Novel Opportunities for Cathepsin S Inhibitors in Cancer Immunotherapy by Nanocarrier-Mediated Delivery


Abstract
1. Introduction
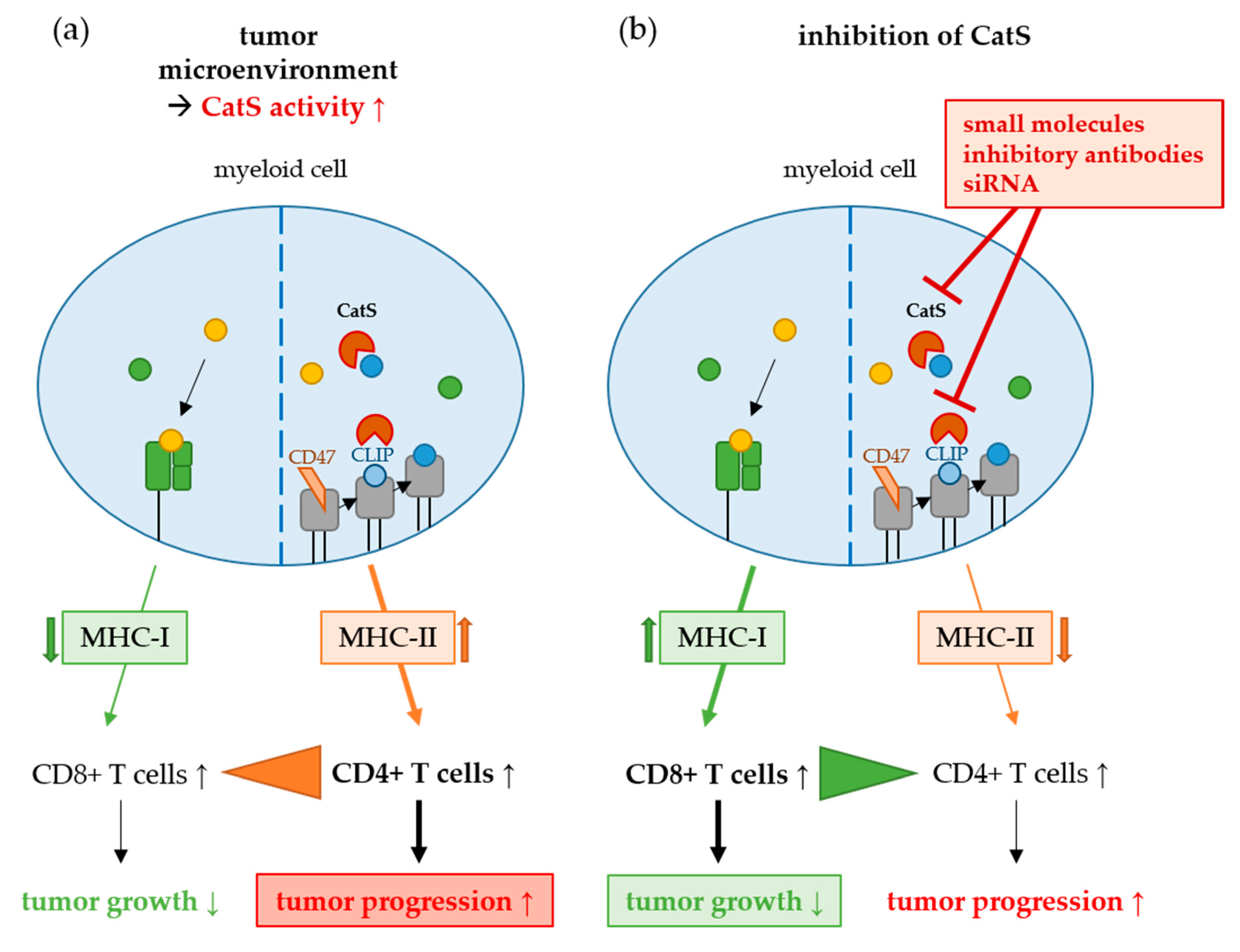
2. Cysteine Cathepsins in Tumor Progression
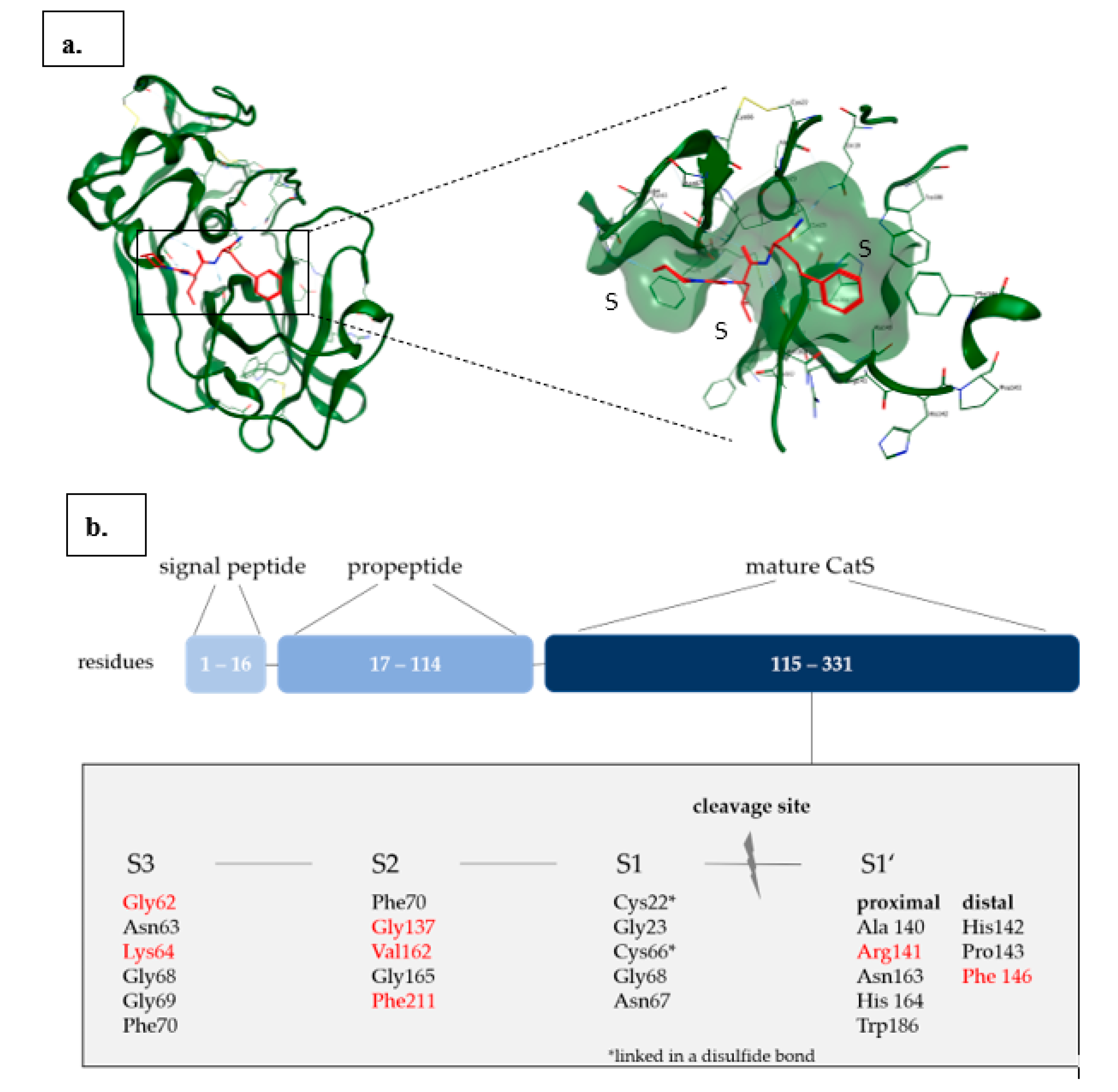
Cathepsin S
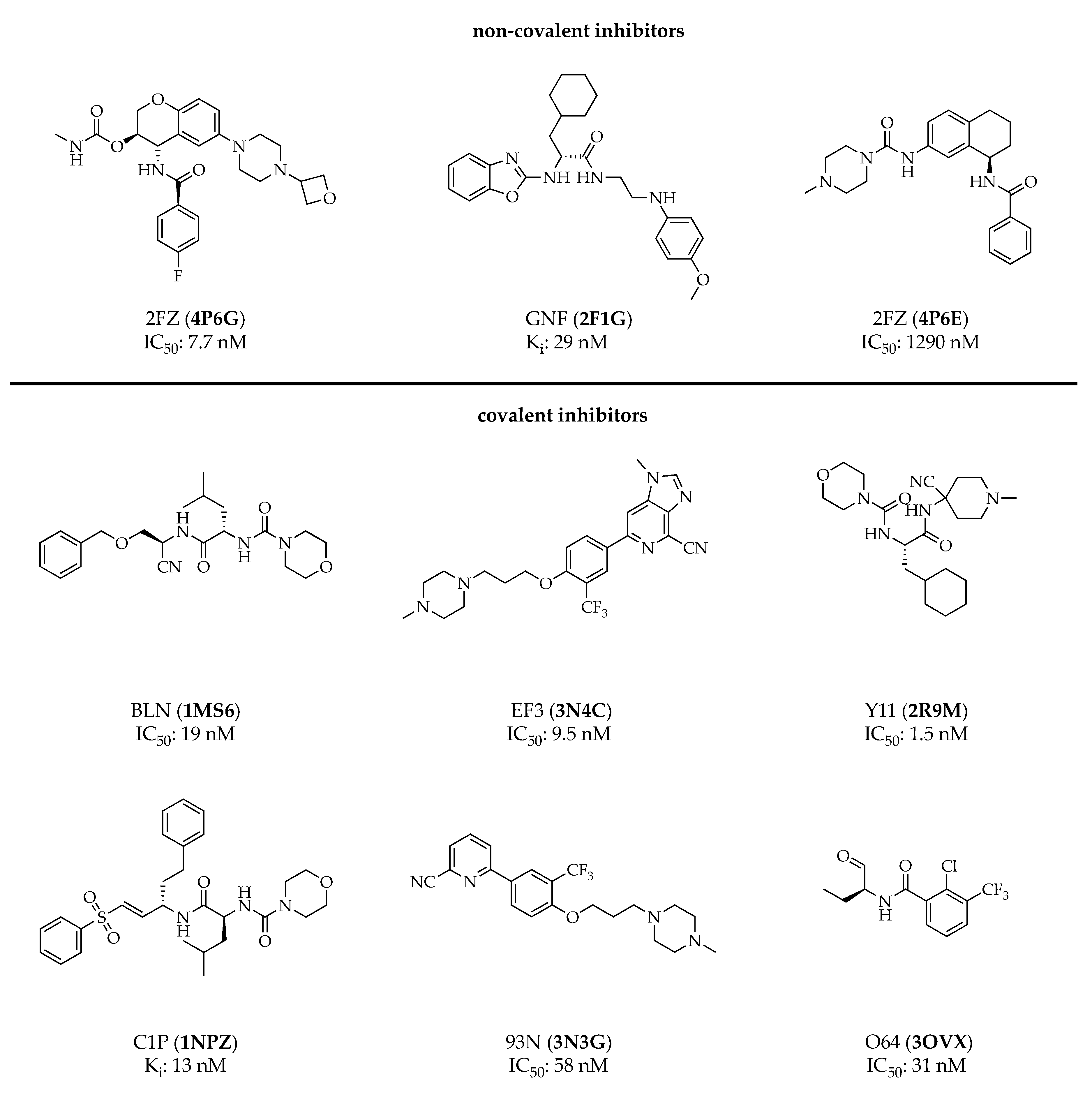
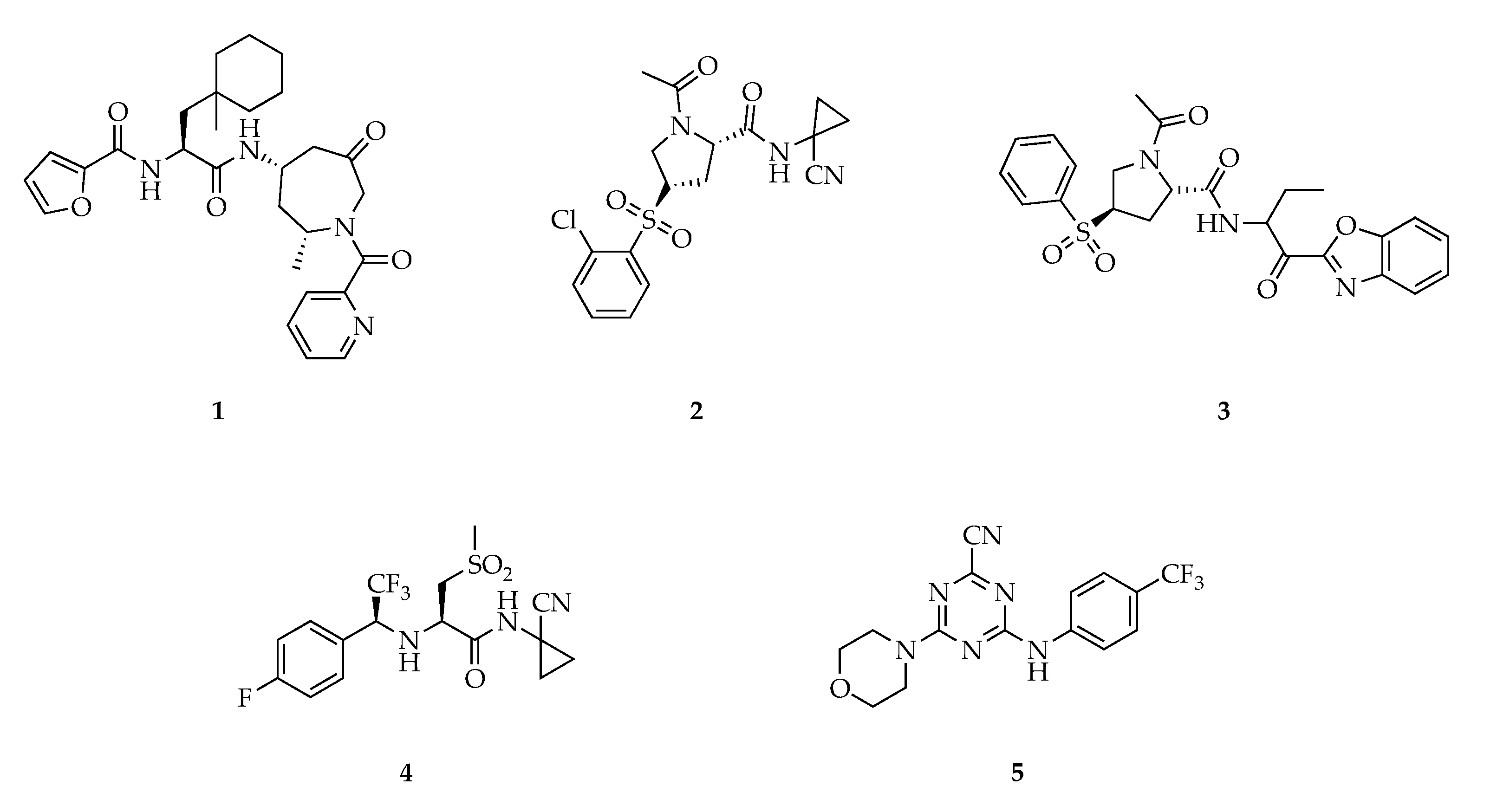
3. Cathepsin S Inhibitors
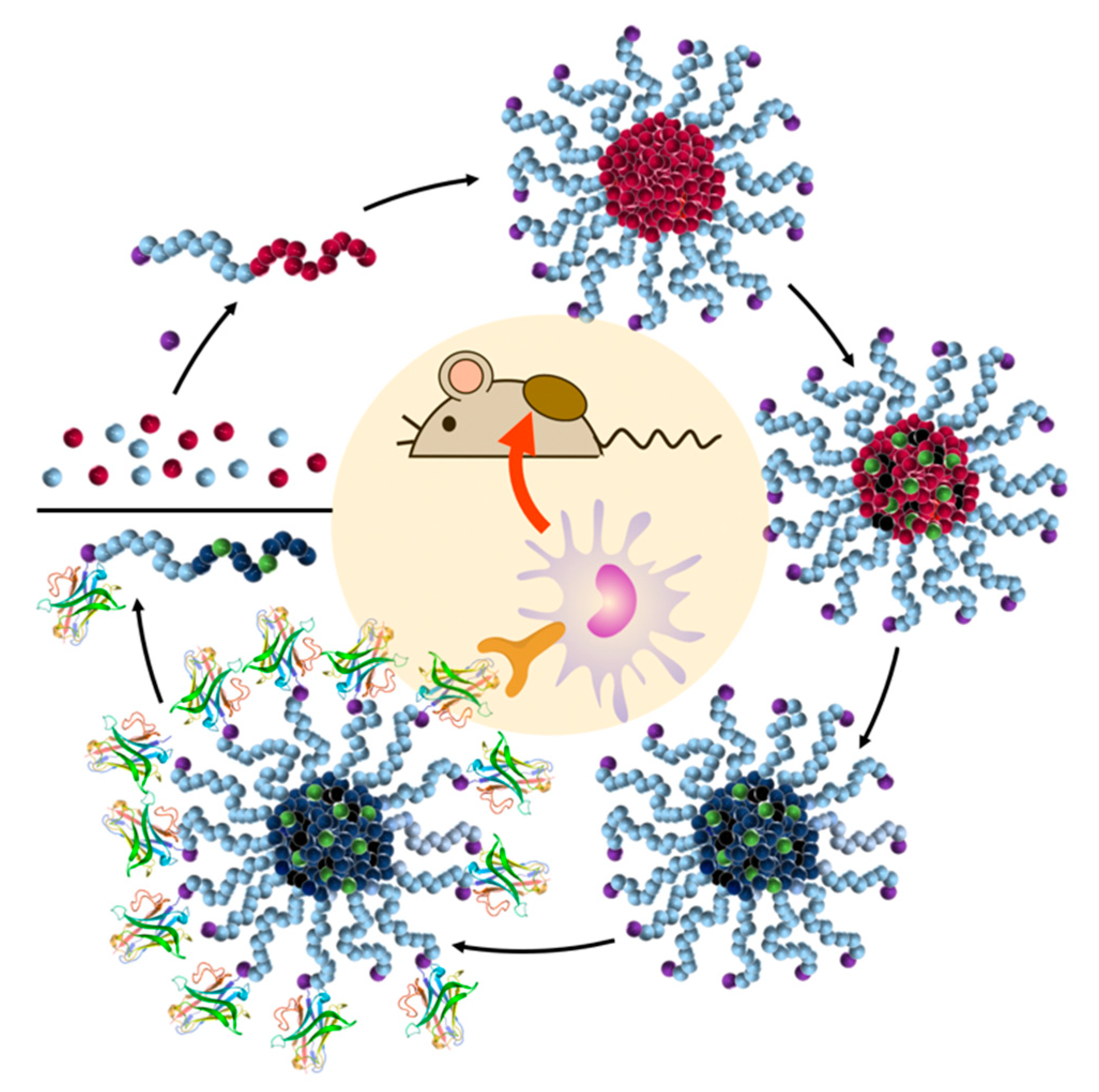
4. Nanocarrier-Mediated Delivery of Cathepsin Inhibitors
5. Conclusion and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cell; |
| Cat | cathepsin; |
| CatB | cathepsin B; |
| CatK | cathepsin K; |
| CatL | cathepsin L; |
| CatS | cathepsin S; |
| CCL2 | CC-chemokine ligand-2; |
| CD206 | cluster of differentiation 206; |
| COX2 | cyclooxygenase 2; |
| DC | dendritic cell; |
| EC | endothelial cell; |
| ECM | extracellular matrix; |
| EPR | enhanced permeability and retention; |
| FDA | Food and Drug Administration; |
| HPMA | poly(2-hydroxypropyl methacrylamide); |
| IL-6 | interleukin 6; |
| JAM-B | junctional adhesion molecule B; |
| LE | ligand efficiency; |
| LHVS | leucine homophenylalanine vinyl sulfone; |
| MC38 | murine colon adenocarcinoma; |
| MCF7 | Michigan Cancer Foundation–7; |
| MDSC | myeloid-derived suppressor cell; |
| MHC | major histocompatibility complex; |
| MMR | mannose receptor; |
| MRI | magnetic resonance imaging; |
| PDB | protein database; |
| PEG | polyethylene glycol; |
| PLGA | PEG-block-poly-lactic-co-glycolic acid; |
| rhBMP-2 | recombinant human bone morphogenetic protein-2; |
| SAR | structure–activity relationship; |
| siRNA | small interfering RNA; |
| TAM | tumor-associated macrophages; |
| TME | tumor microenvironment; |
| Treg | regulatory T cell; |
| ZINC | ZINC is not commercial; |
References
- Bararia, D.; Hildebrand, J.A.; Stolz, S.; Haebe, S.; Alig, S.; Trevisani, C.P.; Osorio-Barrios, F.; Bartoschek, M.D.; Mentz, M.; Pastore, A.; et al. Cathepsin S Alterations Induce a Tumor-Promoting Immune Microenvironment in Follicular Lymphoma. Cell Rep. 2020, 31, 107–522. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, A.C.; Santa-Cruz, F.; Mattos, L.A.R.; Aquino, M.A.R.; Martins, C.R.; Ferraz Álvaro, A.B.; Figueiredo, J.L. Cathepsin S as a target in gastric cancer (Review). Mol. Clin. Oncol. 2019, 12, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Dheilly, E.; Battistello, E.; Katanayeva, N.; Sungalee, S.; Michaux, J.; Duns, G.; Wehrle, S.; Sordet-Dessimoz, J.; Mina, M.; Racle, J.; et al. Cathepsin S Regulates Antigen Processing and T Cell Activity in Non-Hodgkin Lymphoma. Cancer Cell 2020, 37, 674–689.e12. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.; Williams, R.; Scott, C.J.; Burden, R.E.; Williams, R. Cathepsin S: Therapeutic, diagnostic, and prognostic potential. Biol. Chem. 2015, 396, 867–882. [Google Scholar] [CrossRef]
- Jakoš, T.; Pišlar, A.; Jewett, A.; Kos, J. Cysteine Cathepsins in Tumor-Associated Immune Cells. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef]
- McDowell, S.H.; Gallaher, S.A.; Burden, R.E.; Scott, C.J. Leading the invasion: The role of Cathepsin S in the tumour microenvironment. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2020, 1867, 118–781. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2018, 234, 8509–8521. [Google Scholar] [CrossRef]
- Quaranta, V.; Schmid, M.C. Macrophage-Mediated Subversion of Anti-Tumour Immunity. Cells 2019, 8, 747. [Google Scholar] [CrossRef]
