-S.; Zhang, C.; Song, L.; Zhang, P.; Zhang, X.; et al. ULK1/2 Constitute a Bifurcate Node Controlling Glucose Metabolic Fluxes in Addition to Autophagy. Mol. Cell 2016, 62, 359–370. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nature 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2015, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Europe PMC Funders Group Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2018, 284, 3132–3144. [Google Scholar] [CrossRef]
- Ma, L.; Zong, X. Metabolic Symbiosis in Chemoresistance: Refocusing the Role of Aerobic Glycolysis. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yao, L.; Fang, Y.; Yang, K.; Jiang, W.; Huang, W.; Cai, Z. Specifically blocking the fatty acid synthesis to inhibit the malignant phenotype of bladder cancer. Int. J. Biol. Sci. 2019, 15, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1. [Google Scholar] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F. The Tumor Suppressor p53 Down-Regulates Glucose Transporters GLUT1 and GLUT4 Gene Expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose Deprivation Contributes to the Development of KRAS Pathway Mutations in Tumor Cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2009, 2, a001008. [Google Scholar] [CrossRef]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 561–577.e22. [Google Scholar] [CrossRef]
- RajeshKumar, N.V.; Dutta, P.; Yabuuchi, S.; De Wilde, R.F.; Martinez, G.V.; Le, A.; Kamphorst, J.J.; Rabinowitz, J.D.; Jain, S.K.; Hidalgo, M.; et al. Therapeutic Targeting of the Warburg Effect in Pancreatic Cancer Relies on an Absence of p53 Function. Cancer Res. 2015, 75, 3355–3364. [Google Scholar] [CrossRef]
- Han, C.Y.; Patten, D.A.; Lee, S.G.; Parks, R.J.; Chan, D.W.; Harper, M.; Tsang, B.K. p53 Promotes chemoresponsiveness by regulating hexokinase II gene transcription and metabolic reprogramming in epithelial ovarian cancer. Mol. Carcinog. 2019, 58, 2161–2174. [Google Scholar] [CrossRef]
- Wilkie, M.D.; Anaam, E.A.; Lau, A.S.; Rubbi, C.P.; Jones, T.M.; Boyd, M.T.; Vlatković, N. TP53 mutations in head and neck cancer cells determine the Warburg phenotypic switch creating metabolic vulnerabilities and therapeutic opportunities for stratified therapies. Cancer Lett. 2020, 478, 107–121. [Google Scholar] [CrossRef]
- Ko, Y.-H.; Domingo-Vidal, M.; Roche, M.; Lin, Z.; Whitaker-Menezes, D.; Seifert, E.; Capparelli, C.; Tuluc, M.; Birbe, R.C.; Tassone, P.; et al. TP53-inducible Glycolysis and Apoptosis Regulator (TIGAR) Metabolically Reprograms Carcinoma and Stromal Cells in Breast Cancer. J. Biol. Chem. 2016, 291, 26291–26303. [Google Scholar] [CrossRef] [PubMed]
- Harami-Papp, H.; Pongor, L.S.; Munkácsy, G.; Horváth, G.; Nagy, Á.M.; Ambrus, A.; Hauser, P.; Szabó, A.; Tretter, L.; Győrffy, B. TP53 mutation hits energy metabolism and increases glycolysis in breast cancer. Oncotarget 2016, 7, 67183–67195. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. The sweet trap in tumors: Aerobic glycolysis and potential targets for therapy. Oncotarget 2016, 7, 38908–38926. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nature 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Heiden, M.G.V. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nature 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The Regulation of AMPK β1, TSC2, and PTEN Expression by p53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-mTOR Pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.J.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Lin, M.; Zhu, W.; Liang, Y.; Hong, X.; Zhao, Y.; Young, K.H.; Hu, W.; Feng, Z. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget 2014, 5, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. p53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17, 2074. [Google Scholar] [CrossRef]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 Activation in Adipocytes of Obese Mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef]
- Genovese, G.; Carugo, A.; Tepper, J.; Robinson, F.S.; Li, L.; Svelto, M.; Nezi, L.; Corti, D.; Minelli, R.; Pettazzoni, P.; et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 2017, 542, 362–366. [Google Scholar] [CrossRef]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; E Ayer, D. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef]
- Priolo, C.; Pyne, S.; Rose, J.; Regan, E.R.; Zadra, G.; Photopoulos, C.; Cacciatore, S.; Schultz, D.; Scaglia, N.; McDunn, J.; et al. AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Cancer Res. 2014, 74, 7198–7204. [Google Scholar] [CrossRef]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef]
- Mishra, P.; Tang, W.; Putluri, V.; Dorsey, T.H.; Jin, F.; Wang, F.; Zhu, D.; Amable, L.; Deng, T.; Zhang, S.; et al. ADHFE1 is a breast cancer oncogene and induces metabolic reprogramming. J. Clin. Investig. 2017, 128, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Tang, W.; Ambs, S. ADHFE1 is a MYC-linked oncogene that induces metabolic reprogramming and cellular de-differentiation in breast cancer. Mol. Cell. Oncol. 2018, 5, e1432260. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sun, J.; Zhang, Y.; Li, J.; Hu, J.; Li, K.; Gao, L.; Shen, L. c-Myc-driven glycolysis via TXNIP suppression is dependent on glutaminase-MondoA axis in prostate cancer. Biochem. Biophys. Res. Commun. 2018, 504, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, J.Y.; Prager, B.C.; A Flavahan, W.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.; Cavallaro, F.D.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 1–11. [Google Scholar] [CrossRef]
- Xiao, D.; Ren, P.; Su, H.; Yue, M.; Xiu, R.; Hu, Y.; Liu, H.; Qing, G. Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 2015, 6, 40655–40666. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Bott, A.J.; Peng, I.-C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.-M.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef]
