Ubiquitin, Autophagy and Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Neurodegenerative Diseases and Protein Aggregates
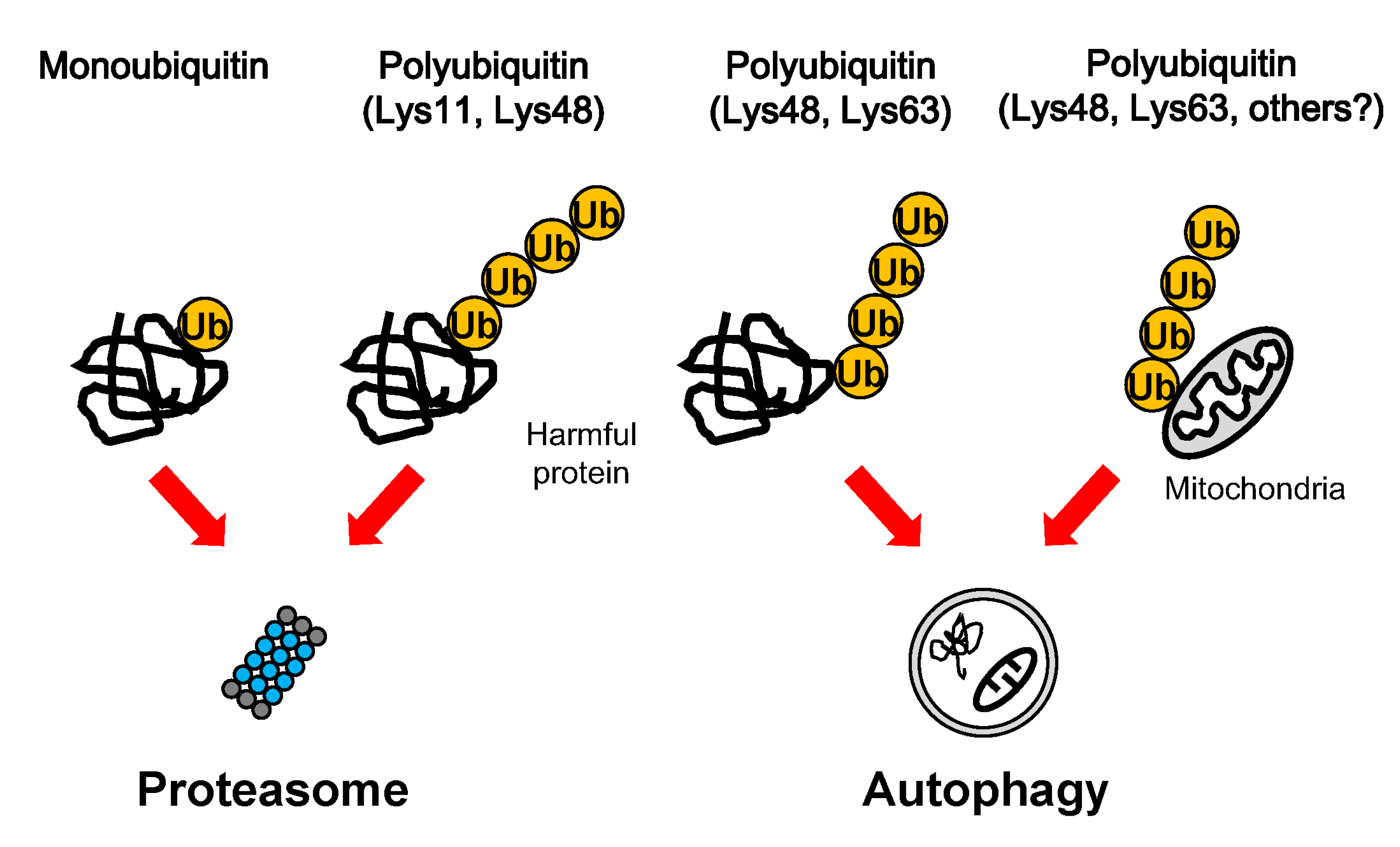
3. Ubiquitination in Protein Degradation
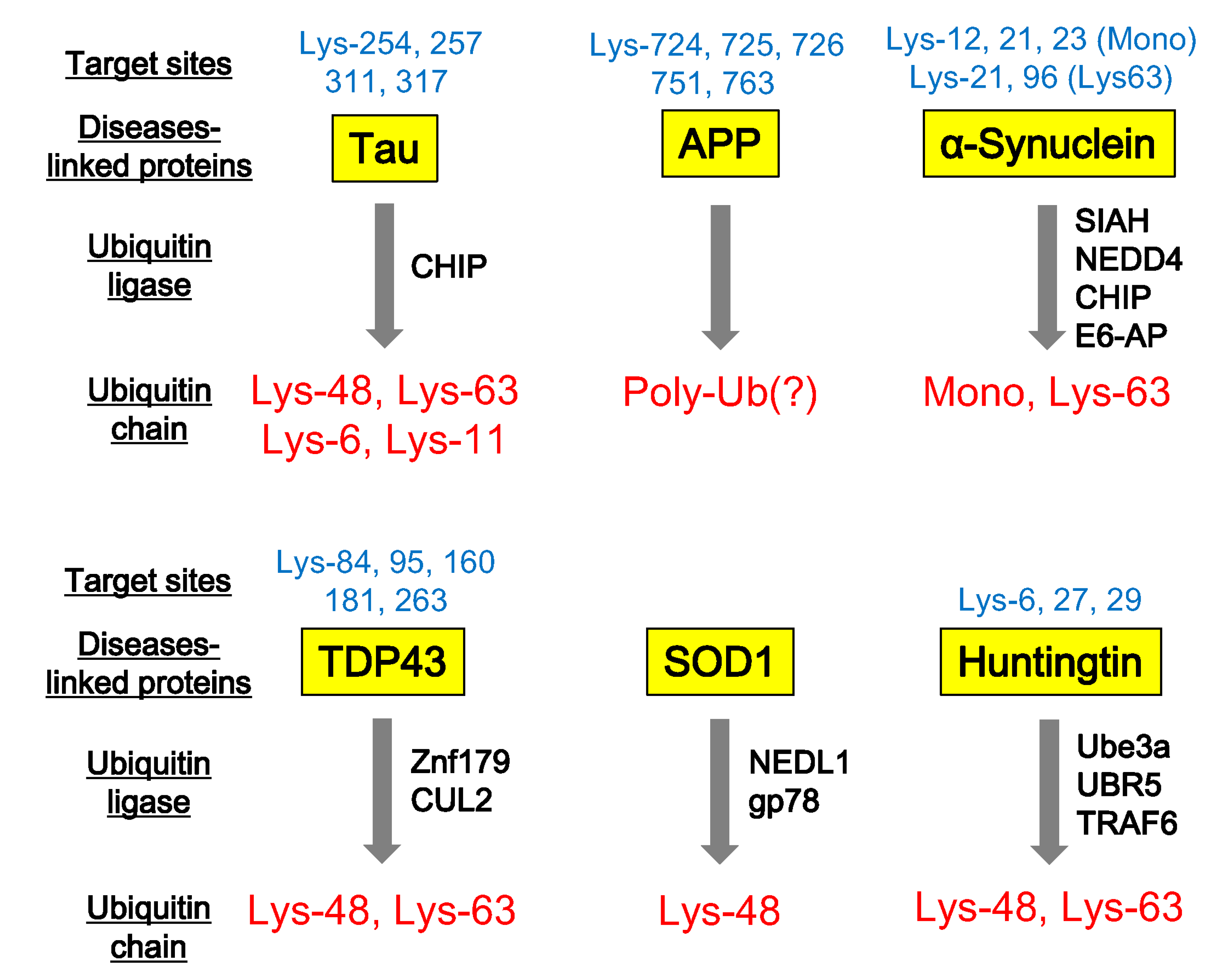
4. Ubiquitination of Neurodegenerative Disease-Associated Proteins
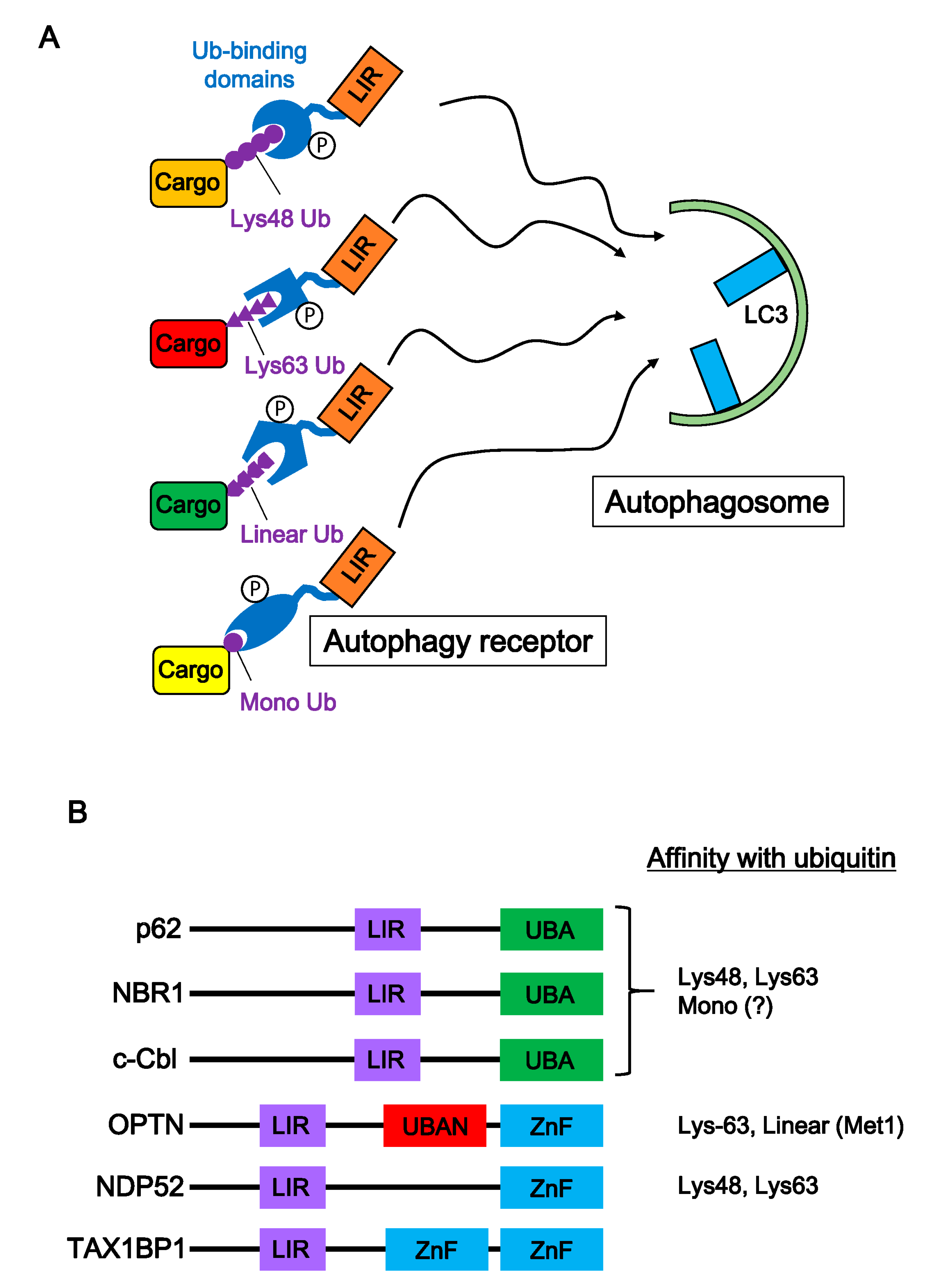
5. Autophagic Degradation of Neurodegenerative Disease-Associated Proteins
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galves, M.; Rathi, R.; Prag, G.; Ashkenazi, A. Ubiquitin Signaling and Degradation of Aggregate-Prone Proteins. Trends Biochem. Sci. 2019, 44, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Ziv, I.; Matiuhin, Y.; Kirkpatrick, D.S.; Erpapazoglou, Z.; Leon, S.; Pantazopoulou, M.; Kim, W.; Gygi, S.P.; Haguenauer-Tsapis, R.; Reis, N.; et al. A perturbed ubiquitin landscape distinguishes between ubiquitin in trafficking and in proteolysis. Mol. Cell. Proteom. 2011, 10, M111.009753. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Esler, W.P.; Wolfe, M.S. A portrait of Alzheimer secretases—New features and familiar faces. Science 2001, 293, 1449–1454. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Sinha, S.; Lieberburg, I. Cellular mechanisms of beta-amyloid production and secretion. Proc. Natl. Acad. Sci. USA 1999, 96, 11049–11053. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Gao, Y.L.; Wang, N.; Sun, F.R.; Cao, X.P.; Zhang, W.; Yu, J.T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef]
- Kosik, K.S. The molecular and cellular biology of tau. Brain Pathol. 1993, 3, 39–43. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Busciglio, J.; Lorenzo, A.; Yeh, J.; Yankner, B.A. Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995, 14, 879–888. [Google Scholar] [CrossRef]
- Avila, J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006, 580, 2922–2927. [Google Scholar] [CrossRef]
- Delacourte, A.; David, J.P.; Sergeant, N.; Buée, L.; Wattez, A.; Vermersch, P.; Ghozali, F.; Fallet-Bianco, C.; Pasquier, F.; Lebert, F.; et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 1999, 52, 1158–1165. [Google Scholar] [CrossRef]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Takeda, A.; Mallory, M.; Sundsmo, M.; Honer, W.; Hansen, L.; Masliah, E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am. J. Pathol. 