- Riese, R.J.; Wolf, P.R.; Brömme, D.; Natkin, L.R.; Villadangos, J.A.; Ploegh, H.L.; Chapman, H.A. Essential Role for Cathepsin S in MHC Class II–Associated Invariant Chain Processing and Peptide Loading. Immunity 1996, 4, 357–366. [Google Scholar] [CrossRef]
- Riese, R.J.; Mitchell, R.N.; Villadangos, J.A.; Shi, G.P.; Palmer, J.T.; Karp, E.R.; De Sanctis, G.T.; Ploegh, H.L.; Chapman, H.A. Cathepsin S activity regulates antigen presentation and immunity. J. Clin. Investig. 1998, 101, 2351–2363. [Google Scholar] [CrossRef]
- Shi, G.-P.; Villadangos, J.A.; Dranoff, G.; Small, C.; Gu, L.; Haley, K.J.; Riese, R.; Ploegh, H.L.; Chapman, H.A. Cathepsin S Required for Normal MHC Class II Peptide Loading and Germinal Center Development. Immunity 1999, 10, 197–206. [Google Scholar] [CrossRef]
- Hsing, L.C.; Rudensky, A.Y. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 2005, 207, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wu, C.; Chen, T.; Santos, M.M.; Liu, C.-L.; Yang, C.; Zhang, L.; Ren, J.; Liao, S.; Guo, H.; et al. Cathepsin S inhibition changes regulatory T-cell activity in regulating bladder cancer and immune cell proliferation and apoptosis. Mol. Immunol. 2017, 82, 66–74. [Google Scholar] [CrossRef]
- Prunk, M.; Kos, J. Nanoparticle Based Delivery of Protease Inhibitors to Cancer Cells. Curr. Med. Chem. 2018, 24, 4816–4837. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Vizin, T.; Fonović, U.P.; Pišlar, A. Intracellular signaling by cathepsin X: Molecular mechanisms and diagnostic and therapeutic opportunities in cancer. Semin. Cancer Biol. 2015, 31, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Lah, T.T. Cysteine proteinases and their endogenous inhibitors: Target proteins for prognosis, diagnosis and therapy in cancer (review). Oncol. Rep. 1998, 5, 1349–1361. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Sudhan, D.; Siemann, D.W. Cathepsin L targeting in cancer treatment. Pharmacol. Ther. 2015, 155, 105–116. [Google Scholar] [CrossRef]
- Gocheva, V.; Wang, H.-W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef]
- Kostoulas, G.; Lang, A.; Nagase, H.; Baici, A. Stimulation of angiogenesis through cathepsin B inactivation of the tissue inhibitors of matrix metalloproteinases. FEBS Lett. 1999, 455, 286–290. [Google Scholar] [CrossRef]
- Brömme, D.; Wilson, S. Role of Cysteine Cathepsins in Extracellular Proteolysis. Extracellular Matrix Degradation 2011, 23–51. [Google Scholar] [CrossRef]
- Wang, B.; Sun, J.; Kitamoto, S.; Yang, M.; Grubb, A.; Chapman, H.A.; Kalluri, R.; Shi, G.-P. Cathepsin S Controls Angiogenesis and Tumor Growth via Matrix-derived Angiogenic Factors. J. Biol. Chem. 2005, 281, 6020–6029. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.M.; Aucher, A.; Alyahya, S.; Elder, M.; Olson, S.T.; Davis, D.M.; Ashton-Rickardt, P.G. Cathepsin B controls the persistence of memory CD8+ T lymphocytes. J. Immunol. 2012, 189, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Gounaris, E.; Tung, C.-H.; Restaino, C.; Maehr, R.; Köhler, R.; Joyce, J.A.; Plough, H.L.; Barrett, T.A.; Weissleder, R.; Khazaie, K. Live Imaging of Cysteine-Cathepsin Activity Reveals Dynamics of Focal Inflammation, Angiogenesis, and Polyp Growth. PLoS ONE 2008, 3, e2916. [Google Scholar] [CrossRef]
- Herroon, M.K.; Rajagurubandara, E.; Rudy, D.L.; Chalasani, A.; Hardaway, A.L.; Podgorski, I. Macrophage cathepsin K promotes prostate tumor progression in bone. Oncogene 2012, 32, 1580–1593. [Google Scholar] [CrossRef]
- Plüger, E.B.E.; Boes, M.; Alfonso, C.; Schröter, C.J.; Kalbacher, H.; Ploegh, H.L.; Driessen, C. Specific role for cathepsin S in the generation of antigenic peptides in vivo. Eur. J. Immunol. 2002, 32, 467–476. [Google Scholar] [CrossRef]