- Liu, W.; Hancock, C.N.; Fischer, J.; Harman, M.; Phang, J.M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: Involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 17206. [Google Scholar] [CrossRef] [PubMed]
- Jancik, S.; Drabek, J.; Radzioch, D.; Hajduch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.C.; Trenary, I.A.; Young, J.D.; Li, M.; Liebler, D.C. Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer. Mol. Cell. Proteom. 2016, 15, 2924–2938. [Google Scholar] [CrossRef]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.-F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Toda, K.; Kawada, K.; Iwamoto, M.; Inamoto, S.; Sasazuki, T.; Shirasawa, S.; Hasegawa, S.; Sakai, Y. Metabolic Alterations Caused by KRAS Mutations in Colorectal Cancer Contribute to Cell Adaptation to Glutamine Depletion by Upregulation of Asparagine Synthetase. Neoplasia 2016, 18, 654–665. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Nosho, K.; Shima, K.; Irahara, N.; Chan, A.T.; Meyerhardt, J.A.; Chung, D.C.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. HIF1A Overexpression Is Associated with Poor Prognosis in a Cohort of 731 Colorectal Cancers. Am. J. Pathol. 2010, 176, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-S.; Chen, X.-H.; Yin, X.; Zhang, B. Prognostic Significance of HIF-1α Expression in Hepatocellular Carcinoma: A Meta-Analysis. PLoS ONE 2013, 8, e65753. [Google Scholar] [CrossRef]
- Chen, L.; Shi, Y.; Yuan, J.; Han, Y.; Qin, R.; Wu, Q.; Jia, B.; Wei, B.; Wei, L.; Dai, G.; et al. HIF-1 Alpha Overexpression Correlates with Poor Overall Survival and Disease-Free Survival in Gastric Cancer Patients Post-Gastrectomy. PLoS ONE 2014, 9, e90678. [Google Scholar] [CrossRef]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation ofglut1mRNA by Hypoxia-inducible Factor-1. J. Biol. Chem. 2000, 276, 9519–9525. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Tian, R.-F.; Liu, W.; Fei, Z.; Long, Q.; Wang, X.-A.; Zhang, X. The expression and significance of HIF-1α and GLUT-3 in glioma. Brain Res. 2009, 1304, 149–154. [Google Scholar] [CrossRef]
- Riddle, S.R.; Ahmad, A.; Ahmad, S.; Deeb, S.S.; Malkki, M.; Schneider, B.K.; Allen, C.B.; White, C.W. Hypoxia induces hexokinase II gene expression in human lung cell line A549. Am. J. Physiol. Cell. Mol. Physiol. 2000, 278, L407–L416. [Google Scholar] [CrossRef]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar]
- Semenza, G.L.; Jiang, B.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase a Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed]
- Firth, J.D.; Ebert, B.L.; Pugh, C.W.; Ratcliffe, P.J. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase a genes: Similarities with the erythropoietin 3′ enhancer. Proc. Natl. Acad. Sci. USA 1994, 91, 6496–6500. [Google Scholar] [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The Plasma Membrane Lactate Transporter MCT4, but Not MCT1, Is Up-regulated by Hypoxia through a HIF-1α-dependent Mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- Firth, J.D.; Ebert, B.L.; Ratcliffe, P.J. Hypoxic Regulation of Lactate Dehydrogenase A. J. Biol. Chem. 1995, 270, 21021–21027. [Google Scholar] [CrossRef]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-Mediated Suppression of Acyl-CoA Dehydrogenases and Fatty Acid Oxidation Is Critical for Cancer Progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.-Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty Acid Synthase Gene Is Up-regulated by Hypoxia via Activation of Akt and Sterol Regulatory Element Binding Protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.; Wigfield, S.; Buffa, F.; Li, J.-L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Soh, H.; Wasa, M.; Fukuzawa, M. Hypoxia upregulates amino acid transport in a human neuroblastoma cell line. J. Pediatr. Surg. 2007, 42, 608–612. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell. Mol. Life Sci. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Takano, N.; Xiang, L.; Gilkes, D.M.; Luo, W.; Semenza, G.L. Hypoxia-inducible factors enhance glutamate signaling in cancer cells. Oncotarget 2014, 5, 8853–8868. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef]
- Lyu, Z.; Mao, Z.; Li, Q.; Xia, Y.; Liu, Y.; He, Q.; Wang, Y.; Zhao, H.; Lu, Z.; Zhou, Q. PPARγ maintains the metabolic heterogeneity and homeostasis of renal tubules. EBioMedicine 2018, 38, 178–190. [Google Scholar] [CrossRef]
- Karmaus, P.W.; Chen, X.; Lim, S.A.; Herrada, A.A.; Nguyen, T.-L.M.; Xu, B.; Dhungana, Y.; Rankin, S.; Chen, W.; Rosencrance, C.; et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 2018, 565, 101–105. [Google Scholar] [CrossRef]
- Dyar, K.A.; Lutter, D.; Artati, A.; Ceglia, N.J.; Liu, Y.; Armenta, D.; Jastroch, M.; Schneider, S.; De Mateo, S.; Cervantes, M.; et al. Atlas of Circadian Metabolism Reveals System-wide Coordination and Communication between Clocks. Cell 2018, 174, 1571–1585.e11. [Google Scholar] [CrossRef] [PubMed]
- Loponte, S.; Lovisa, S.; Deem, A.K.; Carugo, A.; Viale, A. The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind? Cancers 2019, 11, 1574. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 3712–3763. [Google Scholar] [CrossRef]
- Heaster, T.M.; Landman, B.A.; Skala, M.C. Quantitative Spatial Analysis of Metabolic Heterogeneity Across in vivo and in vitro Tumor Models. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Laymon, C.; Oborski, M.; Hou, W.; Wang, L.; Grandis, J.R.; Ferris, R.L.; Mountz, J.M.; Van Houten, B. Quantifying Metabolic Heterogeneity in Head and Neck Tumors in Real Time: 2-DG Uptake Is Highest in Hypoxic Tumor Regions. PLoS ONE 2014, 9, e102452. [Google Scholar] [CrossRef]
- Tran, M.; Latifoltojar, A.; Neves, J.B.; Papoutsaki, M.-V.; Gong, F.; Comment, A.; Costa, A.S.H.; Glaser, M.; Tran-Dang, M.-A.; El Sheikh, S.; et al. First-in-human in vivo non-invasive assessment of intra-tumoral metabolic heterogeneity in renal cell carcinoma. BJR Case Rep. 2019, 5. [Google Scholar] [CrossRef]