1998, 152, 367–372. [Google Scholar] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Forman, M.S.; Lee, V.M.; Trojanowski, J.Q. Nosology of Parkinson’s disease: looking for the way out of a quagmire. Neuron 2005, 47, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Brain region-dependent differential expression of alpha-synuclein. J. Comp. Neurol. 2016, 524, 1236–1258. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tatebe, H.; Miyata, S.; Tokuda, T.; Mizuno, T.; Tanaka, M. Differential expression of alpha-synuclein in hippocampal neurons. PLoS ONE 2014, 9, e89327. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Lee, H.J.; Suk, J.E.; Jung, J.W.; Kim, K.P.; Lee, S.J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar] [CrossRef]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef]
- Fussi, N.; Höllerhage, M.; Chakroun, T.; Nykänen, N.P.; Rösler, T.W.; Koeglsperger, T.; Wurst, W.; Behrends, C.; Höglinger, G.U. Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018, 9, 757. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Gao, F.B.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, 2931–2950. [Google Scholar] [CrossRef] [PubMed]
- Farrawell, N.E.; Lambert-Smith, I.A.; Warraich, S.T.; Blair, I.P.; Saunders, D.N.; Hatters, D.M.; Yerbury, J.J. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 2015, 5, 13416. [Google Scholar] [CrossRef]
- Saberi, S.; Stauffer, J.E.; Schulte, D.J.; Ravits, J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol. Clin. 2015, 33, 855–876. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, H.J. Prion-like Mechanism in Amyotrophic Lateral Sclerosis: are Protein Aggregates the Key? Exp. Neurobiol. 2015, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Guest, W.C.; Yanai, A.; Pokrishevsky, E.; O’Neill, M.A.; Gibbs, E.; Semenchenko, V.; Yousefi, M.; Wishart, D.S.; Plotkin, S.S.; et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16398–16403. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Suzuki, N. Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNA-binding proteins TDP-43 and FUS. Neurology 2011, 77, 1636–1643. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Advani, V.M.; Ivanov, P. Stress granule subtypes: An emerging link to neurodegeneration. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Orrù, S.; Coni, P.; Floris, A.; Littera, R.; Carcassi, C.; Sogos, V.; Brancia, C. Reduced stress granule formation and cell death in fibroblasts with the A382T mutation of TARDBP gene: Evidence for loss of TDP-43 nuclear function. Hum. Mol. Genet. 2016, 25, 4473–4483. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e809. [Google Scholar] [CrossRef]
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim. Biophys. Acta 2015, 1852, 679–684. [Google Scholar] [CrossRef]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef]
- Frontali, M.; Novelletto, A.; Annesi, G.; Jodice, C. CAG repeat instability, cryptic sequence variation and pathogeneticity: evidence from different loci. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1999, 354, 1089–1094. [Google Scholar] [CrossRef][Green Version]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef]
- Yushchenko, T.; Deuerling, E.; Hauser, K. Insights into the Aggregation Mechanism of PolyQ Proteins with Different Glutamine Repeat Lengths. Biophys. J. 2018, 114, 1847–1857. [Google Scholar] [CrossRef]
- Mejia, R.O.S.; Friedlander, R.M. Caspases in Huntington’s disease. Neurosci. Rev. J. 2001, 7, 480–489. [Google Scholar] [CrossRef]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef]
- Cicchetti, F.; Lacroix, S.; Cisbani, G.; Vallières, N.; Saint-Pierre, M.; St-Amour, I.; Tolouei, R.; Skepper, J.N.; Hauser, R.A.; Mantovani, D.; et al. Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann. Neurol. 2014, 76, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.M.P.; Kopito, R.R. Prion-Like Characteristics of Polyglutamine-Containing Proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a024257. [Google Scholar] [CrossRef]
- Watanabe, Y.; Tsujimura, A.; Taguchi, K.; Tanaka, M. HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62-associated proteostasis. Autophagy 2017, 13, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Schuman, E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin–proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Jacobson, A.D. Functions of the 19S complex in proteasomal degradation. Trends Biochem. Sci. 2013, 38, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Shabek, N.; Herman-Bachinsky, Y.; Buchsbaum, S.; Lewinson, O.; Haj-Yahya, M.; Hejjaoui, M.; Lashuel, H.A.; Sommer, T.; Brik, A.; Ciechanover, A. The size of the proteasomal substrate determines whether its degradation will be mediated by mono- or polyubiquitylation. Mol. Cell 2012, 48, 87–97. [Google Scholar] [CrossRef]