- Wilkinson, R.; Magorrian, S.M.; Williams, R.; Young, A.; Small, D.M.; Scott, C.J.; Burden, R.E. CCL2 is transcriptionally controlled by the lysosomal protease cathepsin S in a CD74-dependent manner. Oncotarget 2015, 6, 29725–29739. [Google Scholar] [CrossRef]
- Jevnikar, Z.; Obermajer, N.; Bogyo, M.; Kos, J. The role of cathepsin X in the migration and invasiveness of T lymphocytes. J. Cell Sci. 2008, 121, 2652–2661. [Google Scholar] [CrossRef]
- Somoza, J.R.; Zhan, H.; Bowman, K.K.; Yu, L.; Mortara, K.D.; Palmer, J.T.; Clark, J.M.; McGrath, M.E. Crystal Structure of Human Cathepsin V. Biochemistry 2000, 39, 12543–12551. [Google Scholar] [CrossRef]
- Kopitar, G.; Dolinar, M.; Strukelj, B.; Pungerčar, J.; Turk, V. Folding and Activation of Human Procathepsin S from Inclusion Bodies Produced in Escherichia coli. JBIC J. Biol. Inorg. Chem. 1996, 236, 558–562. [Google Scholar] [CrossRef]
- Shi, G.P.; Munger, J.S.; Meara, J.P.; Rich, D.H.; Chapman, H.A. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J. Biol. Chem. 1992, 267, 7258–7262. [Google Scholar] [PubMed]
- Pauly, T.A.; Sulea, T.; Ammirati, M.; Sivaraman, J.; Danley, D.E.; Griffor, M.C.; Kamath, A.V.; Wang, I.-K.; Laird, E.R.; Seddon, A.P.; et al. Specificity Determinants of Human Cathepsin S Revealed by Crystal Structures of Complexes. Biochemistry 2003, 42, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, H.; Wiederanders, B.; Brömme, D.; Rinne, A. Cathepsin S from bovine spleen. Purification, distribution, intracellular localization and action on proteins. Biochem. J. 1989, 264, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.P.; Webb, A.C.; Foster, K.E.; Knoll, J.H.; Lemere, C.A.; Munger, J.S.; Chapman, H.A. Human cathepsin S: Chromosomal localization, gene structure, and tissue distribution. J. Biol. Chem. 1994, 269, 11530–11536. [Google Scholar]
- Liu, W.-L.; Liu, D.; Cheng, K.; Liu, Y.-J.; Xing, S.; Chi, P.-D.; Liu, X.-H.; Xue, N.; Lai, Y.-Z.; Guo, L.; et al. Evaluating the diagnostic and prognostic value of circulating cathepsin S in gastric cancer. Oncotarget 2016, 7, 28124–28138. [Google Scholar] [CrossRef]
- Yixuan, Y.; Kiat, L.S.; Yee, C.L.; Huiyin, L.; Yunhao, C.; Kuan, C.P.; Hassan, A.; Ting, W.T.; Manuel, S.-T.; Guan, Y.K.; et al. Cathepsin S Mediates Gastric Cancer Cell Migration and Invasion via a Putative Network of Metastasis-Associated Proteins. J. Proteome Res. 2010, 9, 4767–4778. [Google Scholar] [CrossRef]
- Sevenich, L.; Bowman, R.L.; Mason, S.D.; Quail, D.F.; Rapaport, F.; Elie, B.T.; Brogi, E.; Brastianos, P.K.; Hahn, W.C.; Holsinger, L.J.; et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat. Cell Biol. 2014, 16, 876–888. [Google Scholar] [CrossRef]
- Yang, M.; Liu, J.; Shao, J.; Qin, Y.; Ji, Q.; Zhang, X.; Du, J. Cathepsin S-mediated autophagic flux in tumor-associated macrophages accelerate tumor development by promoting M2 polarization. Mol. Cancer 2014, 13, 43. [Google Scholar] [CrossRef]
- Burden, R.E.; Gormley, J.A.; Kuehn, D.; Ward, C.; Kwok, H.F.; Gazdoiu, M.; McClurg, A.; Jaquin, T.J.; Johnston, J.A.; Scott, C.J.; et al. Inhibition of Cathepsin S by Fsn0503 enhances the efficacy of chemotherapy in colorectal carcinomas. Biochemistry 2012, 94, 487–493. [Google Scholar] [CrossRef]
- Burden, R.E.; Gormley, J.A.; Jaquin, T.J.; Small, D.M.; Quinn, D.J.; Hegarty, S.M.; Ward, C.; Walker, B.; Johnston, J.A.; Olwill, S.A.; et al. Antibody-Mediated Inhibition of Cathepsin S Blocks Colorectal Tumor Invasion and Angiogenesis. Clin. Cancer Res. 2009, 15, 6042–6051. [Google Scholar] [CrossRef]