- Xu, H.N.; Zheng, G.; Tchou, J.; Nioka, S.; Li, L.Z. Characterizing the metabolic heterogeneity in human breast cancer xenografts by 3D high resolution fluorescence imaging. Springerplus 2013, 2, 73. [Google Scholar] [CrossRef]
- Wagner, B.A.; Venkataraman, S.; Buettner, G.R. The rate of oxygen utilization by cells. Free Radic. Biol. Med. 2011, 51, 700–712. [Google Scholar] [CrossRef]
- Hai, P.; Imai, T.; Xu, S.; Zhang, R.; Aft, R.L.; Zou, J.; Wang, L.V. High-throughput, label-free, single-cell photoacoustic microscopy of intratumoral metabolic heterogeneity. Nat. Biomed. Eng. 2019, 3, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Okegawa, T.; Morimoto, M.; Nishizawa, S.; Kitazawa, S.; Honda, K.; Araki, H.; Tamura, T.; Ando, A.; Satomi, Y.; Nutahara, K.; et al. Intratumor Heterogeneity in Primary Kidney Cancer Revealed by Metabolic Profiling of Multiple Spatially Separated Samples within Tumors. EBioMedicine 2017, 19, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, T.; Song, X.; Huang, L.; Zang, Q.; Xu, J.; Bi, N.; Jiao, G.; Hao, Y.; Chen, Y.; et al. Spatially resolved metabolomics to discover tumor-associated metabolic alterations. Proc. Natl. Acad. Sci. USA 2018, 116, 52–57. [Google Scholar] [CrossRef]
- Inglese, P.; McKenzie, J.S.; Mroz, A.; Kinross, J.; Veselkov, K.; Holmes, E.; Takats, Z.; Nicholson, J.K.; Glen, R. Deep learning and 3D-DESI imaging reveal the hidden metabolic heterogeneity of cancer. Chem. Sci. 2017, 8, 3500–3511. [Google Scholar] [CrossRef]
- Prade, V.M.; Kunzke, T.; Feuchtinger, A.; Rohm, M.; Luber, B.; Lordick, F.; Buck, A.; Walch, A. De novo discovery of metabolic heterogeneity with immunophenotype-guided imaging mass spectrometry. Mol. Metab. 2020, 36, 100953. [Google Scholar] [CrossRef]
- Wang, X.; Han, J.; Hardie, D.B.; Yang, J.; Pan, J.; Borchers, C.H. Metabolomic profiling of prostate cancer by matrix assisted laser desorption/ionization-Fourier transform ion cyclotron resonance mass spectrometry imaging using Matrix Coating Assisted by an Electric Field (MCAEF). Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 755–767. [Google Scholar] [CrossRef]
- Huang, L.; Mao, X.; Sun, C.; Luo, Z.; Song, X.; Li, X.; Zhang, R.; Lv, Y.; Chen, J.; He, J.; et al. A graphical data processing pipeline for mass spectrometry imaging-based spatially resolved metabolomics on tumor heterogeneity. Anal. Chim. Acta 2019, 1077, 183–190. [Google Scholar] [CrossRef]
- Gooptu, M.; Whitaker-Menezes, D.; Sprandio, J.; Domingo-Vidal, M.; Lin, Z.; Uppal, G.; Gong, J.; Fratamico, R.; Leiby, B.; Dulau-Florea, A.; et al. Mitochondrial and glycolytic metabolic compartmentalization in diffuse large B-cell lymphoma. Semin. Oncol. 2017, 44, 204–217. [Google Scholar] [CrossRef]
- Mikkilineni, L.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Sprandio, J.; Avena, P.; Cotzia, P.; Dulau-Florea, A.; Gong, J.; Uppal, G.; Zhan, T.; et al. Hodgkin lymphoma: A complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin. Oncol. 2017, 44, 218–225. [Google Scholar] [CrossRef]
- Phillips, J.J. Novel Therapeutic Targets in the Brain Tumor Microenvironment. Oncotarget 2012, 3, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Minh, L.B.; Siebzehnrübl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Massari, M.S.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef] [PubMed]
- Bayin, N.S.; Frenster, J.D.; Sen, R.; Si, S.; Modrek, A.S.; Galifianakis, N.; Dolgalev, I.; Ortenzi, V.; Illa-Bochaca, I.; Khahera, A.; et al. Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells. Oncotarget 2017, 8, 64932–64953. [Google Scholar] [CrossRef] [PubMed]
- Shibao, S.; Minami, N.; Koike, N.; Fukui, N.; Yoshida, K.; Saya, H.; Sampetrean, O. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro Oncol. 2018, 20, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Commander, R.; Wei, C.; Sharma, A.; Mouw, J.K.; Burton, L.J.; Summerbell, E.R.; Mahboubi, D.; Peterson, R.J.; Konen, J.; Zhou, W.; et al. Subpopulation targeting of pyruvate dehydrogenase and GLUT1 decouples metabolic heterogeneity during collective cancer cell invasion. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef]
- Vivas-García, Y.; Falletta, P.; Liebing, J.; Louphrasitthiphol, P.; Feng, Y.; Chauhan, J.; Scott, D.A.; Glodde, N.; Chocarro-Calvo, A.; Bonham, S.; et al. Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Mol. Cell 2019, 77, 120–137.e9. [Google Scholar] [CrossRef]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2019, 577, 115–120. [Google Scholar] [CrossRef]
- Jensen, D.H.; Therkildsen, M.H.; Dabelsteen, E. A reverse Warburg metabolism in oral squamous cell carcinoma is not dependent upon myofibroblasts. J. Oral Pathol. Med. 2014, 44, 714–721. [Google Scholar] [CrossRef]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Krętowski, R.; Borzym-Kluczyk, M.; Stypułkowska, A.; Brańska-Januszewska, J.; Ostrowska, H.; Cechowska-Pasko, M. Low glucose dependent decrease of apoptosis and induction of autophagy in breast cancer MCF-7 cells. Mol. Cell. Biochem. 2016, 417, 35–47. [Google Scholar] [CrossRef]
- Huber, A.-L.; Lebeau, J.; Guillaumot, P.; Petrilli, V.; Malek, M.; Chilloux, J.; Fauvet, F.; Payen, L.; Kfoury, A.; Renno, T.; et al. p58IPK-Mediated Attenuation of the Proapoptotic PERK-CHOP Pathway Allows Malignant Progression upon Low Glucose. Mol. Cell 2013, 49, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Püschel, F.; León-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramón, D.; Lucendo, E.; Muñoz-Pinedo, C. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol. Cell. Biol. 2017, 37, e00479–e00516. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 Regulates MYC-Mediated Neuroblastoma Cell Death upon Glutamine Deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Gwangwa, M.V.; Joubert, A.; Visagie, M.H. Effects of glutamine deprivation on oxidative stress and cell survival in breast cell lines. Biol. Res. 2019, 52, 15. [Google Scholar] [CrossRef]