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Reyes-Turcu, F.; Licchesi, J.D.; Odenwaelder, P.; Wilkinson, K.D.; Barford, D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009, 10, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 2020, 11, 477. [Google Scholar] [CrossRef]
- Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Ng, A.C.; Hara, T.; Hipp, M.S.; Lage, K.; Xavier, R.J.; Ryu, K.Y.; Taguchi, K.; et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: A potential role for protein aggregation in autophagic substrate selection. J. Cell Biol. 2010, 191, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Tanaka, M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J. Cell Sci. 2011, 124, 2692–2701. [Google Scholar] [CrossRef]
- Svenning, S.; Johansen, T. Selective autophagy. Essays Biochem. 2013, 55, 79–92. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Inoshita, T.; Hattori, N.; Imai, Y. Lysine 63-linked polyubiquitination is dispensable for Parkin-mediated mitophagy. J. Biol. Chem. 2014, 289, 33131–33136. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Cripps, D.; Thomas, S.N.; Jeng, Y.; Yang, F.; Davies, P.; Yang, A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 2006, 281, 10825–10838. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 1993, 10, 1151–1160. [Google Scholar] [CrossRef]
- Sahara, N.; Murayama, M.; Mizoroki, T.; Urushitani, M.; Imai, Y.; Takahashi, R.; Murata, S.; Tanaka, K.; Takashima, A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005, 94, 1254–1263. [Google Scholar] [CrossRef]
- Williamson, R.L.; Laulagnier, K.; Miranda, A.M.; Fernandez, M.A.; Wolfe, M.S.; Sadoul, R.; Di Paolo, G. Disruption of amyloid precursor protein ubiquitination selectively increases amyloid β (Aβ) 40 levels via presenilin 2-mediated cleavage. J. Biol. Chem. 2017, 292, 19873–19889. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328. [Google Scholar] [CrossRef]
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.S. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 2008, 17, 906–917. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [Google Scholar] [CrossRef]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef] [PubMed]