- Fan, Q.; Wang, X.; Zhang, H.; Li, C.; Fan, J.; Xu, J. Silencing cathepsin S gene expression inhibits growth, invasion and angiogenesis of human hepatocellular carcinoma in vitro. Biochem. Biophys. Res. Commun. 2012, 425, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Lang, Z.; Xu, J.; Li, N.; Ke, Z.; Liu, R.; Maubach, G. Cathepsin S is aberrantly overexpressed in human hepatocellular carcinoma. Mol. Med. Rep. 2009, 2. [Google Scholar] [CrossRef]
- Ryschich, E.; Lizdenis, P.; Ittrich, C.; Benner, A.; Stahl, S.H.; Hamann, A.; Schmidt, J.; Knolle, P.A.; Arnold, B.; Hämmerling, G.J.; et al. Molecular Fingerprinting and Autocrine Growth Regulation of Endothelial Cells in a Murine Model of Hepatocellular Carcinoma. Cancer Res. 2006, 66, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Otto, H.-H.; Schirmeister, T. Cysteine Proteases and Their Inhibitors. Chem. Rev. 1997, 97, 133–172. [Google Scholar] [CrossRef]
- Lecaille, F.; Kaleta, J.; Brömme, D. Human and Parasitic Papain-Like Cysteine Proteases: Their Role in Physiology and Pathology and Recent Developments in Inhibitor Design. Chem. Rev. 2002, 102, 4459–4488. [Google Scholar] [CrossRef]
- Kang, K.; Kim, W. Recent developments of cathepsin inhibitors and their selectivity. Expert Opin. Ther. Pat. 2002, 12, 419–432. [Google Scholar] [CrossRef]
- Leung-Toung, R.; Li, W.; Tam, T.; Kaarimian, K. Thiol-Dependent Enzymes and Their Inhibitors: A Review. Curr. Med. Chem. 2002, 9, 979–1002. [Google Scholar] [CrossRef]
- Leroy, V. Cathepsin S inhibitors. Expert Opin. Ther. Pat. 2004, 14, 301–311. [Google Scholar] [CrossRef]
- Gauthier, J.Y.; Black, W.C.; Courchesne, I.; Cromlish, W.; Desmarais, S.; Houle, R.; Lamontagne, S.; Li, C.S.; Massé, F.; McKay, D.J.; et al. The identification of potent, selective, and bioavailable cathepsin S inhibitors. Bioorganic Med. Chem. Lett. 2007, 17, 4929–4933. [Google Scholar] [CrossRef]
- Lee-Dutra, A.; Wiener, D.K.; Sun, S. Cathepsin S inhibitors: 2004–2010. Expert Opin. Ther. Pat. 2011, 21, 311–337. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.J.M.; Sun, S.; Thurmond, R.L. Recent advances in the design of cathepsin S inhibitors. Curr. Top. Med. Chem. 2010, 10, 717–732. [Google Scholar] [CrossRef]
- Ward, Y.D.; Thomson, D.S.; Frye, L.L.; Cywin, C.L.; Morwick, T.; Emmanuel, M.J.; Zindell, R.; McNeil, D.; Bekkali, Y.; Hrapchak, M.; et al. Design and Synthesis of Dipeptide Nitriles as Reversible and Potent Cathepsin S Inhibitors. J. Med. Chem. 2002, 45, 5471–5482. [Google Scholar] [CrossRef] [PubMed]
- Tully, D.C.; Liu, H.; Alper, P.B.; Chatterjee, A.K.; Epple, R.; Roberts, M.J.; Williams, J.A.; Nguyen, K.T.; Woodmansee, D.H.; Tumanut, C.; et al. Synthesis and evaluation of arylaminoethyl amides as noncovalent inhibitors of cathepsin S. Part 3: Heterocyclic P3. Bioorganic Med. Chem. Lett. 2006, 16, 1975–1980. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Fradera, X.; Van Zeeland, M.; Dempster, M.; Cameron, K.S.; Bennett, D.J.; Robinson, J.; Popplestone, L.; Baugh, M.; Westwood, P.; et al. 4-(3-Trifluoromethylphenyl)-pyrimidine-2-carbonitrile as cathepsin S inhibitors: N3, not N1 is critically important. Bioorganic Med. Chem. Lett. 2010, 20, 4507–4510. [Google Scholar] [CrossRef]
- Jadhav, P.K.; Schiffler, M.A.; Gavardinas, K.; Kim, E.J.; Matthews, D.P.; Staszak, M.A.; Coffey, D.S.; Shaw, B.W.; Cassidy, K.C.; Brier, R.A.; et al. Discovery of Cathepsin S Inhibitor LY3000328 for the Treatment of Abdominal Aortic Aneurysm. ACS Med. Chem. Lett. 2014, 5, 1138–1142. [Google Scholar] [CrossRef]
