Transient Receptor Potential Canonical (TRPC) Channels: Then and Now

 and
and 
Abstract
:1. Introduction and Historical Overview
2. Discovery of TRPC Channels
3. Structure of TRPC Channels
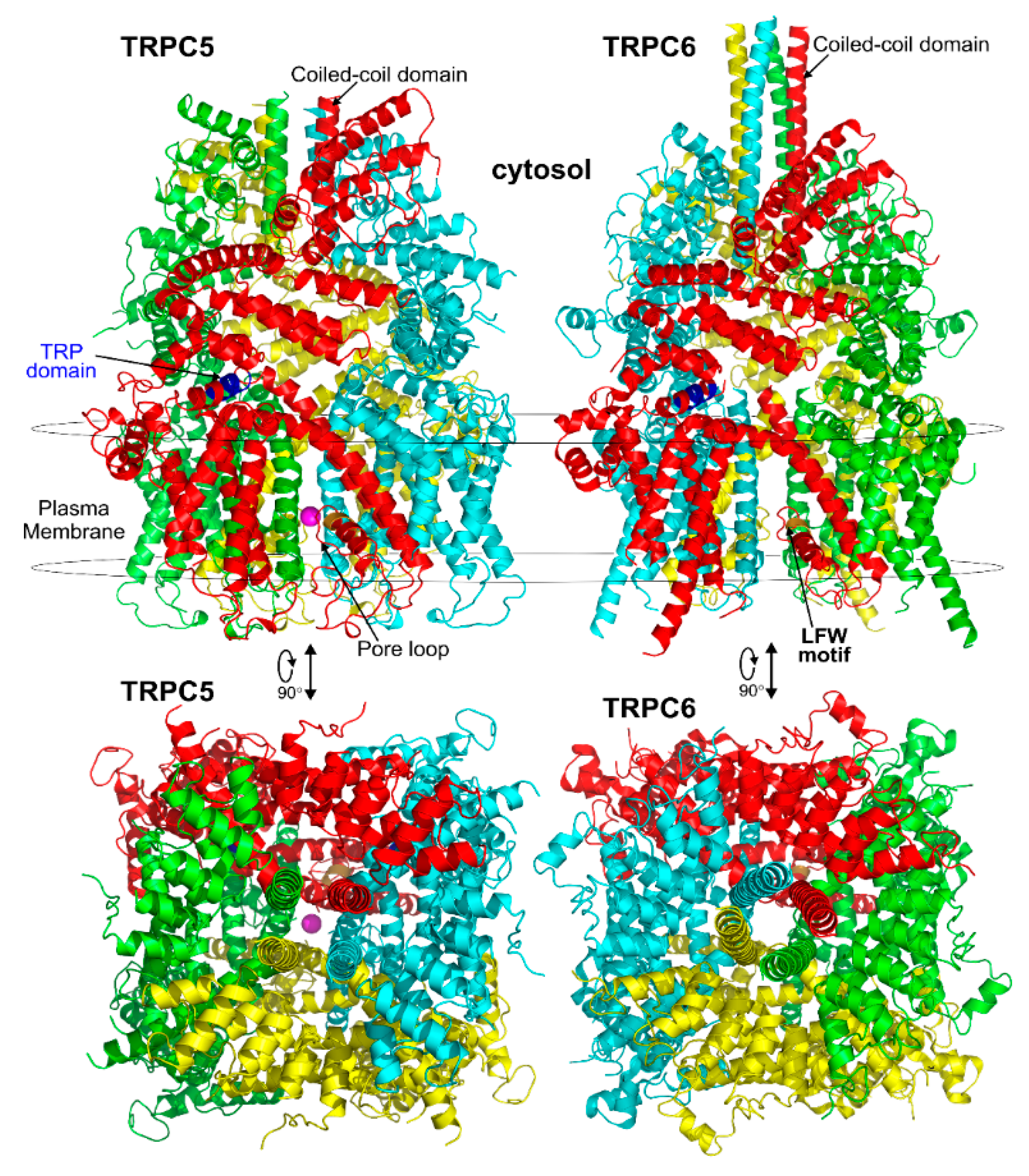
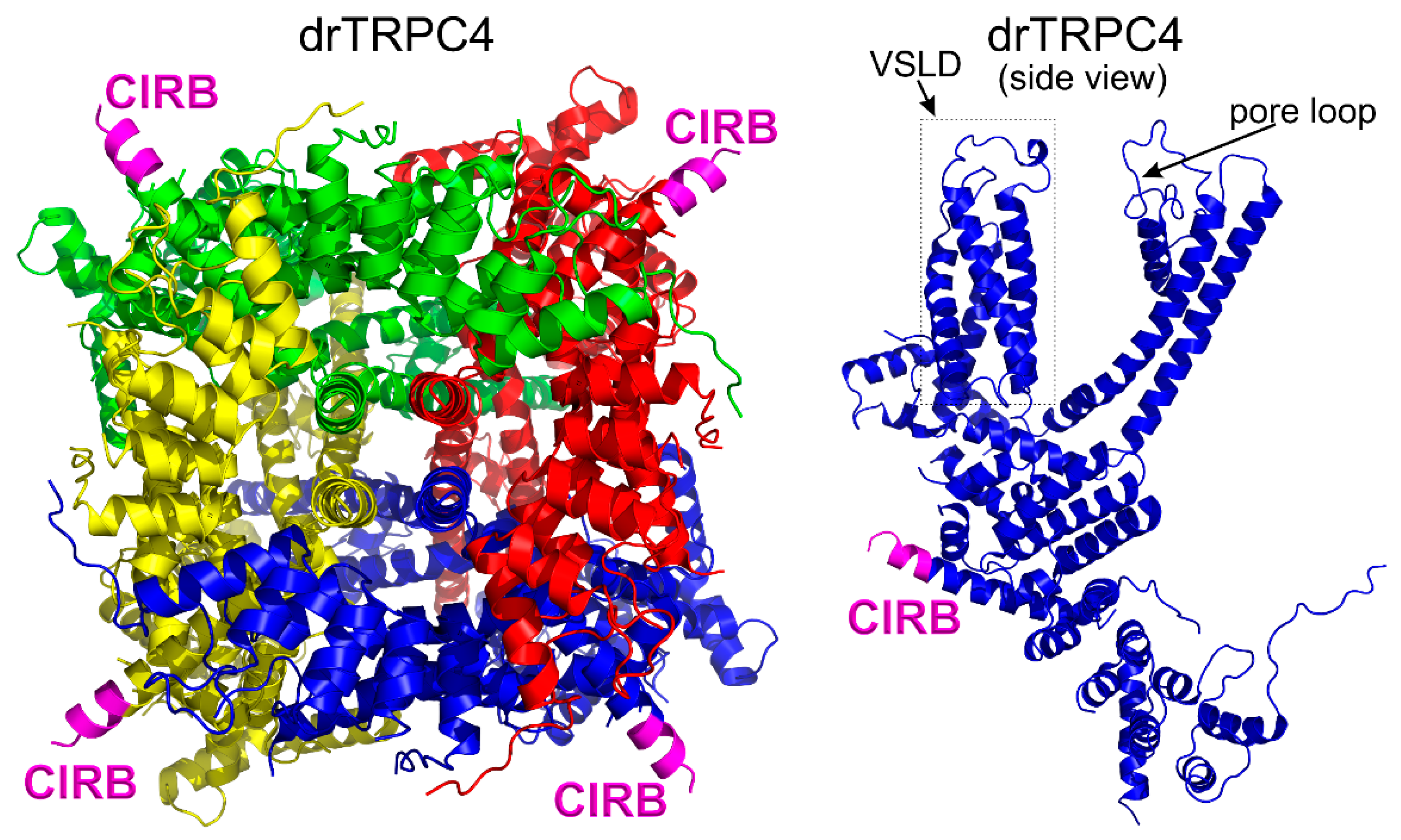
3.1. General Structural Organization of TRPCs
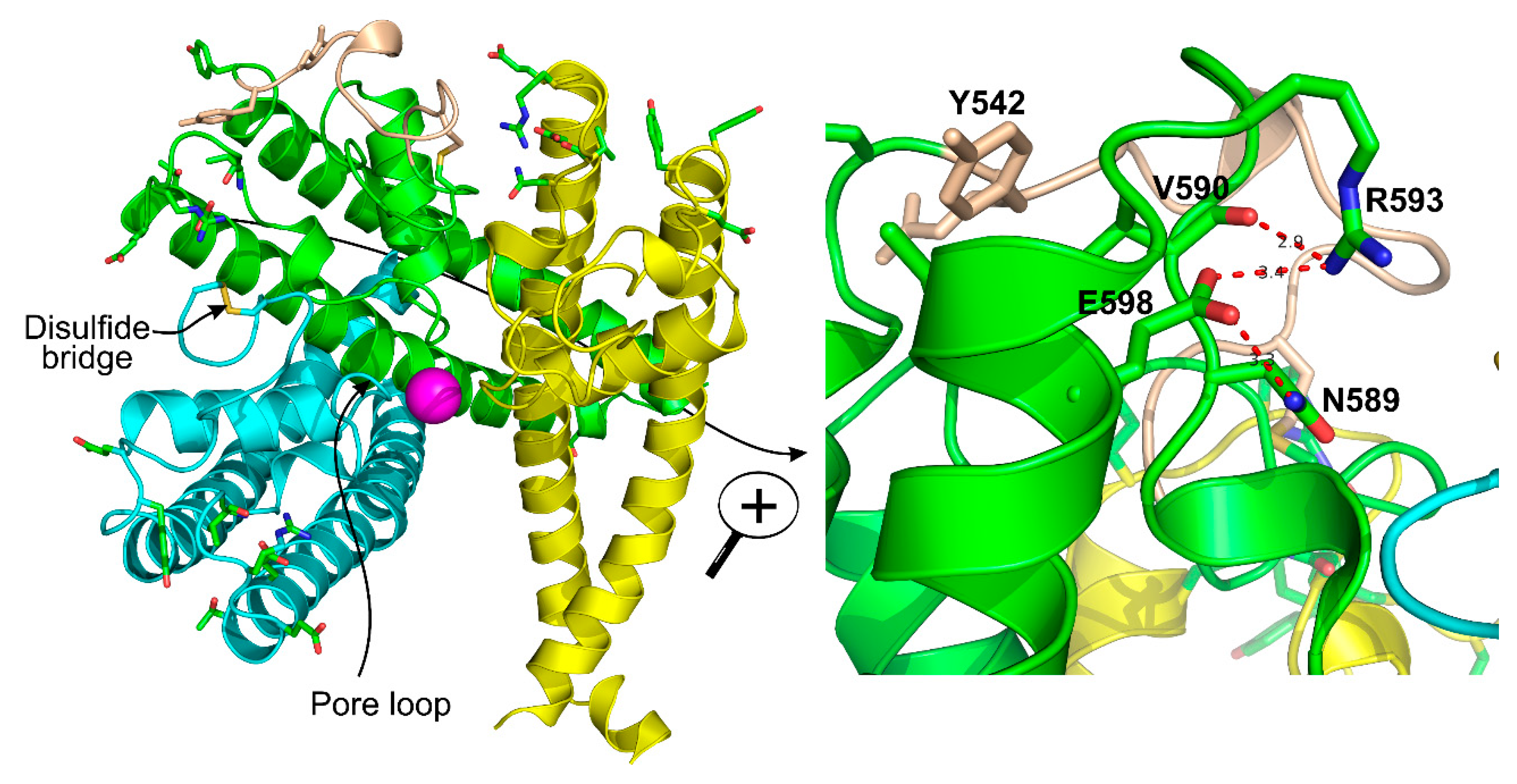
3.2. The Pore Region
3.3. Disulfide Bond
3.4. Cation Binding Sites
3.5. Arg593 Serves as “Molecular Fulcrum” and Is Critical for GPCR-Gq-PLC-Dependent Gating of TRPC5
3.6. The Calmodulin Binding Site on TRPC4
4. Physiological and Pathological Functions of TRPCs Revealed Using Gene Knockout, Knockin and Pharmacological Approaches
4.1. Physiological Activators of TRPC Channels

4.2. TRPC-Mediated Cellular Ca2+ Signaling
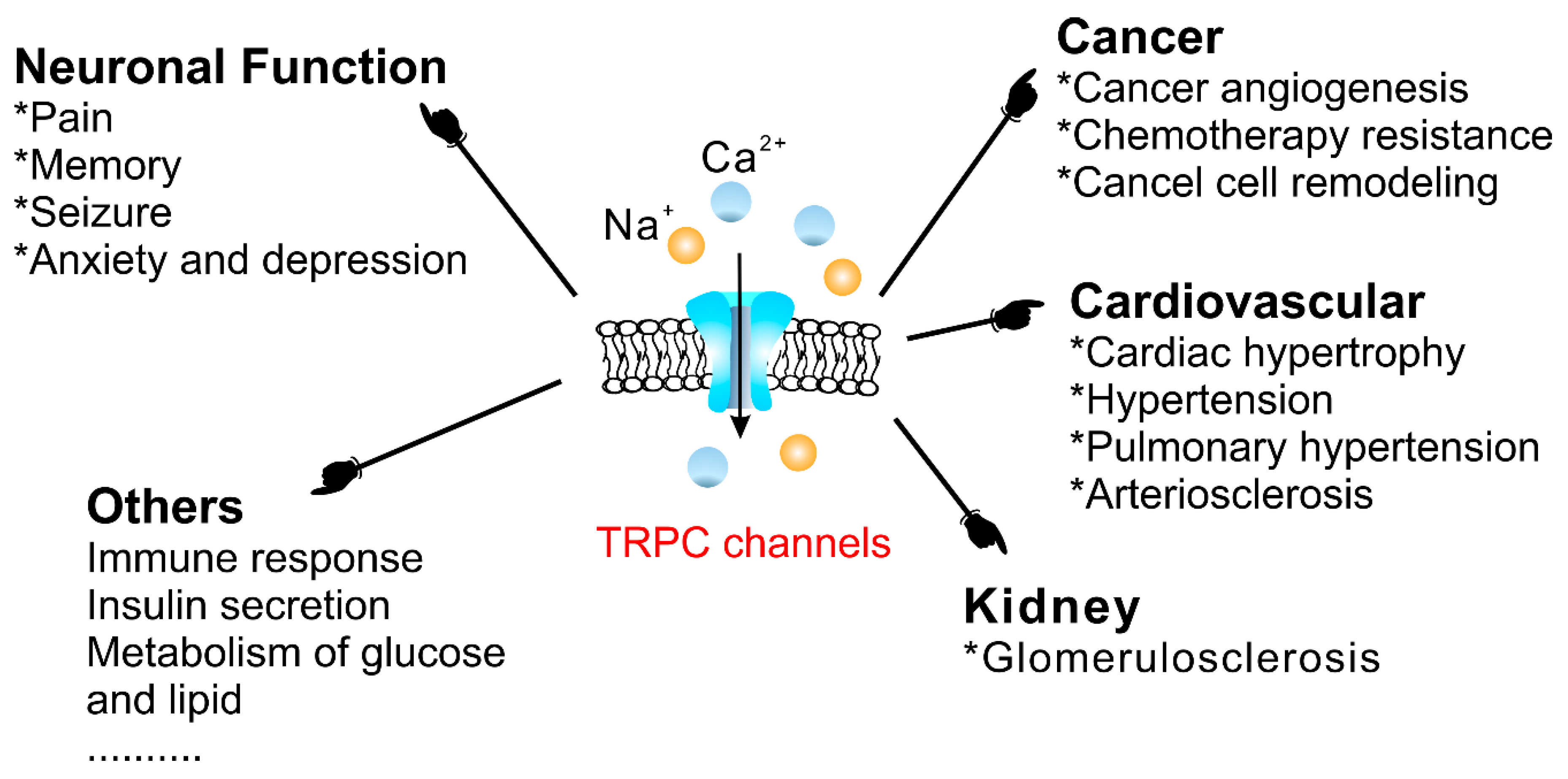
4.3. Physiological and Pathophysiological Functions of TRPC Channels
4.3.1. The Cardiovascular System
4.3.2. Cancer
4.3.3. Diabetes Mellitus
4.3.4. Neuronal Function
4.3.5. The Uterus
4.3.6. The Gastrointestinal Tract
4.3.7. The Kidneys
4.3.8. Others
