Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers


Abstract
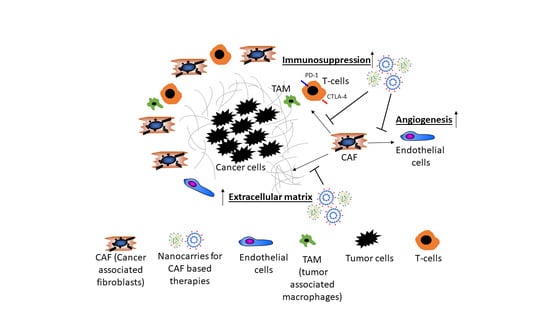
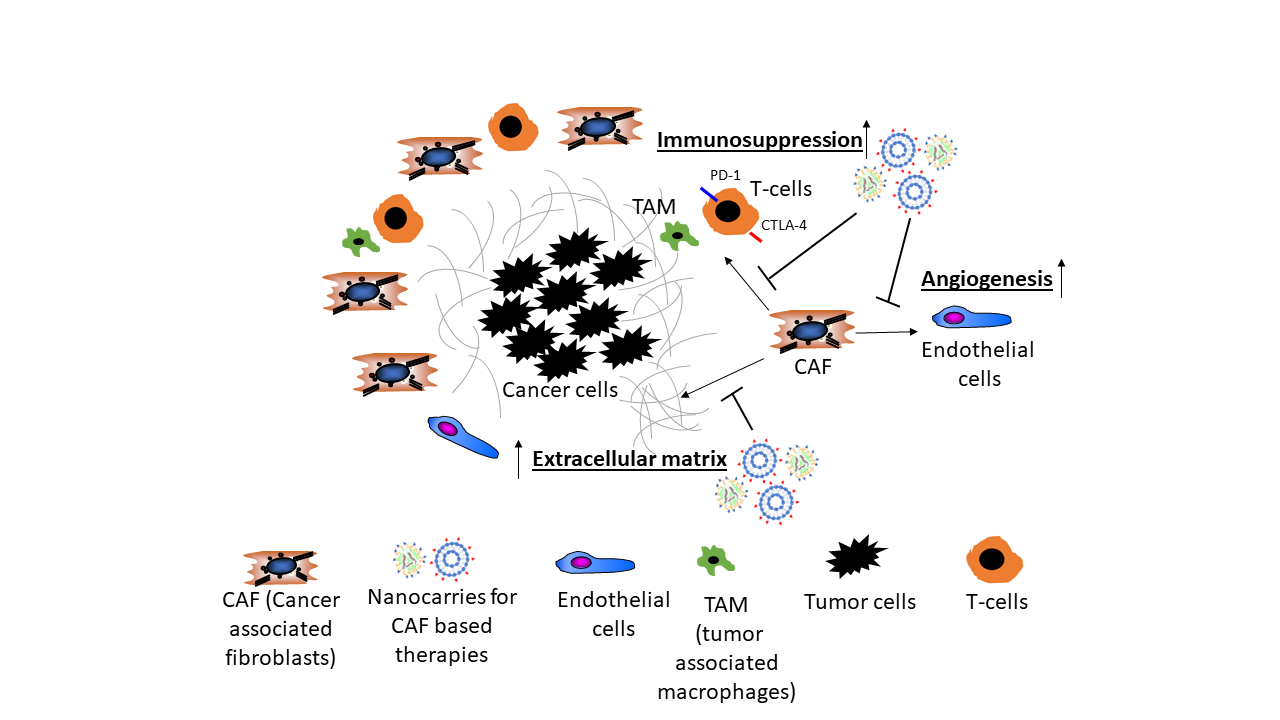
1. Introduction
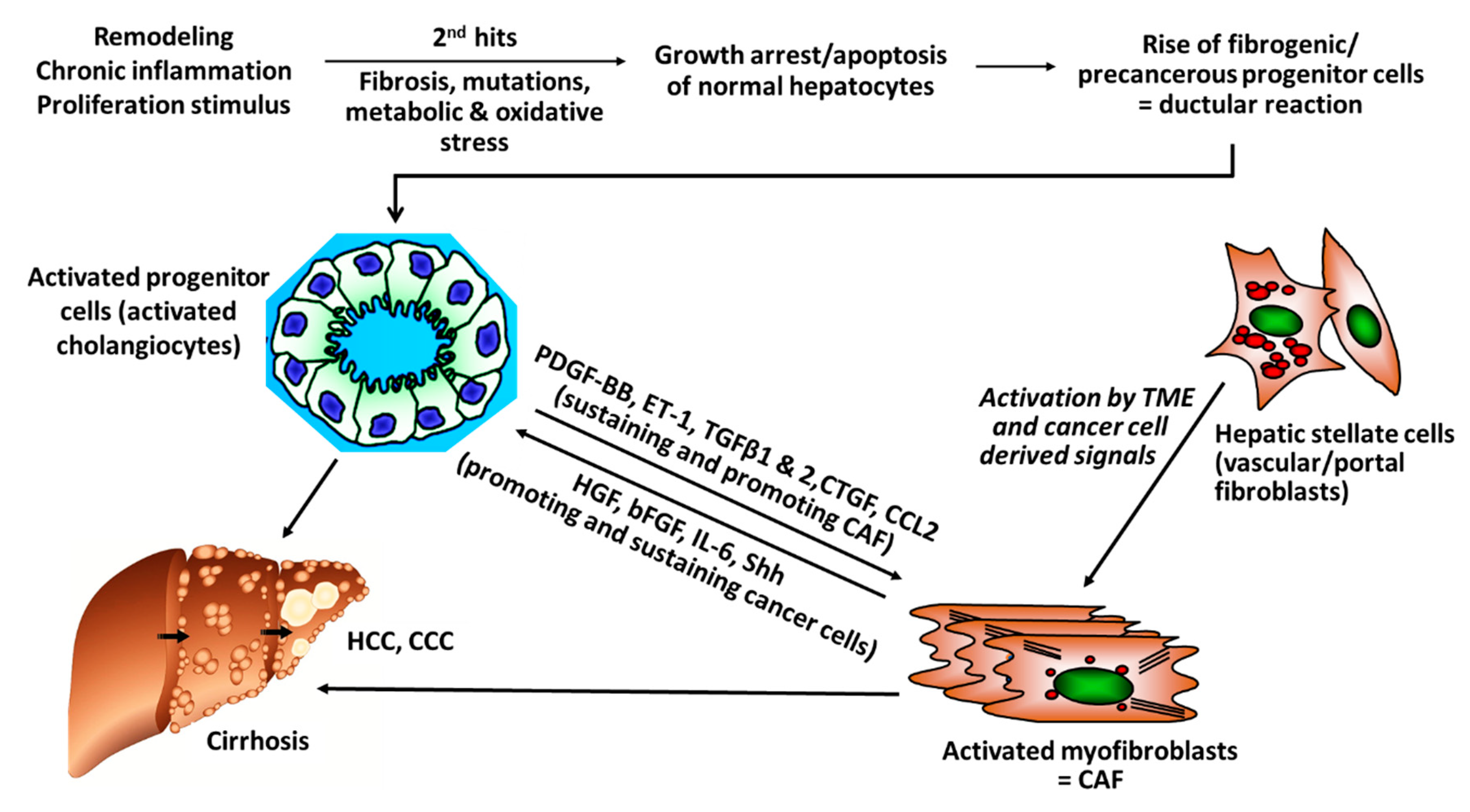
2. Cancer Associated Fibroblasts
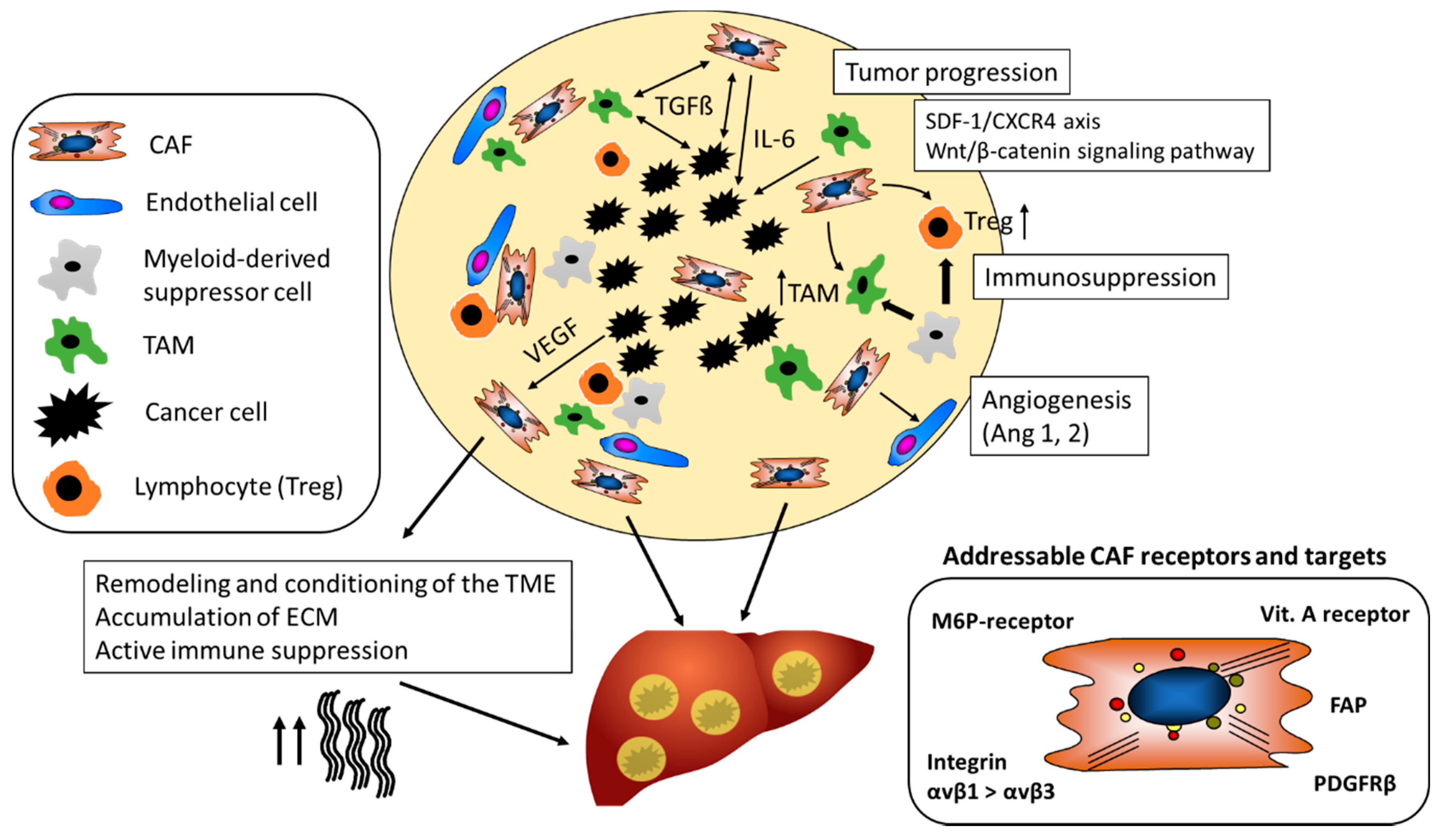
3. Stromal Cells Promote Angiogenesis
4. The Secretome of CAF Supports the Growth of HCC
5. CAF Decrease Immune Surveillance
6. CAF as Target in Anti-Stromal Cancer Therapy
7. Cell Specific Targeting of CAF with Nanoparticles
7.1. M6P/Insulin-Like Growth Factor II (M6P/IGFII) Receptor
7.2. PDGFRβ
7.3. Vitamin A/Retinol Binding Protein Receptor
7.4. Integrin αvβ3
7.5. CXCR4
7.6. Unguided Nanoparticles with HSC/CAF Specific Cargos
8. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACLF | acute-on-chronic-liver failure |
| Arg-1 | arginase-1 |
| AKT | protein kinase B |
| ASO | antisense oligonucleotides |
| a-SMA | alpha-smooth muscle actin |
| bFGF | basic fibroblast growth factor |
| CAF | Cancer-associated fibroblasts |
| CCL2 | CC-Chemokin-Ligand-2 (CCL2) |
| CCl4 | Carbon tetrachloride |
| CXCR4 | chemokine receptor type 4 |
| CTGF | connective tissue growth factor |
| COX2 | Cyclooxygenase-2 (COX-2) |
| c-MET | tyrosine-protein kinase Met |
| COX2-PGE2-EP4 | Cyclooxygenase-2-Prostaglandin E2-Prostaglandin E2 receptor 4 |
| DEN | di-ethyl-nitrosamine |
| ET-1 | Endothelin-1 |
| ERK | Extracellular signal-regulated kinases |
| EPR | enhanced permeability retention |
| Erk1/2 | extracellular signal-regulated kinases |
| FAP | Fibroblast activation protein |
| FLT3 | FMS-like tyrosine kinase 3 |
| FRA1 | Fos-related antigen 1 |
| HCC | Hepatocellular carcinoma |
| HGF | hepatocyte growth factor |
| HEY1 | hairy/enhancer-of-split related with YRPW motif protein 1 |
| IGFRII | insulin-like growth factor type II receptor |
| IL-6 | Interleukin 6 |
| IL-4R | Interleukin 4 receptor |
| iNOS | Nitric oxide synthases |
| IFNγ | Interferon gamma |
| JAK | janus kinase |
| M6P | mannose-6-phosphate |
| MAPK | mitogen-activated protein kinase |
| MDR2 | multidrug resistance protein 2 |
| MDSC | Myeloid-derived suppressor cells |
| MMP-9 | Matrix metallopeptidase 9 |
| TAM | Tumor-associated macrophages (TAMs) |
| mTOR | mechanistic Target of rapamycin |
| PDAC | pancreatic ductal adenocarcinoma |
| PDGFR(β) | platelet-derived growth factor receptor β |
| PDGF-BB/AB | platelet-derived growth factor BB/AB |
| PI3K | phosphoinositide 3-kinase |
| PGE2-EP4 | Prostaglandin E2-Prostaglandin E2 receptor 4 |
| PD-L1 | Programmed cell death 1 ligand 1 |
| SDF-1 | stromal cell-derived factor 1 |
| Shh | hedgehog signaling pathway |
| shRNA | Small hairpin RNA |
| siRNA | silencing RNA |
| STAT3 | signal transducer and activator of transcription 3 |
| TGFβ1 | transforming growth factor beta 1 |
| Tie2 | angiopoietin receptor 2 |
| TIMP-1 | tissue inhibitor metalloproteinase 1 |
| TKI | tyrosine kinase inhibitor |
| TNF-a | Tumor necrosis factor alpha |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
References
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.B.; Hansen, N.-U.B.; Bay-Jensen, A.-C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol. Liver Physiol. 2015, 308, 807–830. [Google Scholar] [CrossRef] [PubMed]
- The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 1, 43–56.
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Sevic, I.; Spinelli, F.M.; Cantero, M.J.; Reszegi, A.; Kovalszky, I.; García, M.G.; Alaniz, L. The Role of the Tumor Microenvironment in the Development and Progression of Hepatocellular Carcinoma. In Hepatocellular Carcinoma; Codon Publications: Brisbane, Australia, 2019; pp. 29–45. [Google Scholar]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Feng, G.; Hanley, K.L.; Liang, Y.; Lin, X. Improving the Efficacy of Liver Cancer Immunotherapy: The Power of Combined Preclinical and Clinical Studies. Hepatology 2020. [Google Scholar] [CrossRef]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Yurchenco, P.D.; Iozzo, R.V. The nature and biology of basement membranes. Matrix Biol. 2017, 57, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kostourou, V.; Papalazarou, V. Non-collagenous ECM proteins in blood vessel morphogenesis and cancer. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef]