- Hilpert, H.; Mauser, H.; Humm, R.; Anselm, L.; Kuehne, H.; Hartmann, G.; Gruener, S.; Banner, D.W.; Benz, J.; Gsell, B.; et al. Identification of Potent and Selective Cathepsin S Inhibitors Containing Different Central Cyclic Scaffolds. J. Med. Chem. 2013, 56, 9789–9801. [Google Scholar] [CrossRef]
- Ahmad, S.; Bhagwati, S.; Kumar, S.; Banerjee, D.; Siddiqi, M.I. Molecular modeling assisted identification and biological evaluation of potent cathepsin S inhibitors. J. Mol. Graph. Model. 2020, 96, 107512. [Google Scholar] [CrossRef]
- Kerns, J.K.; Nie, H.; Bondinell, W.; Widdowson, K.L.; Yamashita, D.S.; Rahman, A.; Podolin, P.L.; Carpenter, D.C.; Jin, Q.; Riflade, B.; et al. Azepanone-based inhibitors of human cathepsin S: Optimization of selectivity via the P2 substituent. Bioorganic Med. Chem. Lett. 2011, 21, 4409–4415. [Google Scholar] [CrossRef]
- Kim, M.; Jeon, J.; Song, J.; Suh, K.H.; Kim, Y.H.; Min, K.-H.; Lee, K.-O. Synthesis of proline analogues as potent and selective cathepsin S inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 3140–3144. [Google Scholar] [CrossRef]
- Wilkinson, R.; Young, A.; Burden, R.E.; Williams, R.; Scott, C.J. A bioavailable cathepsin S nitrile inhibitor abrogates tumor development. Mol. Cancer 2016, 15, 29. [Google Scholar] [CrossRef]
- Tber, Z.; Wartenberg, M.; Jacques, J.-E.; Roy, V.; Lecaille, F.; Warszycki, D.; Bojarski, A.J.; Lalmanach, G.; Agrofoglio, L.A. Selective inhibition of human cathepsin S by 2,4,6-trisubstituted 1,3,5-triazine analogs. Bioorganic Med. Chem. 2018, 26, 4310–4319. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of Nanotechnology on Drug Delivery. Acs Nano 2009, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanoparticle Delivery of Cancer Drugs. Annu. Rev. Med. 2012, 63, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef]
- Toth, I.; Skwarczynski, M. The immune system likes nanotechnology. Nanomedicine 2014, 9, 2607–2609. [Google Scholar] [CrossRef]
- Lepeltier, E.; Nuhn, L.; Lehr, C.; Zentel, R. Not just for tumor targeting: Unmet medical needs and opportunities for nanomedicine. Nanomedicine 2015, 10, 3147–3166. [Google Scholar] [CrossRef]
- Irvine, D.J.; Dane, E.L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving cancer immunotherapy using nanomedicines: Progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Lybaert, L.; Vermaelen, K.; de Geest, B.G.; Nuhn, L. Immunoengineering through cancer vaccines—A personalized and multi-step vaccine approach towards precise cancer immunity. J. Control. Release 2018. [Google Scholar] [CrossRef]
- Duncan, R.; Gaspar, R.S. Nanomedicine(s) under the Microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of proteins and liposomes: A powerful and flexible strategy to improve the drug delivery. Curr. Drug Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef]
- Kim, T.-Y. Phase I and Pharmacokinetic Study of Genexol-PM, a Cremophor-Free, Polymeric Micelle-Formulated Paclitaxel, in Patients with Advanced Malignancies. Clin. Cancer Res. 2004, 10, 3708–3716. [Google Scholar] [CrossRef]
- Sah, H.; Thoma, L.A.; Desu, H.R.; Sah, E.; Wood, G.C. Concepts and practices used to develop functional PLGA-based nanoparticulate systems. Int. J. Nanomed. 2013, 8, 747–765. [Google Scholar] [CrossRef]
- Cogo, F.; Williams, R.; Burden, R.E.; Scott, C.J. Application of nanotechnology to target and exploit tumour associated proteases. Biochemistry 2019, 166, 112–131. [Google Scholar] [CrossRef] [PubMed]