- Papandreou, I.; Krishna, C.; Kaper, F.; Cai, D.; Giaccia, A.J.; Denko, N.C. Anoxia Is Necessary for Tumor Cell Toxicity Caused by a Low-Oxygen Environment. Cancer Res. 2005, 65, 3171–3178. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Glucose starvation induces LKB1-AMPK-mediated MMP-9 expression in cancer cells. Sci. Rep. 2018, 8, 10122. [Google Scholar] [CrossRef]
- Jo, H.; Lee, J.; Jeon, J.; Kim, S.Y.; Chung, J.-I.; Ko, H.Y.; Lee, M.; Yun, M. The critical role of glucose deprivation in epithelial-mesenchymal transition in hepatocellular carcinoma under hypoxia. Sci. Rep. 2020, 10, 1538. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438.e5. [Google Scholar] [CrossRef]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.; Soto, M.; Wagner, M.; Turgeon, M.-O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Shen, X.; Xue, Y.; Si, Y.; Wang, Q.; Wang, Z.; Yuan, J.; Zhang, X. The unfolded protein response potentiates epithelial-to-mesenchymal transition (EMT) of gastric cancer cells under severe hypoxic conditions. Med. Oncol. 2014, 32. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Mir, K.B.; Seligson, N.D.; Nayak, D.; Kumar, R.; Goswami, A. Integration of EMT and cellular survival instincts in reprogramming of programmed cell death to anastasis. Cancer Metastasis Rev. 2020, 39, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef]
- Meyer-Schaller, N.; Cardner, M.; Diepenbruck, M.; Saxena, M.; Tiede, S.; Lüönd, F.; Ivanek, R.; Beerenwinkel, N.; Christofori, G. A Hierarchical Regulatory Landscape during the Multiple Stages of EMT. Dev. Cell 2019, 48, 539–553.e6. [Google Scholar] [CrossRef]
- Liu, M.; Quek, L.-E.; Sultani, G.; Turner, N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. 2016, 4, 19. [Google Scholar] [CrossRef]
- Yang, L.; Hou, Y.; Yuan, J.; Tang, S.; Zhang, H.; Zhu, Q.; Du, Y.-E.; Zhou, M.; Wen, S.; Xu, L.; et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget 2015, 6, 25755–25769. [Google Scholar] [CrossRef] [PubMed]
- Kondaveeti, Y.; Reed, I.K.G.; White, B.A. Epithelial–mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett. 2015, 364, 44–58. [Google Scholar] [CrossRef]
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplancke, B.; E Frawley, L.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014, 2, 11. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Y.; Chai, L.; Bu, H.; Yang, Y.; Huang, H.; Ran, J.; Zhu, Y.; Li, L.; Chen, F.; et al. FRK plays an oncogenic role in non-small cell lung cancer by enhancing the stemness phenotype via induction of metabolic reprogramming. Int. J. Cancer 2019, 146, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hancock, S.; Sultani, G.; Wilkins, B.P.; Ding, E.; Osborne, B.; Quek, L.-E.; Turner, N. Snail-Overexpression Induces Epithelial-mesenchymal Transition and Metabolic Reprogramming in Human Pancreatic Ductal Adenocarcinoma and Non-tumorigenic Ductal Cells. J. Clin. Med. 2019, 8, 822. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Cha, Y.; Lee, J.; Lee, S.-H.; Yang, J.H.; Yun, J.S.; Cho, E.S.; Zhang, X.; Nam, M.; Kim, N.; et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat. Commun. 2017, 8, 14374. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, T.-E.; Chen, M.; Xu, D.; Zhu, Y.; Hu, B.-Y.; Lin, Z.-F.; Pan, J.-J.; Wang, X.; Wu, C.; et al. MFN1-dependent alteration of mitochondrial dynamics drives hepatocellular carcinoma metastasis by glucose metabolic reprogramming. Br. J. Cancer 2019, 122, 209–220. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat. Commun. 2016, 7, 13041. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, W.; Zhang, T.; Zhou, Q.; Liu, J.; Liu, Y.; Kong, D.; Yu, W.; Liu, R.; Hai, C. TGF-β1 induces epithelial-to-mesenchymal transition via inhibiting mitochondrial functions in A549 cells. Free Radic. Res. 2018, 52, 1432–1444. [Google Scholar] [CrossRef]
- Lunetti, P.; Di Giacomo, M.; Vergara, D.; De Domenico, S.; Maffia, M.; Zara, V.; Capobianco, L.; Ferramosca, A. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J. 2019, 286, 688–709. [Google Scholar] [CrossRef]
- Sun, H.-F.; Yang, X.-L.; Zhao, Y.; Tian, Q.; Chen, M.-T.; Zhao, Y.-Y.; Jin, W. Loss of TMEM126A promotes extracellular matrix remodeling, epithelial-to-mesenchymal transition, and breast cancer metastasis by regulating mitochondrial retrograde signaling. Cancer Lett. 2019, 440–441, 189–201. [Google Scholar] [CrossRef]
- Jiang, Y.; Tang, Y.; Wang, S.; Wu, J.; Zhang, M.; Pang, X.; Wu, J.; Chen, Y.; Tang, Y.; Liang, X. PRRX1-induced epithelial-to-mesenchymal transition in salivary adenoid cystic carcinoma activates the metabolic reprogramming of free fatty acids to promote invasion and metastasis. Cell Prolif. 2019, 53, e12705. [Google Scholar] [CrossRef]
- Xu, C.-Y.; Liu, S.-Q.; Qin, M.; Zhuge, C.-F.; Lai, M.-Y.; Huang, J.-A.; Qin, L.; Qin, N. SphK1 modulates cell migration and EMT-related marker expression by regulating the expression of p-FAK in colorectal cancer cells. Int. J. Mol. Med. 2017, 39, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Xie, Y.; Yin, J.; Lu, W.; Fang, S. SphK1 promotes tumor cell migration and invasion in colorectal cancer. Tumor Biol. 2015, 37, 6831–6836. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Duan, P.; Zhu, H.; Rao, D. miR-613 inhibits bladder cancer proliferation and migration through targeting SphK1. Am. J. Transl. Res. 2017, 9, 1213–1221. [Google Scholar] [PubMed]
- Xiao, W.; Wang, X.; Wang, T.; Chen, B.; Xing, J. HAO2 inhibits malignancy of clear cell renal cell carcinoma by promoting lipid catabolic process. J. Cell. Physiol. 2019, 234, 23005–23016. [Google Scholar] [CrossRef]
- Sánchez-Martínez, R.; Cruz-Gil, S.; De Cedrón, M.G.; Alvarez-Fernández, M.; Vargas, T.; Molina, S.; García, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar] [CrossRef]
- Hanai, J.-I.; Doro, N.; Sasaki, A.T.; Kobayashi, S.; Cantley, L.C.; Seth, P.; Sukhatme, V.P. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein Kinase (MAPK) and Phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J. Cell. Physiol. 2012, 227, 1709–1720. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, H.; Li, J.; Fang, X.; Pan, H.; Yuan, X.; Zhang, P. Up-Regulated FASN Expression Promotes Transcoelomic Metastasis of Ovarian Cancer Cell through Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2014, 15, 11539–11554. [Google Scholar] [CrossRef]