- Arteel, G.E.; Naba, A. The liver matrisome, looking beyond collagens. Jhep Rep. 2020, 2, 100115. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef]
- Foerster, F.; Hess, M.; Gerhold-Ay, A.; Marquardt, J.U.; Becker, D.; Galle, P.R.; Schuppan, D.; Binder, H.; Bockamp, E. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci. Rep. 2018, 8, 5351. [Google Scholar] [CrossRef]
- Flier, J.S.; Underhill, L.H.; Dvorak, H.F. Tumors: Wounds That Do Not Heal. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Hirata, E.; Sahai, E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb. Perspect. Med. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta-Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, M.; Kohara, M.; Ohshima, K.; Tahara, S.; Hori, Y.; Nojima, S.; Wada, N.; Ikeda, J.; Miyamura, K.; Ito, M.; et al. Origin of cancer-associated fibroblasts and tumor-associated macrophages in humans after sex-mismatched bone marrow transplantation. Commun. Biol. 2018, 1, 131. [Google Scholar] [CrossRef] [PubMed]
- Baglieri, J.; Brenner, D.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.-L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Seo, H.R. Roles of Tumor Microenvironment in Hepatocelluar Carcinoma. Curr. Cancer Rev. 2015, 11, 82–93. [Google Scholar] [CrossRef]
- Ju, M.-J.; Qiu, S.-J.; Fan, J.; Xiao, Y.-S.; Gao, Q.; Zhou, J.; Li, Y.-W.; Tang, Z.-Y. Peritumoral Activated Hepatic Stellate Cells Predict Poor Clinical Outcome in Hepatocellular Carcinoma After Curative Resection. Am. J. Clin. Pathol. 2009, 131, 498–510. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosisin Liver Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 153–186. [Google Scholar] [CrossRef] [PubMed]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediat. Inflamm. 2017, 2017, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, Q.; Alam, A.; Cui, J.; Suen, K.C.; Soo, A.P.; Eguchi, S.; Gu, J.; Ma, D. The role of osteopontin in the progression of solid organ tumour. Cell Death Dis. 2018, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, M.; Sakamoto, M.; Kanetaka, K.; Chuuma, M.; Hirohashi, S. Overexpression of osteopontin in hepatocellular carcinoma. Pathol. Int. 2002, 52, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.W.; Ou, Y.H.; Peng, S.Y.; Liu, S.H.; Lai, P.L.; Lee, P.H.; Sheu, J.C.; Chen, C.L.; Hsu, H.C. Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular cacinoma. Cancer 2003, 98, 119–127. [Google Scholar] [CrossRef]
- Ye, Q.H.; Qin, L.X.; Forgues, M.; He, P.; Kim, J.W.; Peng, A.C.; Simon, R.; Li, Y.; Robles, A.I.; Chen, Y.; et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat. Med. 2003, 9, 416–423. [Google Scholar] [CrossRef]
- Huang, J.; Sheng, H.-H.; Shen, T.; Hu, Y.-J.; Xiao, H.-S.; Zhang, Q.; Zhang, Q.-H.; Han, Z.-G. Correlation between genomic DNA copy number alterations and transcriptional expression in hepatitis B virus-associated hepatocellular carcinoma. Febs Lett. 2006, 580, 3571–3581. [Google Scholar] [CrossRef]
- Kim, J.; Ki, S.S.; Lee, S.D.; Han, C.J.; Kim, Y.C.; Park, S.H.; Cho, S.Y.; Hong, Y.-J.; Park, H.Y.; Lee, M.; et al. Elevated Plasma Osteopontin Levels in Patients with Hepatocellular Carcinoma. Am. J. Gastroenterol. 2006, 101, 2051–2059. [Google Scholar] [CrossRef]
- Zhang, H.; Ye, Q.-H.; Ren, N.; Zhao, L.; Wang, Y.-F.; Wu, X.; Sun, H.-C.; Wang, L.; Zhang, B.-H.; Liu, Y.-K.; et al. The prognostic significance of preoperative plasma levels of osteopontin in patients with hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2006, 132, 709–717. [Google Scholar] [CrossRef]