5. A Brief Guide to Small Molecular Modulators for TRPC Channels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agonists | Chemical Structure | TRPC Channel (EC50/IC50) | Characteristics | Reference |
|---|---|---|---|---|
| GSK1702934A |  | TRPC3 (0.08 μm) TRPC6 (0.44 μm) | No effect on TRPV4, TRPA1, TRPM1, TRPM4, CaV1.2, hERG, NaV1.5, or CXCR5 receptors at a concentration of 10 μmol/L | [305] |
| Pyrazolo-pyrimidines |  | TRPC6 (0.89–6.28 µm) TRPC3 (0.02–0.45 μm) | Potency order: TRPC3 > TRPC7 > TRPC6 | [306] |
| OptoDArG |  | TRPC6 TRPC3 (30 μm was tested) | Photoswitchable DAG analogue containing two azobenzene photoswitchable moieties; active in cis-form at 365 nm and inactive at 430 nm | [50] |
| OptoBI-1 |  | TRPC6 TRPC3 TRPC7 (10 and 30 μm were tested) | Photoswitchable azobenzene analogue of GSK1702934 A; active in the cis-form | [307] |
| PhoDAGs |  | TRPC6 TRPC2 (5 and 50 μm were tested) | Photoswitchable DAG analogues; contain one photoswitchable moiety; active in cis-form at 370 nM and inactive at 460 nm | [308] |