- Singh, R.; Yadav, V.; Kumar, S.; Saini, N. MicroRNA-195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci. Rep. 2015, 5, 17454. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, F.; Wang, X.; Tsai, Y.; Chuang, K.-H.; Keng, P.C.; Lee, S.O.; Chen, Y. A FASN-TGF-β1-FASN regulatory loop contributes to high EMT/metastatic potential of cisplatin-resistant non-small cell lung cancer. Oncotarget 2016, 7, 55543–55554. [Google Scholar] [CrossRef]
- Zielinska, H.; Holly, J.; Bahl, A.; Perks, C.M. Inhibition of FASN and ERα signalling during hyperglycaemia-induced matrix-specific EMT promotes breast cancer cell invasion via a caveolin-1-dependent mechanism. Cancer Lett. 2018, 419, 187–202. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-O, M.; DeBerardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2014, 34, 3908–3916. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Esposito, C.; Stephen, J.M.; Costa, A.S.H.; Frezza, C.; Blacker, T.S.; Szabadkai, G.; Ashcroft, M. CHCHD4 regulates tumour proliferation and EMT-related phenotypes, through respiratory chain-mediated metabolism. Cancer Metab. 2019, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hardie, R.-A.; Hoy, A.J.; Van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Ulanet, D.B.; Couto, K.; Jha, A.; Choe, S.; Wang, A.; Woo, H.-K.; Steadman, M.; DelaBarre, B.; Gross, S.; Driggers, E.; et al. Mesenchymal Phenotype Predisposes Lung Cancer Cells to Impaired Proliferation and Redox Stress in Response to Glutaminase Inhibition. PLoS ONE 2014, 9, e115144. [Google Scholar] [CrossRef][Green Version]
- Yang, L.; Moss, T.J.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.N.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Jeong, E.K.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget 2016, 7, 7925–7939. [Google Scholar] [CrossRef]
- Liu, G.; Zhu, J.; Yu, M.; Cai, C.; Zhou, Y.; Yu, M.; Fu, Z.; Gong, Y.; Yang, B.; Li, Y.; et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J. Transl. Med. 2015, 13, 144. [Google Scholar] [CrossRef]
- Ramirez-Peña, E.; Arnold, J.; Shivakumar, V.; Joseph, R.; Vijay, G.V.; Hollander, P.D.; Bhangre, N.; Allegakoen, P.; Prasad, R.; Conley, Z.; et al. The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers 2019, 11, 1610. [Google Scholar] [CrossRef]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Cayrefourcq, L.; Mazard, T.; Maudelonde, T.; Assenat, E.; Assou, S. Molecular Portrait of Metastasis-Competent Circulating Tumor Cells in Colon Cancer Reveals the Crucial Role of Genes Regulating Energy Metabolism and DNA Repair. Clin. Chem. 2017, 63, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Piantadosi, C.A. Prevention of H2O2 generation by monoamine oxidase protects against CNS O2 toxicity. J. Appl. Physiol. 1991, 71, 1057–1061. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Cuevas, S.; Villar, V.A.; Escano, C.; Asico, L.D.; Yu, P.; Grandy, D.K.; Felder, R.A.; Armando, I.; et al. Paraoxonase 2 decreases renal reactive oxygen species production, lowers blood pressure, and mediates dopamine D2 receptor-induced inhibition of NADPH oxidase. Free Radic. Biol. Med. 2012, 53, 437–446. [Google Scholar] [CrossRef]
- Veal, E.A.; Toone, W.M.; Jones, N.; Morgan, B.A. Distinct Roles for GlutathioneS-Transferases in the Oxidative Stress Response inSchizosaccharomyces pombe. J. Biol. Chem. 2002, 277, 35523–35531. [Google Scholar] [CrossRef]
- Chen, J.; Ye, C.; Dong, J.; Cao, S.; Hu, Y.; Situ, B.; Xi, X.; Qin, S.; Xu, J.; Cai, Z.; et al. Metabolic classification of circulating tumor cells as a biomarker for metastasis and prognosis in breast cancer. J. Transl. Med. 2020, 18, 14–59. [Google Scholar] [CrossRef]
- Chen, J.; Cao, S.; Si-Tu, B.; Zhong, J.; Hu, Y.; Li, S.; Huang, J.; Xu, J.; Wu, S.; Lin, J.; et al. Metabolic reprogramming-based characterization of circulating tumor cells in prostate cancer. J. Exp. Clin. Cancer Res. 2018, 37, 127. [Google Scholar] [CrossRef]
- Chiu, T.-K.; Lei, K.F.; Hsieh, J.C.-H.; Hsiao, H.-B.; Wang, H.-M.; Wu, M.-H. Development of a Microfluidic-Based Optical Sensing Device for Label-Free Detection of Circulating Tumor Cells (CTCs) Through Their Lactic Acid Metabolism. Sensors 2015, 15, 6789–6806. [Google Scholar] [CrossRef]
- Brisotto, G.; Biscontin, E.; Rossi, E.; Bulfoni, M.; Piruska, A.; Spazzapan, S.; Poggiana, C.; Vidotto, R.; Steffan, A.; Colombatti, A.; et al. Dysmetabolic Circulating Tumor Cells Are Prognostic in Metastatic Breast Cancer. Cancers 2020, 12, 1005. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e14. [Google Scholar] [CrossRef] [PubMed]
- Gunasinghe, N.P.A.D.; Wells, A.; Thompson, E.W.; Hugo, H. Mesenchymal–epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Monkman, J.; Blick, T.; Duijf, P.H.G.; Nagaraj, S.H.; Thompson, E.W. Multi-Omics Characterization of the Spontaneous Mesenchymal-Epithelial Transition in the PMC42 Breast Cancer Cell Lines. J. Clin. Med. 2019, 8, 1253. [Google Scholar] [CrossRef] [PubMed]
- Pouyafar, A.; Heydarabad, M.Z.; Abdolalizadeh, J.; Rahbarghazi, R.; Talebi, M.; Zade, J.A. Modulation of lipolysis and glycolysis pathways in cancer stem cells changed multipotentiality and differentiation capacity toward endothelial lineage. Cell Biosci. 2019, 9, 30. [Google Scholar] [CrossRef]
- Funasaka, T.; Hu, H.; Yanagawa, T.; Hogan, V.; Raz, A. Down-Regulation of Phosphoglucose Isomerase/Autocrine Motility Factor Results in Mesenchymal-to-Epithelial Transition of Human Lung Fibrosarcoma Cells. Cancer Res. 2007, 67, 4236–4243. [Google Scholar] [CrossRef] [PubMed]