- Shang, S.; Plymoth, A.; Ge, S.; Feng, Z.; Rosen, H.R.; Sangrajrang, S.; Hainaut, P.; Marrero, J.A.; Beretta, L. Identification of osteopontin as a novel marker for early hepatocellular carcinoma. Hepatology 2012, 55, 483–490. [Google Scholar] [CrossRef]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Chen, Z.; Lu, Y.; Li, Y.; Hu, K.; Xu, R. Role of activated hepatic stellate cells in proliferation and metastasis of hepatocellular carcinoma. Hepatol. Res. 2015, 45, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.; De Minicis, S.; Seki, E.; Hatano, E.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Miura, K.; Ikai, I.; Uemoto, S.; et al. Hepatic Stellate Cells Secrete Angiopoietin 1 That Induces Angiogenesis in Liver Fibrosis. Gastroenterology 2008, 135, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Cameno, P.; Martín-Vílchez, S.; Lara-Pezzi, E.; Borque, M.J.; Salmerón, J.; De Rueda, P.M.; Solís, J.A.; López-Cabrera, M.; Moreno-Otero, R. Hepatitis B virus promotes angiopoietin-2 expression in liver tissue: Role of HBV X protein. Am. J. Pathol. 2006, 169, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Geng, Z.; Li, Q.; Li, W.; Zheng, J.; Shah, V. Activated Human Hepatic Stellate Cells Promote Growth of Human Hepatocellular Carcinoma in a Subcutaneous Xenograft Nude Mouse Model. Cell Biochem. Biophys. 2014, 70, 337–347. [Google Scholar] [CrossRef]
- Olaso, E.; Salado, C.; Egilegor, E.; Gutierrez, V.; Santisteban, A.; Sancho-Bru, P.; Friedman, S.L.; Vidal-Vanaclocha, F. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology 2003, 37, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Yaqoob, U.; Geng, Z.; Bloch, K.; Liu, C.; Gomez, T.; Billadeau, D.; Shah, V. Focal adhesion assembly in myofibroblasts fosters a microenvironment that promotes tumor growth. Am. J. Pathol. 2010, 177, 1888–1900. [Google Scholar] [CrossRef]
- Sugimachi, K.; Tanaka, S.; Taguchi, K.; Aishima, S.; Shimada, M.; Sugimachi, K.; Tsuneyoshi, M. Angiopoietin switching regulates angiogenesis and progression of human hepatocellular carcinoma. J. Clin. Pathol. 2003, 56, 854–860. [Google Scholar] [CrossRef]
- Torimura, T.; Ueno, T.; Kin, M.; Harada, R.; Taniguchi, E.; Nakamura, T.; Sakata, R.; Hashimoto, O.; Sakamoto, M.; Kumashiro, R.; et al. Overexpression of angiopoietin-1 and angiopoietin-2 in hepatocellular carcinoma. J. Hepatol. 2004, 40, 799–807. [Google Scholar] [CrossRef]
- Thompson, A.I.; Conroy, K.P.; Henderson, N.C. Hepatic stellate cells: Central modulators of hepatic carcinogenesis. Bmc Gastroenterol. 2015, 15, 63. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Araki, K.; Kuwano, H.; Shirabe, K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6841–6850. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-β as Multifaceted Orchestrator in HCC Progression: Signaling, EMT, Immune Microenvironment, and Novel Therapeutic Perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar]
- Fabregat, I.; Caballero-Díaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef]
- Yoshida, S.; Kornek, M.; Ikenaga, N.; Schmelzle, M.; Masuzaki, R.; Csizmadia, E.; Wu, Y.; Robson, S.C.; Schuppan, D. Sublethal heat treatment promotes epithelial-mesenchymal transition and enhances the malignant potential of hepatocellular carcinoma. Hepatology 2013, 58, 1667–1680. [Google Scholar] [CrossRef]
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma 2018, 5, 61–73. [Google Scholar] [CrossRef]
- Wang, H.; Rao, B.; Lou, J.; Li, J.; Liu, Z.; Li, A.; Cui, G.; Ren, Z.; Yu, Z. The Function of the HGF/c-MET Axis in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2020, 8, 55. [Google Scholar] [CrossRef]
- Schuppan, D.; Schmid, M.; Somasundaram, R.; Ackermann, R.; Ruehl, M.; Nakamura, T.; Riecken, E.O. Collagens in the liver extracellular matrix bind hepatocyte growth factor. Gastroenterology 1998, 114, 139–152. [Google Scholar] [CrossRef]
- Cramer, T.; Schuppan, D.; Bauer, M.; Pfander, D.; Neuhaus, P.; Herbst, H. Hepatocyte growth factor and c-Met expression in rat and human liver fibrosis. Liver Int. 2004, 24, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Muzumdar, M.D.; Zhu, A.X. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, H. Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp. Mol. Med. 2017, 49, e307. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, W.; Yang, F.; Feng, T.; Zhou, M.; Yu, Y.; Yu, X.; Zhao, W.; Yi, F.; Tang, W.; et al. Interleukin-6-stimulated progranulin expression contributes to the malignancy of hepatocellular carcinoma cells by activating mTOR signaling. Sci. Rep. 2016, 6, 21260. [Google Scholar] [CrossRef]