- Cegnar, M.; Kos, J.; Kristl, J. Cystatin incorporated in poly(lactide-co-glycolide) nanoparticles: Development and fundamental studies on preservation of its activity. Eur. J. Pharm. Sci. 2004, 22, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Cegnar, M.; Premzl, A.; Zavašnik-Bergant, V.; Kristl, J.; Kos, J. Poly(lactide-co-glycolide) nanoparticles as a carrier system for delivering cysteine protease inhibitor cystatin into tumor cells. Exp. Cell Res. 2004, 301, 223–231. [Google Scholar] [CrossRef]
- Cegnar, M.; Kos, J.; Kristl, J. Intracellular delivery of cysteine protease inhibitor cystatin by polymeric nanoparticles. J. Nanosci. Nanotechnol. 2006, 6, 3087–3094. [Google Scholar] [CrossRef]
- Varshosaz, J.; Fard, M.M.; Mirian, M.; Hassanzadeh, F. Targeted Nanoparticles for Co-delivery of 5-FU and Nitroxoline, a Cathepsin B Inhibitor, in HepG2 Cells of Hepatocellular Carcinoma. Anti-Cancer Agents Med. Chem. 2020, 20, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Fathi, A.; Murphy, C.M.; Mikulec, K.; Peacock, L.; Cantrill, L.C.; Dehghani, F.; Little, D.G.; Schindeler, A. Local co-delivery of rh BMP -2 and cathepsin K inhibitor L006235 in poly(d,l -lactide- co -glycolide) nanospheres. J. Biomed. Mater. Res. Part B Appl. Biomater. 2015, 105, 136–144. [Google Scholar] [CrossRef]
- Kumar, V.; Wang, L.; Riebe, M.; Tung, H.-H.; Prud’Homme, R.K. Formulation and Stability of Itraconazole and Odanacatib Nanoparticles: Governing Physical Parameters. Mol. Pharm. 2009, 6, 1118–1124. [Google Scholar] [CrossRef]
- Mikhaylov, G.; Mikac, U.; Magaeva, A.A.; Itin, V.I.; Naiden, E.P.; Psakhye, I.; Babes, L.; Reinheckel, T.; Peters, C.; Zeiser, R.; et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nat. Nanotechnol. 2011, 6, 594–602. [Google Scholar] [CrossRef]
- Mikhaylov, G.; Klimpel, D.; Schaschke, N.; Mikac, U.; Vizovišek, M.; Fonović, M.; Turk, V.; Turk, B.; Vasiljeva, O. Selective Targeting of Tumor and Stromal Cells By a Nanocarrier System Displaying Lipidated Cathepsin B Inhibitor**. Angew. Chem. Int. Ed. 2014, 53, 10077–10081. [Google Scholar] [CrossRef]
- Bratovš, A.; Kramer, L.; Mikhaylov, G.; Vasiljeva, O.; Turk, B. Stefin A-functionalized liposomes as a system for cathepsins S and L-targeted drug delivery. Biochemistry 2019, 166, 94–102. [Google Scholar] [CrossRef]
- Tabish, T.A.; Pranjol, Z.I.; Whatmore, J.L.; Zhang, S. Status and Future Directions of Anti-metastatic Cancer Nanomedicines for the Inhibition of Cathepsin L. Front. Nanotechnol. 2020, 2, 1–10. [Google Scholar] [CrossRef]
- Junior, J.C.Q.; Carlos, F.D.R.R.; Montanari, A.; Leitão, A.; Mignone, V.W.; Arruda, M.A.; Turyanska, L.; Bradshaw, T.D. Apoferritin encapsulation of cysteine protease inhibitors for cathepsin L inhibition in cancer cells. RSC Adv. 2019, 9, 36699–36706. [Google Scholar] [CrossRef]
- Wang, N.; Pechar, M.; Li, W.; Kopečková, P.; Brömme, D.; Kopeček, J. Inhibition of Cathepsin K with Lysosomotropic Macromolecular Inhibitors. Biochemistry 2002, 41, 8849–8859. [Google Scholar] [CrossRef]
- Wang, D.; Li, W.; Pechar, M.; Kopečková, P.; Brömme, D.; Kopeček, J. Cathepsin K inhibitor–polymer conjugates: Potential drugs for the treatment of osteoporosis and rheumatoid arthritis. Int. J. Pharm. 2004, 277, 73–79. [Google Scholar] [CrossRef]
- Hemmelmann, M.; Mohr, K.; Fischer, K.; Zentel, R.; Schmidt, M. Interaction of pHPMA–pLMA Copolymers with Human Blood Serum and Its Components. Mol. Pharm. 2013, 10, 3769–3775. [Google Scholar] [CrossRef] [PubMed]