- Tiraby, C.; Hazen, B.C.; Gantner, M.L.; Kralli, A. Estrogen-related receptor gamma promotes mesenchymal-to-epithelial transition and suppresses breast tumor growth. Cancer Res. 2011, 71, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Moen, I.; Øyan, A.M.; Kalland, K.-H.; Tronstad, K.J.; Akslen, L.A.; Chekenya, M.; Sakariassen, P.; Øystein, R.R.K.; Stuhr, L.E.B. Hyperoxic Treatment Induces Mesenchymal-to-Epithelial Transition in a Rat Adenocarcinoma Model. PLoS ONE 2009, 4, e6381. [Google Scholar] [CrossRef]
- Bigagli, E.; Cinci, L.; D’Ambrosio, M.; Luceri, C. Transcriptomic Characterization, Chemosensitivity and Regulatory Effects of Exosomes in Spontaneous EMT/MET Transitions of Breast Cancer Cells. Cancer Genom. Proteom. 2019, 16, 163–173. [Google Scholar] [CrossRef]
- Tachtsidis, A.; Le, A.V.-P.; Blick, T.; Gunasinghe, D.; De Sousa, E.; Waltham, M.; Dobrovic, A.; Thompson, E.W. Human-specific RNA analysis shows uncoupled epithelial-mesenchymal plasticity in circulating and disseminated tumour cells from human breast cancer xenografts. Clin. Exp. Metastasis 2019, 36, 393–409. [Google Scholar] [CrossRef]
- Reka, A.K.; Kurapati, H.; Narala, V.R.; Bommer, G.T.; Chen, J.; Standiford, T.J.; Keshamouni, V.G. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol. Cancer Ther. 2010, 9, 3221–3232. [Google Scholar] [CrossRef]
- Lim, J.C.W.; Kwan, Y.P.; Tan, M.S.; Teo, M.H.Y.; Chiba, S.; Wahli, W.; Wang, X. The Role of PPARβ/δ in Melanoma Metastasis. Int. J. Mol. Sci. 2018, 19, 2860. [Google Scholar] [CrossRef]
- Budczies, J.; Von Winterfeld, M.; Klauschen, F.; Bockmayr, M.; Lennerz, J.K.; Denkert, C.; Wolf, T.; Warth, A.; Dietel, M.; Anagnostopoulos, I.; et al. The landscape of metastatic progression patterns across major human cancers. Oncotarget 2014, 6, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Croucher, P. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Heaster, T.M.; Walsh, A.J.; Zhao, Y.; Hiebert, S.W.; Skala, M.C. Autofluorescence imaging identifies tumor cell-cycle status on a single-cell level. J. Biophotonics 2017, 11, e201600276. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2002, 278, 8516–8525. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Israels, S.J.; Gibson, S.; Kong, J. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007, 120, 4155–4166. [Google Scholar] [CrossRef]
- Albayrak, T.; Scherhammer, V.; Schoenfeld, N.; Braziulis, E.; Mund, T.; Bauer, M.K.; Scheffler, I.E.; Grimm, S. The Tumor Suppressor cybL, a Component of the Respiratory Chain, Mediates Apoptosis Induction. Mol. Biol. Cell 2003, 14, 3082–3096. [Google Scholar] [CrossRef] [PubMed]
- Rusu, P.; Shao, C.; Neuerburg, A.; Acikgöz, A.A.; Wu, Y.; Zou, P.; Phapale, P.; Shankar, T.S.; Döring, K.; Dettling, S.; et al. GPD1 Specifically Marks Dormant Glioma Stem Cells with a Distinct Metabolic Profile. Cell Stem Cell 2019, 25, 241–257.e8. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, R.W.; Blais, E.M.; Cowan, C.R.; Sherman, N.E.; Grigera, P.R.; Jeffery, E.D.; Fox, J.W.; Blackman, B.R.; Tschumperlin, D.J.; A Papin, J.; et al. Matrix Rigidity Regulates Cancer Cell Growth by Modulating Cellular Metabolism and Protein Synthesis. PLoS ONE 2012, 7, e37231. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Hampsch, R.A.; Wells, J.D.; Traphagen, N.A.; Mccleery, C.F.; Fields, J.L.; Shee, K.; Dillon, L.M.; Pooler, D.B.; Lewis, L.D.; Demidenko, E.; et al. AMPK Activation by Metformin Promotes Survival of Dormant ER+ Breast Cancer Cells. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Ornelas, A.; McCullough, C.R.; Lu, Z.; Zacharias, N.M.; Kelderhouse, L.E.; Gray, J.; Yang, H.; Engel, B.J.; Wang, Y.; Mao, W.; et al. Induction of autophagy by ARHI (DIRAS3) alters fundamental metabolic pathways in ovarian cancer models. BMC Cancer 2016, 16. [Google Scholar] [CrossRef]
- Fabian, A.; Stegner, S.; Miarka, L.; Zimmermann, J.; Lenk, L.; Rahn, S.; Buttlar, J.; Viol, F.; Knaack, H.; Esser, D.; et al. Metastasis of pancreatic cancer: An uninflamed liver micromilieu controls cell growth and cancer stem cell properties by oxidative phosphorylation in pancreatic ductal epithelial cells. Cancer Lett. 2019, 453, 95–106. [Google Scholar] [CrossRef]
- Cavnar, S.P.; Xiao, A.; Gibbons, A.E.; Rickelmann, A.D.; Neely, T.; Luker, K.E.; Takayama, S.; Luker, G.D. Imaging Sensitivity of Quiescent Cancer Cells to Metabolic Perturbations in Bone Marrow Spheroids. Tomogr. 2016, 2, 146–157. [Google Scholar] [CrossRef]
- Vincent, Z.; Urakami, K.; Maruyama, K.; Yamaguchi, K.; Kusuhara, M. CD133-positive cancer stem cells from colo205 human colon adenocarcinoma cell line show resistance to chemotherapy and display a specific metabolomic profile. Genes Cancer 2014, 5, 250. [Google Scholar] [CrossRef]
- Correa, R.J.M.; Peart, T.; Valdes, Y.R.; DiMattia, G.E.; Shepherd, T.G. Modulation of AKT activity is associated with reversible dormancy in ascites-derived epithelial ovarian cancer spheroids. Carcinogenesis 2011, 33, 49–58. [Google Scholar] [CrossRef]
- Peart, T.; Valdes, Y.R.; Correa, R.J.M.; Fazio, E.; Bertrand, M.; McGee, J.; Préfontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget 2015, 6, 22424–22438. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-H.; Park, M.S.; Nandu, T.; Gadad, S.; Kim, S.-C.; Kim, M.-Y. GALNT14 promotes lung-specific breast cancer metastasis by modulating self-renewal and interaction with the lung microenvironment. Nat. Commun. 2016, 7, 13796. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Watters, A.; Cheng, N.; Perry, C.E.; Xu, K.; Alicea, G.M.; Parris, J.L.; Baraban, E.; Ray, P.; Nayak, A.; et al. Polyunsaturated Fatty Acids from Astrocytes Activate PPARγ Signaling in Cancer Cells to Promote Brain Metastasis. Cancer Discov. 2019, 9, 1720–1735. [Google Scholar] [CrossRef]
- Davis, R.; Blake, K.; Ma, D.; Gabra, M.B.I.; Hernandez, G.A.; Phung, A.T.; Yang, Y.; Maurer, D.; Lefebvre, A.E.Y.T.; Alshetaiwi, H.; et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nature 2020, 22, 310–320. [Google Scholar] [CrossRef]