- Thiele, N.D.; Wirth, J.W.; Steins, D.; Koop, A.C.; Ittrich, H.; Lohse, A.W.; Kluwe, J. TIMP-1 is upregulated, but not essential in hepatic fibrogenesis and carcinogenesis in mice. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Scott, E.; Lu, R.; Xu, Y.; Oh, W.K.; Yu, Q. TIMP-1 Promotes Accumulation of Cancer Associated Fibroblasts and Cancer Progression. PLoS ONE 2013, 8, e77366. [Google Scholar] [CrossRef]
- Song, T.; Dou, C.; Jia, Y.; Tu, K.; Zheng, X. TIMP-1 activated carcinoma-associated fibroblasts inhibit tumor apoptosis by activating SDF1/CXCR4 signaling in hepatocellular carcinoma. Oncotarget 2015, 6, 12061–12079. [Google Scholar] [CrossRef]
- Kuramitsu, K.; Sverdlov, D.Y.; Liu, S.B.; Csizmadia, E.; Burkly, L.; Schuppan, D.; Hanto, D.W.; Otterbein, L.E.; Popov, Y. Failure of Fibrotic Liver Regeneration in Mice Is Linked to a Severe Fibrogenic Response Driven by Hepatic Progenitor Cell Activation. Am. J. Pathol. 2013, 183, 182–194. [Google Scholar] [CrossRef]
- Popov, Y.; Schuppan, D. Epithelial-to-mesenchymal transition in liver fibrosis: Dead or alive? Gastroenterology 2010, 139, 722–725. [Google Scholar] [CrossRef]
- Peng, Z.W.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Dixit, R.; Weinreb, P.H.; Violette, S.; Sheppard, D.; Schuppan, D.; et al. Integrin αvβ6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016, 63, 217–232. [Google Scholar] [CrossRef]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 10, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Patsenker, E.; Popov, Y.; Stickel, F.; Schneider, V.; Ledermann, M.; Sägesser, H.; Niedobitek, G.; Goodman, S.L.; Schuppan, D. Pharmacological inhibition of integrin αvβ3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology 2009, 50, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, L.; Yin, Z.; Su, W.; Ren, G.; Zhou, C.; You, J.; Fan, J.; Wang, X. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int. J. Cancer 2011, 29, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, W.; Xu, J.; Li, J.; Hong, Z.; Yin, Z.; Wang, X. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget 2016, 7, 8866–8878. [Google Scholar] [CrossRef]
- Ji, J.; Eggert, T.; Budhu, A.; Forgues, M.; Takai, A.; Dang, H.; Ye, Q.; Lee, J.-S.; Kim, J.H.; Greten, T.F.; et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 2015, 62, 481–495. [Google Scholar] [CrossRef]
- Höchst, B.; Schildberg, F.A.; Sauerborn, P.; Gäbel, Y.A.; Gevensleben, H.; Goltz, D.; Heukamp, L.C.; Türler, A.; Ballmaier, M.; Gieseke, F.; et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J. Hepatol. 2013, 59, 528–535. [Google Scholar] [CrossRef]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef]
- Shu, Q.-H.; Ge, Y.-S.; Ma, H.-X.; Gao, X.-Q.; Pan, J.-J.; Liu, D.; Xu, G.-L.; Ma, J.-L.; Jia, W.-D. Prognostic value of polarized macrophages in patients with hepatocellular carcinoma after curative resection. J. Cell. Mol. Med. 2016, 20, 1024–1035. [Google Scholar] [CrossRef]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-Associated Macrophages Produce Interleukin 6 and Signal via STAT3 to Promote Expansion of Human Hepatocellular Carcinoma Stem Cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Gores, G.J.; Shah, V.H. Hepatic stellate cells: Partners in crime for liver metastases? Hepatology 2011, 54, 707–713. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, X.; Lu, J.; Salfenmoser, M.; Wirsik, N.M.; Schleussner, N.; Imle, A.; Freire Valls, A.; Radhakrishnan, P.; Liang, J.; et al. Reduction of Liver Metastasis Stiffness Improves Response to Bevacizumab in Metastatic Colorectal Cancer. Cancer Cell 2020, 37, 800–817. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, A.A.; Weiner, L.M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020. [Google Scholar] [CrossRef]
- Keane, F.M.; Yao, T.W.; Seelk, S.; Gall, M.G.; Chowdhury, S.; Poplawski, S.E.; Lai, J.H.; Li, Y.; Wu, W.; Farrell, P.; et al. Quantitation of fibroblast activation protein (FAP)-specific protease activity in mouse, baboon and human fluids and organs. Febs Open Bio 2014, 4, 43–54. [Google Scholar] [CrossRef]