- Alberg, I.; Kramer, S.; Schinnerer, M.; Hu, Q.; Seidl, C.; Leps, C.; Drude, N.; Möckel, D.; Rijcken, C.; Lammers, T.; et al. Polymeric Nanoparticles: Polymeric Nanoparticles with Neglectable Protein Corona (Small 18/2020). Small 2020, 16. [Google Scholar] [CrossRef]
- Talelli, M.; Barz, M.; Rijcken, C.J.; Kiessling, F.; Hennink, W.E.; Lammers, T. Core-crosslinked polymeric micelles: Principles, preparation, biomedical applications and clinical translation. Nano Today 2015, 10, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Leber, N.; Nuhn, L.; Zentel, R. Cationic Nanohydrogel Particles for Therapeutic Oligonucleotide Delivery. Macromol. Biosci. 2017, 17, 1700092. [Google Scholar] [CrossRef]
- Stickdorn, J.; Nuhn, L. Reactive-ester derived polymer nanogels for cancer immunotherapy. Eur. Polym. J. 2020, 124, 109481. [Google Scholar] [CrossRef]
- Nuhn, L.; Braun, L.; Overhoff, I.; Kelsch, A.; Schaeffel, D.; Koynov, K.; Zentel, R. Degradable Cationic Nanohydrogel Particles for Stimuli-Responsive Release of siRNA. Macromol. Rapid Commun. 2014, 35, 2057–2064. [Google Scholar] [CrossRef]
- Nuhn, L.; Van Herck, S.; Best, A.; Deswarte, K.; Kokkinopoulou, M.; Lieberwirth, I.; Koynov, K.; Lambrecht, B.N.; De Geest, B.G. FRET Monitoring of Intracellular Ketal Hydrolysis in Synthetic Nanoparticles. Angew. Chem. Int. Ed. 2018, 57, 10760–10764. [Google Scholar] [CrossRef]
- Nuhn, L.; Vanparijs, N.; De Beuckelaer, A.; Lybaert, L.; Verstraete, G.; Deswarte, K.; Lienenklaus, S.; Shukla, N.M.; Salyer, A.C.D.; Lambrecht, B.N.; et al. pH-degradable imidazoquinoline-ligated nanogels for lymph node-focused immune activation. Proc. Natl. Acad. Sci. USA 2016, 113, 8098–8103. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, L.; Van Hoecke, L.; Deswarte, K.; Schepens, B.; Li, Y.; Lambrecht, B.N.; De Koker, S.; David, S.A.; Saelens, X.; De Geest, B.G. Potent anti-viral vaccine adjuvant based on pH-degradable nanogels with covalently linked small molecule imidazoquinoline TLR7/8 agonist. Biomaterials 2018, 178, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, L.; De Koker, S.; Van Lint, S.; Zhong, Z.; Catani, J.P.; Combes, F.; Deswarte, K.; Li, Y.; Lambrecht, B.N.; Lienenklaus, S.; et al. Nanoparticle-Conjugate TLR7/8 Agonist Localized Immunotherapy Provokes Safe Antitumoral Responses. Adv. Mater. 2018, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, L.; Bolli, E.; Massa, S.; Vandenberghe, I.; Movahedi, K.; Devreese, B.; Van Ginderachter, J.A.; De Geest, B.G.; Boli, E. Targeting Protumoral Tumor-Associated Macrophages with Nanobody-Functionalized Nanogels through Strain Promoted Azide Alkyne Cycloaddition Ligation. Bioconjugate Chem. 2018, 29, 2394–2405. [Google Scholar] [CrossRef]
- Obermajer, N.; Kocbek, P.; Repnik, U.; KuŽNik, A.; Cegnar, M.; Kristl, J.; Kos, J. Immunonanoparticles—An effective tool to impair harmful proteolysis in invasive breast tumor cells. FEBS J. 2007, 274, 4416–4427. [Google Scholar] [CrossRef]
- Kos, J.; Obermajer, N.; Doljak, B.; Kocbek, P.; Kristl, J. Inactivation of harmful tumour-associated proteolysis by nanoparticulate system. Int. J. Pharm. 2009, 381, 106–112. [Google Scholar] [CrossRef]
- Tuettenberg, A.; Steinbrink, K.; Schuppan, D. Myeloid cells as orchestrators of the tumor microenvironment: Novel targets for nanoparticular cancer therapy. Nanomedicne 2016, 11, 2735–2751. [Google Scholar] [CrossRef]
- Schupp, J.; Krebs, F.K.; Zimmer, N.; Trzeciak, E.; Schuppan, D.; Tuettenberg, A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell. Immunol. 2019, 343, 103713. [Google Scholar] [CrossRef]