- Mulherkar, S.A.; Sharma, J.; Jana, N.R. The ubiquitin ligase E6-AP promotes degradation of alpha-synuclein. J. Neurochem. 2009, 110, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Huang, W.C.; Lin, J.H.; Kao, T.J.; Lin, H.C.; Lee, K.H.; Shen, C.J.; Chang, W.C.; Huang, C.C. Znf179 E3 ligase-mediated TDP-43 polyubiquitination is involved in TDP-43- ubiquitinated inclusions (UBI) (+)-related neurodegenerative pathology. J. Biomed. Sci. 2018, 25, 76. [Google Scholar] [CrossRef]
- Uchida, T.; Tamaki, Y.; Ayaki, T.; Shodai, A.; Kaji, S.; Morimura, T.; Banno, Y.; Nishitsuji, K.; Sakashita, N.; Maki, T.; et al. CUL2-mediated clearance of misfolded TDP-43 is paradoxically affected by VHL in oligodendrocytes in ALS. Sci. Rep. 2016, 6, 19118. [Google Scholar] [CrossRef]
- Hans, F.; Eckert, M.; von Zweydorf, F.; Gloeckner, C.J.; Kahle, P.J. Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2018, 293, 16083–16099. [Google Scholar] [CrossRef]
- Miyazaki, K.; Fujita, T.; Ozaki, T.; Kato, C.; Kurose, Y.; Sakamoto, M.; Kato, S.; Goto, T.; Itoyama, Y.; Aoki, M.; et al. NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J. Biol. Chem. 2004, 279, 11327–11335. [Google Scholar] [CrossRef]
- Fang, S.; Ferrone, M.; Yang, C.; Jensen, J.P.; Tiwari, S.; Weissman, A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 14422–14427. [Google Scholar] [CrossRef]
- Ying, Z.; Wang, H.; Fan, H.; Zhu, X.; Zhou, J.; Fei, E.; Wang, G. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum. Mol. Genet. 2009, 18, 4268–4281. [Google Scholar] [CrossRef]
- Bhat, K.P.; Yan, S.; Wang, C.E.; Li, S.; Li, X.J. Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc. Natl. Acad. Sci. USA 2014, 111, 5706–5711. [Google Scholar] [CrossRef]
- Koyuncu, S.; Saez, I.; Lee, H.J.; Gutierrez-Garcia, R.; Pokrzywa, W.; Fatima, A.; Hoppe, T.; Vilchez, D. The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat. Commun. 2018, 9, 2886. [Google Scholar] [CrossRef]
- Zucchelli, S.; Marcuzzi, F.; Codrich, M.; Agostoni, E.; Vilotti, S.; Biagioli, M.; Pinto, M.; Carnemolla, A.; Santoro, C.; Gustincich, S.; et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) associates with huntingtin protein and promotes its atypical ubiquitination to enhance aggregate formation. J. Biol. Chem. 2011, 286, 25108–25117. [Google Scholar] [CrossRef]