- Zou, B.; Liu, X.; Zhang, B.; Gong, Y.; Cai, C.; Li, P.; Chen, J.; Xing, S.; Chen, J.; Peng, S.; et al. The Expression of FAP in Hepatocellular Carcinoma Cells is Induced by Hypoxia and Correlates with Poor Clinical Outcomes. J. Cancer 2018, 9, 3278–3286. [Google Scholar] [CrossRef]
- Niedermeyer, J.; Kriz, M.; Hilberg, F.; Garin-Chesa, P.; Bamberger, U.; Lenter, M.C.; Park, J.; Viertel, B.; Püschner, H.; Mauz, M.; et al. Targeted Disruption of Mouse Fibroblast Activation Protein. Mol. Cell. Biol. 2000, 20, 1089–1094. [Google Scholar] [CrossRef]
- Wang, X.M.; Holz, L.E.; Chowdhury, S.; Cordoba, S.P.; Evans, K.A.; Gall, M.G.; De Ribeiro, A.J.V.; Zheng, Y.Z.; Levy, M.T.; Yu, D.M.T.; et al. The pro-fibrotic role of dipeptidyl peptidase 4 in carbon tetrachloride-induced experimental liver injury. Immunol. Cell Biol. 2017, 95, 443–453. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, Q.; Liu, Z.; Luo, X.; Fan, Y.; Liu, Y.; Zhang, Y.; Hua, S.; Fu, Q.; Zhao, M.; et al. Downregulation of FAP suppresses cell proliferation and metastasis through PTEN/PI3K/AKT and Ras-ERK signaling in oral squamous cell carcinoma. Cell Death Dis. 2014, 5, 1155. [Google Scholar] [CrossRef] [PubMed]
- Aimes, R.; Zijlstra, A.; Hooper, J.; Ogbourne, S.; Sit, M.-L.; Fuchs, S.; Gotley, D.; Quigley, J.; Antalis, T. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb. Haemost. 2003, 89, 561–572. [Google Scholar] [CrossRef]
- Gao, L.-M.; Wang, F.; Zheng, Y.; Fu, Z.-Z.; Zheng, L.; Chen, L.-L. Roles of Fibroblast Activation Protein and Hepatocyte Growth Factor Expressions in Angiogenesis and Metastasis of Gastric Cancer. Pathol. Oncol. Res. 2019, 25, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Shipley, J.M.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypetrophic chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef]
- Hofheinz, R.-D.; Al-Batran, S.-E.; Hartmann, F.; Hartung, G.; Jäger, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients with Metastatic Colorectal Cancer. Oncol. Res. Treat. 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGFβ1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Lara, P.N.; Le Chevalier, T.; Breton, J.L.; Bonomi, P.; Sandler, A.B.; Socinski, M.A.; Delbaldo, C.; McHenry, B.; Lebwohl, D.; et al. Phase II clinical trial of the epothilone B analog, ixabepilone, in patients with non-small-cell lung cancer whose tumors have failed first-line platinum-based chemotherapy. J. Clin. Oncol. 2007, 25, 3448–3455. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Segersten, U.; Leiss, L.W.; Malmström, P.U.; Hatina, J.; Östman, A.; Strell, C. Fibroblasts in urothelial bladder cancer define stroma phenotypes that are associated with clinical outcome. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.F.; Wei, W.; Gupta, S.; Smithy, J.W.; Zelterman, D.; Kluger, H.M.; Rimm, D.L. Multiplex quantitative analysis of cancer-associated fibroblasts and immunotherapy outcome in metastatic melanoma. J. Immunother. Cancer 2019, 7, 194. [Google Scholar] [CrossRef] [PubMed]
- Kilvaer, T.K.; Khanehkenari, M.R.; Hellevik, T.; Al-Saad, S.; Paulsen, E.E.; Bremnes, R.M.; Busund, L.T.; Donnem, T.; Martinez, I.Z. Cancer associated fibroblasts in stage I-IIIA NSCLC: Prognostic impact and their correlations with tumor molecular markers. PLoS ONE 2015, 10, e0134965. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.S.; Datta, P.K. Targeting the transforming growth factor-β signaling pathway in human cancer. Expert Opin. Investig. Drugs 2010, 19, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L.; Molema, G.; Schuppan, D.; Geerts, A.; De Bleser, P.J.; Weert, B.; Meijer, D.K.F.; Poelstra, K. Successful targeting to rat hepatic stellate cells using albumin modified with cyclic peptides that recognize the collagen type VI receptor. J. Biol. Chem. 2000, 275, 12743–12751. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Frohn, F.; Magdaleno, F.; Reker-Smit, C.; Schierwagen, R.; Schierwagen, I.; Uschner, F.E.; van Dijk, F.; Fürst, D.O.; Djudjaj, S.; et al. Rho-kinase inhibitor coupled to peptide-modified albumin carrier reduces portal pressure and increases renal perfusion in cirrhotic rats. Sci. Rep. 2019, 9, 2256. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, F.; Teekamp, N.; Beljaars, L.; Post, E.; Zuidema, J.; Steendam, R.; Kim, Y.O.; Frijlink, H.W.; Schuppan, D.; Poelstra, K.; et al. Pharmacokinetics of a sustained release formulation of PDGFβ-receptor directed carrier proteins to target the fibrotic liver. J. Control. Release 2018, 269, 258–265. [Google Scholar] [CrossRef]
- Deshmukh, M.; Nakagawa, S.; Higashi, T.; Vincek, A.; Venkatesh, A.; Ruiz de Galarreta, M.; Koh, A.P.; Goossens, N.; Hirschfield, H.; Bian, C.B.; et al. Cell type-specific pharmacological kinase inhibition for cancer chemoprevention. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 317–325. [Google Scholar] [CrossRef]
- Bansal, R.; Prakash, J.; De Ruiter, M.; Poelstra, K. Interferon gamma peptidomimetic targeted to hepatic stellate cells ameliorates acute and chronic liver fibrosis in vivo. J. Control. Release 2014, 179, 18–24. [Google Scholar] [CrossRef]
- Bansal, R.; Tomar, T.; Östman, A.; Poelstra, K.; Prakash, J. Selective targeting of interferon γ to stromal fibroblasts and pericytes as a novel therapeutic approach to inhibit angiogenesis and tumor growth. Mol. Cancer 2012, 11, 2419–2428. [Google Scholar] [CrossRef]
- Bansal, R.; Prakash, J.; Post, E.; Beljaars, L.; Schuppan, D.; Poelstra, K. Novel engineered targeted interferon-gamma blocks hepatic fibrogenesis in mice. Hepatology 2011, 54, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Gonzalo, T.; Kok, R.J.; Sancho-Bru, P.; Van Beuge, M.; Swart, J.; Prakash, J.; Temming, K.; Fondevila, C.; Beljaars, L.; et al. Reduction of advanced liver fibrosis by short-term targeted delivery of an angiotensin receptor blocker to hepatic stellate cells in rats. Hepatology 2010, 51, 942–952. [Google Scholar] [CrossRef] [PubMed]
- van Beuge, M.M.; Prakash, J.; Lacombe, M.; Post, E.; Reker-Smit, C.; Beljaars, L.; Poelstra, K. Enhanced Effectivity of an ALK5-Inhibitor after Cell-Specific Delivery to Hepatic Stellate Cells in Mice with Liver Injury. PLoS ONE 2013, 8, e56442. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, Q.; Wang, J.; Zhan, C.; Xie, C.; Lu, W. Effects of interferon-gamma liposomes targeted to platelet-derived growth factor receptor–beta on hepatic fibrosis in rats. J. Control. Release 2012, 159, 261–270. [Google Scholar] [CrossRef]
- Jia, Z.; Gong, Y.; Pi, Y.; Liu, X.; Gao, L.; Kang, L.; Wang, J.; Yang, F.; Tang, J.; Lu, W.; et al. pPB Peptide-Mediated siRNA-Loaded Stable Nucleic Acid Lipid Nanoparticles on Targeting Therapy of Hepatic Fibrosis. Mol. Pharm. 2018, 15, 53–62. [Google Scholar] [CrossRef]
- Sato, Y.; Murase, K.; Kato, J.; Kobune, M.; Sato, T.; Kawano, Y.; Takimoto, R.; Takada, K.; Miyanishi, K.; Matsunaga, T.; et al. Resolution of liver cirrhosis using vitamin A–coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 2008, 26, 431–442. [Google Scholar] [CrossRef]
- Qiao, J.-B.; Fan, Q.-Q.; Zhang, C.-L.; Lee, J.; Byun, J.; Xing, L.; Gao, X.-D.; Oh, Y.-K.; Jiang, H.-L. Hyperbranched lipoid-based lipid nanoparticles for bidirectional regulation of collagen accumulation in liver fibrosis. J. Control. Release 2020, 321, 629–640. [Google Scholar] [CrossRef]
- Li, Y.; Pu, S.; Liu, Q.; Li, R.; Zhang, J.; Wu, T.; Chen, L.; Li, H.; Yang, X.; Zou, M.; et al. An integrin-based nanoparticle that targets activated hepatic stellate cells and alleviates liver fibrosis. J. Control. Release 2019, 303, 77–90. [Google Scholar] [CrossRef]
- Sung, Y.C.; Liu, Y.C.; Chao, P.H.; Chang, C.C.; Jin, P.R.; Lin, T.T.; Lin, J.A.; Cheng, H.T.; Wang, J.; Lai, C.P.; et al. Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development. Theranostics 2018, 8, 894–905. [Google Scholar] [CrossRef]
- Leber, N.; Kaps, L.; Aslam, M.; Schupp, J.; Brose, A.; Schäffel, D.; Fischer, K.; Diken, M.; Strand, D.; Koynov, K.; et al. SiRNA-mediated in vivo gene knockdown by acid-degradable cationic nanohydrogel particles. J. Control. Release 2017, 248, 10–23. [Google Scholar] [CrossRef]