| Hyp9 |  | TRPC6 (1.26 μm) | A derivative of nonselective activator of TRPC3, TRPC6, and TRPC7 | [305] |
| Artemisinin |  | TRPC3 (33 µm) | May inhibit TRPC6 | [309] |
| (−)-Englerin A |  | TRPC4 (11.2 nm) and TRPC5 (7.6 nm) | Selective activator | [296] |
| Antagonists | Chemical Structure | TRPC Channel (EC50/IC50) | Characteristics | Reference |
|---|---|---|---|---|
| Pyrazolo [1,5-a] pyrimidine (14a) |  | TRPC6 (1 μm) | Inhibits TRPC3/6/7 (TRPC6 > C7 > C3) with a very weak effect on TRPC4 and no effect on other TRP channels. | [197] |
| Pyrazole 3 (Pyr3) |  | TRPC3 (0.5 μm) TRPC6 (> 10 μm) | Also inhibits STIM/Orai | [274] |
| Pyrazole 10 (Pyr10) |  | TRPC (0.72 μm) TRPC6 (> 10 μm) | More selective than Pyr3; does not inhibit STIM/Orai | [160] |
| BTDM |  | TRPC3 (0.01 μm) TRPC6 (0.01 μm) | The exact BTDM binding site in TRPC6 was defined by cryo-EM; wedges between the S5-S6 pore domain and voltage sensor-like domain to inhibit channel opening | [99] |
| GSK503A |  | TRPC3 (0.003 μm) TRPC6 (0.021 μm) | Anilino thiazoles; good selectivity over other TRPA1,TRPV1, TRPV4, CaV1.2, hERG, and NaV1.5; in rodent models not orally bioavailable; high clearance, more suitable as in vitro tool | [141] |