- Buchberger, A. From UBA to UBX: New words in the ubiquitin vocabulary. Trends Cell Biol. 2002, 12, 216–221. [Google Scholar] [CrossRef]
- Waters, S.; Marchbank, K.; Solomon, E.; Whitehouse, C.; Gautel, M. Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett. 2009, 583, 1846–1852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Raasi, S.; Fushman, D. Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J. Mol. Biol. 2008, 377, 162–180. [Google Scholar] [CrossRef]
- Swanson, K.A.; Hicke, L.; Radhakrishnan, I. Structural basis for monoubiquitin recognition by the Ede1 UBA domain. J. Mol. Biol. 2006, 358, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xu, D.; Wang, Y.; Zhou, Z.; Liu, J.; Hu, S.; Gong, Y.; Yuan, J.; Pan, L. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 2018, 14, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Gleason, C.E.; Ordureau, A.; Gourlay, R.; Arthur, J.S.; Cohen, P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon β. J. Biol. Chem. 2011, 286, 35663–35674. [Google Scholar] [CrossRef]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef]
- Xie, X.; Li, F.; Wang, Y.; Lin, Z.; Cheng, X.; Liu, J.; Chen, C.; Pan, L. Molecular basis of ubiquitin recognition by the autophagy receptor CALCOCO2. Autophagy 2015, 11, 1775–1789. [Google Scholar] [CrossRef]
- Von Muhlinen, N.; Thurston, T.; Ryzhakov, G.; Bloor, S.; Randow, F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy 2010, 6, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.K.; Kumar, A. TRAF6 coordinates the activation of autophagy and ubiquitin–proteasome systems in atrophying skeletal muscle. Autophagy 2011, 7, 555–556. [Google Scholar] [CrossRef]
- Walinda, E.; Morimoto, D.; Sugase, K.; Konuma, T.; Tochio, H.; Shirakawa, M. Solution structure of the ubiquitin-associated (UBA) domain of human autophagy receptor NBR1 and its interaction with ubiquitin and polyubiquitin. J. Biol. Chem. 2014, 289, 13890–13902. [Google Scholar] [CrossRef]
- Sánchez-Martín, P.; Komatsu, M. p62/SQSTM1—Steering the cell through health and disease. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef]
- Nicot, A.S.; Lo Verso, F.; Ratti, F.; Pilot-Storck, F.; Streichenberger, N.; Sandri, M.; Schaeffer, L.; Goillot, E. Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy 2014, 10, 1036–1053. [Google Scholar] [CrossRef]