- Kaps, L.; Nuhn, L.; Aslam, M.; Brose, A.; Foerster, F.; Rosigkeit, S.; Renz, P.; Heck, R.; Kim, Y.O.; Lieberwirth, I.; et al. In Vivo Gene-Silencing in Fibrotic Liver by siRNA-Loaded Cationic Nanohydrogel Particles. Adv. Healthc. Mater. 2015, 4, 2809–2815. [Google Scholar] [CrossRef] [PubMed]
- Jiménez Calvente, C.; Sehgal, A.; Popov, Y.; Kim, Y.O.; Zevallos, V.; Sahin, U.; Diken, M.; Schuppan, D. Specific hepatic delivery of procollagen α1(I) small interfering RNA in lipid-like nanoparticles resolves liver fibrosis. Hepatology 2015, 62, 1285–1297. [Google Scholar] [CrossRef]
- Adrian, J.E.; Kamps, J.A.A.M.; Scherphof, G.L.; Meijer, D.K.F.; van Loenen-Weemaes, A.; Reker-Smit, C.; Terpstra, P.; Poelstra, K. A novel lipid-based drug carrier targeted to the non-parenchymal cells, including hepatic stellate cells, in the fibrotic livers of bile duct ligated rats. Biochim. Biophys. Acta-Biomembr. 2007, 1768, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; MacDonald, R.G.; Thinakaran, G.; Kar, S. Insulin-Like Growth Factor-II/Cation-Independent Mannose 6-Phosphate Receptor in Neurodegenerative Diseases. Mol. Neurobiol. 2017, 54, 2636–2658. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatology 1999, 29, 1486–1493. [Google Scholar] [CrossRef]
- Prakash, J.; Beljaars, L.; Harapanahalli, A.K.; Zeinstra-Smith, M.; de Jager-Krikken, A.; Hessing, M.; Steen, H.; Poelstra, K. Tumor-targeted intracellular delivery of anticancer drugs through the mannose-6-phosphate/insulin-like growth factor II receptor. Int. J. Cancer 2010, 126, 1966–1981. [Google Scholar] [CrossRef]
- Greupink, R.; Bakker, H.I.; Van Goor, H.; De Borst, M.H.; Beljaars, L.; Poelstra, K. Mannose-6-phosphate/insulin-like growth factor-II receptors may represent a target for the selective delivery of mycophenolic acid to fibrogenic cells. Pharm. Res. 2006, 23, 1827–1834. [Google Scholar] [CrossRef]
- Hagens, W.I.; Mattos, A.; Greupink, R.; De Jager-Krikken, A.; Reker-Smit, C.; Van Loenen-Weemaes, A.; Gouw, A.S.H.; Poelstra, K.; Beljaars, L. Targeting 15d-prostaglandin J2 to hepatic stellate cells: Two options evaluated. Pharm. Res. 2007, 24, 566–574. [Google Scholar] [CrossRef]
- Adrian, J.E.; Poelstra, K.; Scherphof, G.L.; Molema, G.; Meijer, D.K.F.; Reker-Smit, C.; Morselt, H.W.M.; Kamps, J.A.A.M. Interaction of targeted liposomes with primary cultured hepatic stellate cells: Involvement of multiple receptor systems. J. Hepatol. 2006, 4, 560–567. [Google Scholar] [CrossRef]
- Beljaars, L.; Weert, B.; Geerts, A.; Meijer, D.K.F.; Poelstra, K. The preferential homing of a platelet derived growth factor receptor-recognizing macromolecule to fibroblast-like cells in fibrotic tissue. Biochem. Pharm. 2003, 66, 1307–1317. [Google Scholar] [CrossRef]
- Haubner, R.; Finsinger, D.; Kessler, H. Stereoisomeric Peptide Libraries and Peptidomimetics for Designing Selective Inhibitors of theαvβ3 Integrin for a New Cancer Therapy. Angew. Chem. Int. Ed. Engl. 1997, 36, 1374–1389. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 69, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, L.; Hirsch, M.; Krieg, B.; Koynov, K.; Fischer, K.; Schmidt, M.; Helm, M.; Zentel, R. Cationic nanohydrogel particles as potential siRNA carriers for cellular delivery. Acs Nano 2012, 6, 2198–2214. [Google Scholar] [CrossRef] [PubMed]
- Kaps, L.; Leber, N.; Klefenz, A.; Choteschovsky, N.; Zentel, R.; Nuhn, L.; Schuppan, D. In Vivo siRNA Delivery to Immunosuppressive Liver Macrophages by α-Mannosyl-Functionalized Cationic Nanohydrogel Particles. Cells 2020, 9, 1905. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Addressed Target | Cell-Specific Ligand | In Vivo Cellular Uptake | Coupled/ Encapsulated Drug | In Vivo Therapeutic Effect | Nano Carrier | Size (Zeta Potential) | Reference |
|---|---|---|---|---|---|---|---|
| Collagen type VI receptors | Cyclic peptide | Activated HSC | Not reported | Not reported | HSA | Not reported | [106] |