- Shinde, A.; Wilmanski, T.; Chen, H.; Teegarden, D.; Wendt, M.K. Pyruvate carboxylase supports the pulmonary tropism of metastatic breast cancer. Breast Cancer Res. 2018, 20, 76. [Google Scholar] [CrossRef]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; Van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.M.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.-L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Lee, C.-K.; Jeong, S.-H.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2016, 541, 41–45. [Google Scholar] [CrossRef]
- Wang, R.; Tao, B.; Fan, Q.; Wang, S.; Chen, L.; Zhang, J.; Hao, Y.; Dong, S.; Wang, Z.; Wang, W.; et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333-Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine 2019, 45, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Lin, K.; Zhao, Y.; Wu, Q.; Chen, D.; Wang, J.; Liang, Y.; Li, J.; Hu, J.; Wang, H.; et al. Adipocytes fuel gastric cancer omental metastasis via PITPNC1-mediated fatty acid metabolic reprogramming. Theranostics 2018, 8, 5452–5468. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, N.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.M.; Jalali, A.; Kircher, D.A.; Lee, W.-C.; McQuade, J.L.; Haydu, L.E.; Joon, A.Y.; Reuben, A.; De Macedo, M.P.; Carapeto, F.C.L.; et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov. 2019, 9, 628–645. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nature 2015, 17, 183–194. [Google Scholar] [CrossRef]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar] [CrossRef]
- Nieman, K.M.; A Kenny, H.; Penicka, C.V.; Ladányi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.-Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262.e4. [Google Scholar] [CrossRef]
- Turner, N.C.; Reis-Filho, J.S. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012, 13, e178–e185. [Google Scholar] [CrossRef]
- Klein, C.A. Selection and adaptation during metastatic cancer progression. Nature 2013, 501, 365–372. [Google Scholar] [CrossRef]
- Von Stechow, L.; Ruiz-Aracama, A.; Van De Water, B.; Peijnenburg, A.; Danen, E.H.J.; Lommen, A. Identification of Cisplatin-Regulated Metabolic Pathways in Pluripotent Stem Cells. PLoS ONE 2013, 8, e76476. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arunachalam, S.; Pichiah, P.T.; Achiraman, S. Doxorubicin treatment inhibits PPARγ and may induce lipotoxicity by mimicking a type 2 diabetes-like condition in rodent models. FEBS Lett. 2012, 587, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; De Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Jiang, W.; Lu, W.; Zhang, H.; Yu, T.; Tian, J.; Wen, S.; Garcia-Manero, G.; Huang, P.; Hu, Y. Metabolic reprogramming and redox adaptation in sorafenib-resistant leukemia cells: Detected by untargeted metabolomics and stable isotope tracing analysis. Cancer Commun. 2019, 39, 17. [Google Scholar] [CrossRef]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.A.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-Fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef]
- Cruz-Bermúdez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; García-Grande, A.; Coronado, M.J.; Laine-Menéndez, S.; Palacios-Zambrano, S.; Moreno-Villa, M.R.; Ruiz-Valdepeñas, A.M.; Lendinez, C.; et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS inhibition. Free Radic. Biol. Med. 2019, 135, 167–181. [Google Scholar] [CrossRef]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef]
- Sirois, I.; Aguilar-Mahecha, A.; LaFleur, J.; Fowler, E.; Vu, V.; Scriver, M.; Buchanan, M.; Chabot, C.; Ramanathan, A.; Balachandran, B.; et al. A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability. Mol. Cancer Res. 2019, 17, 2492–2507. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef]
- Qian, X.; Xu, W.; Xu, J.; Shi, Q.; Li, J.; Weng, Y.; Jiang, Z.; Feng, L.; Wang, X.; Zhou, J.; et al. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget 2017, 8, 47691–47708. [Google Scholar] [CrossRef]
- Johnson, J.M.; Lai, S.Y.; Cotzia, P.; Cognetti, D.; Luginbuhl, A.; Pribitkin, E.A.; Zhan, T.; Mollaee, M.; Domingo-Vidal, M.; Chen, Y.; et al. Mitochondrial Metabolism as a Treatment Target in Anaplastic Thyroid Cancer. Semin. Oncol. 2015, 42, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Cheng, Q. Inhibiting 6-phosphogluconate dehydrogenase reverses doxorubicin resistance in anaplastic thyroid cancer via inhibiting NADPH-dependent metabolic reprogramming. Biochem. Biophys. Res. Commun. 2018, 498, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Hultsch, S.; Kankainen, M.; Paavolainen, L.; Kovanen, R.-M.; Ikonen, E.; Kangaspeska, S.; Pietiäinen, V.; Kallioniemi, O. Association of tamoxifen resistance and lipid reprogramming in breast cancer. BMC Cancer 2018, 18, 850. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wu, G.; Song, L.; Cao, L.; Tan, Z.; Tang, M.; Li, Z.; Shi, D.; Zhang, S.; Li, J. NKX2-8 deletion-induced reprogramming of fatty acid metabolism confers chemoresistance in epithelial ovarian cancer. EBioMedicine 2019, 43, 238–252. [Google Scholar] [CrossRef]
- Obrist, F.; Michels, J.; Durand, S.; Chery, A.; Pol, J.G.; Levesque, S.; Joseph, A.; Astesana, V.; Pietrocola, F.; Wu, G.S.; et al. Metabolic vulnerability of cisplatin-resistant cancers. EMBO J. 2018, 37, e98597. [Google Scholar] [CrossRef]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef]
- Masamha, C.P.; Lafontaine, P. Molecular targeting of glutaminase sensitizes ovarian cancer cells to chemotherapy. J. Cell. Biochem. 2018, 119, 6136–6145. [Google Scholar] [CrossRef]
- Chu, J.; Niu, X.; Chang, J.; Shao, M.; Peng, L.; Xi, Y.; Lin, A.; Wang, C.; Cui, Q.; Luo, Y.; et al. Metabolic remodeling by TIGAR overexpression is a therapeutic target in esophageal squamous-cell carcinoma. Theranostics 2020, 10, 3488–3502. [Google Scholar] [CrossRef]
- Hudson, C.D.; Savadelis, A.; Nagaraj, A.B.; Joseph, P.; Avril, S.; DiFeo, A.; Avril, N. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget 2016, 7, 41637–41649. [Google Scholar] [CrossRef]
- Pallavi, R.; Giorgio, M.; Pelicci, P.G. Insights into the beneficial effect of caloric/ dietary restriction for a healthy and prolonged life. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef]