- Deng, Z.; Purtell, K.; Lachance, V.; Wold, M.S.; Chen, S.; Yue, Z. Autophagy Receptors and Neurodegenerative Diseases. Trends Cell Biol. 2017, 27, 491–504. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Nixon, R.A. Rapamycin induces autophagic flux in neurons. Proc. Natl. Acad. Sci. USA 2010, 107, E181. [Google Scholar] [CrossRef]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Tatebe, H.; Taguchi, K.; Endo, Y.; Tokuda, T.; Mizuno, T.; Nakagawa, M.; Tanaka, M. p62/SQSTM1-dependent autophagy of Lewy body-like α-synuclein inclusions. PLoS ONE 2012, 7, e52868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014, 10, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Goedert, M. Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy 2012, 8, 1686–1687. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847. [Google Scholar] [CrossRef]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Cano-Cuenca, N.; Solís-García del Pozo, J.E.; Jordán, J. Evidence for the efficacy of latrepirdine (Dimebon) treatment for improvement of cognitive function: A meta-analysis. J. Alzheimer’s Dis. 2014, 38, 155–164. [Google Scholar] [CrossRef]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Suresh, S.N.; Chakravorty, A.; Giridharan, M.; Garimella, L.; Manjithaya, R. Pharmacological Tools to Modulate Autophagy in Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2822–2842. [Google Scholar] [CrossRef]
- Lim, K.H.; Joo, J.Y.; Baek, K.H. The potential roles of deubiquitinating enzymes in brain diseases. Ageing Res. Rev. 2020, 61, 101088. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, Y.; Taguchi, K.; Tanaka, M. Ubiquitin, Autophagy and Neurodegenerative Diseases. Cells 2020, 9, 2022. https://doi.org/10.3390/cells9092022
Watanabe Y, Taguchi K, Tanaka M. Ubiquitin, Autophagy and Neurodegenerative Diseases. Cells. 2020; 9(9):2022. https://doi.org/10.3390/cells9092022
Chicago/Turabian StyleWatanabe, Yoshihisa, Katsutoshi Taguchi, and Masaki Tanaka. 2020. "Ubiquitin, Autophagy and Neurodegenerative Diseases" Cells 9, no. 9: 2022. https://doi.org/10.3390/cells9092022
APA StyleWatanabe, Y., Taguchi, K., & Tanaka, M. (2020). Ubiquitin, Autophagy and Neurodegenerative Diseases. Cells, 9(9), 2022. https://doi.org/10.3390/cells9092022