| PDGFRβ | Cyclic peptide | HSC | ROCK-inhibitor Y-27632 | Lowers portal pressure | HSA | Not reported | [107] |
| PDGFRβ | Cyclic peptide | Not reported | None but suitable for delivery of protein-based drugs | Not reported | HSA—polymeric microspheres | ~22 μm | [108] |
| PDGFRβ | Cyclic peptide | Myofibroblasts | Erlotinib (epidermal growth factor receptor inhibitor) | Improved antitumor activity, reduced in vivo toxicity (hepatotoxicity) | Silica nanoparticles | ~200 nm | [109] |
| PDGFRβ | Cyclic peptide | HSC, myofibroblasts | IFNγ peptidomimetic (without extracellular recognition domain) | Improved antifibrotic effect (compared to free IFNγ in CCl4 fibrotic mice) | IFNγ coupled to cyclic peptide | Not reported | [110] |
| PDGFRβ | Cyclic peptide | Tumor pericytes | IFNγ | Improved anti-tumor effect via inhibition of angiogenesis (compared to unguided IFNγ) | HSA | Not reported | [111] |
| PDGFRβ | Cyclic peptide | HSC | IFNγ | Improved antifibrotic effect (compared to free IFNγ in CCl4 fibrotic mice) | IFNγ coupled to cyclic peptide | Not reported | [112] |
| M6P receptor | M6P | HSCs, Myofibroblasts | losartan | Antifibrotic effect vs. free losartan | HSA | Not reported | [113] |
| M6P receptor | M6P | HSC, >LSEC; no colocalization with macrophages | TGFβ receptor 1 (ALK5) inhibitor LY-36494 | Improved antifibrotic activity (reduction of collagen III and fibronectin) | HSA | Not reported | [114] |
| Addressed Target | Cell-Specific Ligand | In Vivo Cellular Uptake | Encapsulated Drug | In Vivo Therapeutic Effect | Nano Carrier | Size (Zeta Potential) | Reference |
|---|---|---|---|---|---|---|---|
| PDGFRβ | Cyclic peptide | HSC | IFNγ | Improved antifibrotic effect vs untargeted IFNγ | Liposomes | ~83.5 nm | [115] |
| PDGFRβ | Cyclic peptide | HSC | antiHSP 47 siRNA | Improved antifibrotic effect vs control siRNA/carriers in TAA fibrotic mice | Lipoplexes | ~110 nm (~0 mV) | [116] |
| Receptors for retinol binding protein | Vitamin A | HSC, (macrophages) | antiHSP47 siRNA | Improved antifibrotic effect vs control siRNA/carriers in liver fibrotic rats | Liposomes | ~150 nm | [117] |
| Receptors for retinol binding protein | Vitamin A | HSC | anticol1a1 and antiTIMP-1 siRNA | Antifibrotic effect vs scrambled siRNA loaded carriers in CCl4 fibrotic mice | Lipoplexes | ~140 nm (~−12.9 mV) | [118] |
| Integrin αvβ3 | Cyclic RGD peptide | Activated HSC (+++), Kupfer cells and LSECs (++), biliary cells (+), hepatocytes (+) | Vismodegib (hedgehog inhibitor) | Improved antifibrotic effect vs control drug/empty carrier in BDL fibrotic mice | Liposomes | ~80 nm, (~−24.8 mV) | [119] |
| CXCR4 | CXCR4 antagonistic peptide (CTCE9908) | HSC | Sorafenib/multi-tyrosine-kinase inhibitor | Antifibrotic and antitumor effect in mice with CCl4-induced fibrosis, HCC and PDAC | Liposomes | ~140 nm | [120] |
| No specific target | Unguided carriers | HSC (+++), Kupffer cells and LSEC (++), hepatocytes (+) | Anti-col1a1 siRNA | Antifibrotic effect vs scrambled siRNA loaded carriers in CCl4 fibrotic mice | Cationic nanohydrogel particles | ~40 nm (~0 mV) | [121,122] |
| No specific target | Unguided carriers | HSC (+++), Kupffer cells/macrophages (++), hepatocytes (+), LSEC (+) | Anti-col1a1 siRNA | Antifibrotic effect vs scramble siRNA loaded nano- carriers in CCl4 and MDR2-/- fibrotic mice | Lipid cationic nanoparticles | [123] | |
| M6P receptor | M6P | HSC > Kupffer cells > LSEC | No drug reported | No effect reported | Liposomes | ~102 nm | [124] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaps, L.; Schuppan, D. Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers. Cells 2020, 9, 2027. https://doi.org/10.3390/cells9092027
Kaps L, Schuppan D. Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers. Cells. 2020; 9(9):2027. https://doi.org/10.3390/cells9092027
Chicago/Turabian StyleKaps, Leonard, and Detlef Schuppan. 2020. "Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers" Cells 9, no. 9: 2027. https://doi.org/10.3390/cells9092027
APA StyleKaps, L., & Schuppan, D. (2020). Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers. Cells, 9(9), 2027. https://doi.org/10.3390/cells9092027