- Leber, N.; Kaps, L.; Yang, A.; Aslam, M.; Giardino, M.; Klefenz, A.; Choteschovsky, N.; Rosigkeit, S.; Mostafa, A.; Nuhn, L.; et al. α-Mannosyl-Functionalized Cationic Nanohydrogel Particles for Targeted Gene Knockdown in Immunosuppressive Macrophages. Macromol. Biosci. 2019, 19, e1900162. [Google Scholar] [CrossRef]
- Scodeller, P.; Simón-Gracia, L.; Kopanchuk, S.; Tobi, A.; Kilk, K.; Säälik, P.; Kurm, K.; Squadrito, M.L.; Kotamraju, V.R.; Rinken, A.; et al. Precision Targeting of Tumor Macrophages with a CD206 Binding Peptide. Sci. Rep. 2017, 7, 14655. [Google Scholar] [CrossRef] [PubMed]
- Lepland, A.; Asciutto, E.K.; Malfanti, A.; Simón-Gracia, L.; Sidorenko, V.; Vicent, M.J.; Teesalu, T.; Scodeller, P. Targeting Pro-Tumoral Macrophages in Early Primary and Metastatic Breast Tumors with the CD206-Binding mUNO Peptide. Mol. Pharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wagener, K.; Bros, M.; Krumb, M.; Langhanki, J.; Pektor, S.; Worm, M.; Schinnerer, M.; Montermann, E.; Miederer, M.; Frey, H.; et al. Targeting of Immune Cells with Trimannosylated Liposomes. Adv. Ther. 2020. [Google Scholar] [CrossRef]
- Wong, C.S.; Hoogendoorn, S.; Van Der Marel, G.A.; Overkleeft, H.S.; Codée, J.D.C. Targeted Delivery of Fluorescent High-Mannose-Type Oligosaccharide Cathepsin Inhibitor Conjugates. ChemPlusChem 2015, 80, 928–937. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CatB | CatK | CatL | CatS | CatX | |
|---|---|---|---|---|---|
| Physiological occurrence [14] | ubiquitous | ubiquitous; predominantly in bone tissue | ubiquitous, | ubiquitous, more prominent in M2-type macrophages than T cells, EC, epithelia | predominantly in immune cells [15] |
| Modified/additional occurrence in tumor tissue [14] | cytoplasm [16,17], plasma membrane, secreted in ECM | - | nucleus, secreted in ECM [18] | TAM > MDSC > angiogenic EC, TM epithelia [5,19] | - |
| Mechanisms of tumor progression | angiogenesis [20] | angiogenesis, bone metastasis [21] | metastasis, cell proliferation [18] | ECM turnover, angiogenesis [22], suppression of anti-tumor immune responses ↓ [3,5] | additive effects of CatB + CatX [15] |
| Functions in immune response [5] | CD8+ cell apoptosis [23], MDSC promotion [24] | secretion of IL-6 [19], enhanced expression of COX2 + CatB (via CCL2) [25] | antigen presentation + processing [12] | antigen presentation + processing [26], M2-type macrophage polarization toward TAM [27] | enhanced migration of T lymphocytes [28] |
| Influence on anti-tumor immune response | ↓ | ↓↑ | ↑ | ↓ | ↑ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchs, N.; Meta, M.; Schuppan, D.; Nuhn, L.; Schirmeister, T. Novel Opportunities for Cathepsin S Inhibitors in Cancer Immunotherapy by Nanocarrier-Mediated Delivery. Cells 2020, 9, 2021. https://doi.org/10.3390/cells9092021
Fuchs N, Meta M, Schuppan D, Nuhn L, Schirmeister T. Novel Opportunities for Cathepsin S Inhibitors in Cancer Immunotherapy by Nanocarrier-Mediated Delivery. Cells. 2020; 9(9):2021. https://doi.org/10.3390/cells9092021
Chicago/Turabian StyleFuchs, Natalie, Mergim Meta, Detlef Schuppan, Lutz Nuhn, and Tanja Schirmeister. 2020. "Novel Opportunities for Cathepsin S Inhibitors in Cancer Immunotherapy by Nanocarrier-Mediated Delivery" Cells 9, no. 9: 2021. https://doi.org/10.3390/cells9092021
APA StyleFuchs, N., Meta, M., Schuppan, D., Nuhn, L., & Schirmeister, T. (2020). Novel Opportunities for Cathepsin S Inhibitors in Cancer Immunotherapy by Nanocarrier-Mediated Delivery. Cells, 9(9), 2021. https://doi.org/10.3390/cells9092021