- Lévesque, S.; Le Naour, J.; Pietrocola, F.; Paillet, J.; Kremer, M.; Castoldi, F.; Baracco, E.E.; Wang, Y.; Vacchelli, E.; Stoll, G.; et al. A synergistic triad of chemotherapy, immune checkpoint inhibitors, and caloric restriction mimetics eradicates tumors in mice. OncoImmunology 2019, 8, e1657375. [Google Scholar] [CrossRef] [PubMed]
- Simone, B.A.; Palagani, A.; Strickland, K.; Ko, K.; Jin, L.; Lim, M.K.; Dan, T.; Sarich, M.; Monti, D.A.; Cristofanilli, M.; et al. Caloric restriction counteracts chemotherapy-induced inflammation and increases response to therapy in a triple negative breast cancer model. Cell Cycle 2018, 17, 1536–1544. [Google Scholar] [CrossRef]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Guidi, N.; Longo, V.D. Periodic fasting starves cisplatin-resistant cancers to death. EMBO J. 2018, 37, e99815. [Google Scholar] [CrossRef]
- Di Tano, M.; Raucci, F.; Vernieri, C.; Caffa, I.; Buono, R.; Fanti, M.; Brandhorst, S.; Curigliano, G.; Nencioni, A.; De Braud, F.; et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Safdie, F.; Brandhorst, S.; Wei, M.; Wang, W.; Lee, C.; Hwang, S.; Conti, P.S.; Chen, T.C.; Longo, V.D. Fasting Enhances the Response of Glioma to Chemo- and Radiotherapy. PLoS ONE 2012, 7, e44603. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, Ş.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Dinardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Stein, E.M.; De Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib inIDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.-Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.-J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.J.; Agip, A.-N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Kanakkanthara, A.; Kurmi, K.; Ekstrom, T.L.; Hou, X.; Purfeerst, E.R.; Heinzen, E.P.; Correia, C.; Huntoon, C.J.; O’Brien, D.; Hendrickson, A.E.W.; et al. BRCA1 Deficiency Upregulates NNMT, Which Reprograms Metabolism and Sensitizes Ovarian Cancer Cells to Mitochondrial Metabolic Targeting Agents. Cancer Res. 2019, 79, 5920–5929. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.C.; Rustandy, F.D.; Tiwari, P.; et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019, 568, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.-M.; Moroni, C.; Lane, H.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058.e4. [Google Scholar] [CrossRef]
- Pathria, G.; A Scott, D.; Feng, Y.; Lee, J.S.; Fujita, Y.; Zhang, G.; Sahu, A.D.; Ruppin, E.; Herlyn, M.; Osterman, A.L.; et al. Targeting the Warburg effect via LDHA inhibition engages ATF 4 signaling for cancer cell survival. EMBO J. 2018, 37, e99735. [Google Scholar] [CrossRef]
- Gonzalez, P.S.; O’Prey, J.; Cardaci, S.; Barthet, V.J.; Sakamaki, J.-I.; Beaumatin, F.; Roseweir, A.; Gay, D.M.; Mackay, G.; Malviya, G.; et al. Mannose impairs tumour growth and enhances chemotherapy. Nature 2018, 563, 719–723. [Google Scholar] [CrossRef]
- Nguyen, H.A.; Su, Y.; Zhang, J.Y.; Antanasijevic, A.; Caffrey, M.; Schalk, A.M.; Liu, L.; Rondelli, D.; Oh, A.; Mahmud, D.L.; et al. A Novel l-Asparaginase with low l-Glutaminase Coactivity Is Highly Efficacious against Both T- and B-cell Acute Lymphoblastic LeukemiasIn Vivo. Cancer Res. 2018, 78, 1549–1560. [Google Scholar] [CrossRef]
- Pieters, R.; Hunger, S.P.; Boos, J.; Rizzari, C.; Pui, C. L-asp treatment in ALL: A focus on erwinia. Cancer 2012, 117, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nature 2018, 20, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J., Jr.; Coffey, J.C.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, G.; Wang, Y.; Cai, L.; Qian, K.; Ju, L.; Liu, X.; Xiao, Y.; Wang, X. Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARγ-mediated pathway in bladder cancer. Clin. Sci. 2019, 133, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 726–734. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2009, 120, 142–156. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, D.; He, J.; Song, L.; Chen, H.; Zhang, Z.; Tan, N. Inhibition of fatty acid synthesis arrests colorectal neoplasm growth and metastasis: Anti-cancer therapeutical effects of natural cyclopeptide RA-XII. Biochem. Biophys. Res. Commun. 2019, 512, 819–824. [Google Scholar] [CrossRef]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate Dependence of Tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Suzuki, K.; Sawamoto, J.; Tokizawa, Y.; Iwase, Y.; Yumita, N.; Ikeda, T. Inhibition of Fatty Acid Synthesis Induces Apoptosis of Human Pancreatic Cancer Cells. Anticancer. Res. 2016, 36, 4655–4660. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Yoo, Y.J.; Kim, H.; Lee, H.; Chung, H.; Nam, M.-H.; Moon, J.-Y.; Lee, H.S.; Yoon, S.; Kim, W.-Y. Clinical and biochemical relevance of monounsaturated fatty acid metabolism targeting strategy for cancer stem cell elimination in colon cancer. Biochem. Biophys. Res. Commun. 2019, 519, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Fargnoli, J.; Henley, B.J.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; Wong, T.W.; Rupnow, B.A.; et al. Cancer Cell Dependence on Unsaturated Fatty Acids Implicates Stearoyl-CoA Desaturase as a Target for Cancer Therapy. Mol. Cancer Res. 2011, 9, 1551–1561. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roda, N.; Gambino, V.; Giorgio, M. Metabolic Constrains Rule Metastasis Progression. Cells 2020, 9, 2081. https://doi.org/10.3390/cells9092081
Roda N, Gambino V, Giorgio M. Metabolic Constrains Rule Metastasis Progression. Cells. 2020; 9(9):2081. https://doi.org/10.3390/cells9092081
Chicago/Turabian StyleRoda, Niccolo’, Valentina Gambino, and Marco Giorgio. 2020. "Metabolic Constrains Rule Metastasis Progression" Cells 9, no. 9: 2081. https://doi.org/10.3390/cells9092081
APA StyleRoda, N., Gambino, V., & Giorgio, M. (2020). Metabolic Constrains Rule Metastasis Progression. Cells, 9(9), 2081. https://doi.org/10.3390/cells9092081