| DS88790512 |  | TRPC6 (0.011 μm) | Novel blocker of TRPC6; cyclohexanone derivative; excellent selectivity against hERG and hNaV1.5 channels | [310] |
| larixyl acetate |  | TRPC6 (0.1–0.6 μm) | Larch-derived labdane-type diterpenes; 12- and 5-fold selectivity compared with TRPC3 and TRPC7 | [177] |
| BI749327 |  | TRPC6 (13 nm) | BI 749327 is 85-fold more selective for mouse TRPC6 than TRPC3 (IC50 = 1100 nm) and 42-fold versus TRPC7 | [311] |
| Pico145 (HC-608) |  | TRPC4 (63 pM) TRPC5 (1.3 nm) | Pico145 potency ranges from 9 to 1300 pM depending on the TRPC1/4/5 subtype while a range of other TRPC channels were unaffected | [312] |
| HC-070 |  | TRPC4 (46.0 ± 3.9 nm) TRPC5 (9.3 ± 0.9 nm) | HC-070 inhibits recombinant TRPC4 and TRPC5 homomultimers in heterologous expression systems with nanomolar potency | [220] |
| AC-1903 |  | TRPC5 (14.7 μm) | AC1903 selectively blocks TRPC5 ion channels | [265] |
| ML204 |  | TRPC4 (0.99 μm) TRPC5 (9.2 μm) | ML204 exhibited modest inhibitory effects on TRPC6 | [174] |
| Galangin |  | TRPC5 (0.45 μm) | Galangin is a natural product from the ginger family and a TRPC5 inhibitor depending on the substitution patterns of both the chromone core and the phenyl ring | [313] |
| SAR7334 |  | TRPC6 (7.9 nm) | SAR7334 inhibited TRPC3 and TRPC7-mediated Ca2+ influx into cells with IC50 s of 282 nm and 226 nm | [314] |
| SH045 |  | TRPC6(~5.8 nm) | IC50 for TRPC3 and TRPC7 are 0.84 μm and 0.22 μm, respectively | [315] |
| Bromoenol lactone (BEL) |  | TRPC5 TRPC6 TRPC1–TRPC5 Cav1.2, SOCE | TRPC5: 10.6 μm TRPC6: 7.2 μm Cav1.2: 7.6 μm | [55] |
6. Conclusions
Funding
Conflicts of Interest
References
- Minke, B.; Wu, C.; Pak, W.L. Induction of photoreceptor voltage noise in the dark in Drosophila mutant. Nature 1975, 258, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Cosens, D.J.; Manning, A. Abnormal electroretinogram from a Drosophila mutant. Nature 1969, 224, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.; Hokanson, K.M.; Chang, L.T. Molecular basis of an inherited retinal defect in Drosophila. Invest. Ophthalmol. Vis. Sci 1985, 26, 243–246. [Google Scholar]
- Montell, C.; Rubin, G.M. Molecular characterization of the Drosophila trp locus: A putative integral membrane protein required for phototransduction. Neuron 1989, 2, 1313–1323. [Google Scholar] [CrossRef]
- Wong, F.; Schaefer, E.L.; Roop, B.C.; LaMendola, J.N.; Johnson-Seaton, D.; Shao, D. Proper function of the Drosophila trp gene product during pupal development is important for normal visual transduction in the adult. Neuron 1989, 3, 81–94. [Google Scholar] [CrossRef]
- Hardie, R.C.; Minke, B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron 1992, 8, 643–651. [Google Scholar] [CrossRef]
- Phillips, A.M.; Bull, A.; Kelly, L.E. Identification of a Drosophila gene encoding a calmodulin-binding protein with homology to the trp phototransduction gene. Neuron 1992, 8, 631–642. [Google Scholar] [CrossRef]
- Niemeyer, B.A.; Suzuki, E.; Scott, K.; Jalink, K.; Zuker, C.S. The Drosophila light-activated conductance is composed of the two channels TRP and TRPL. Cell 1996, 85, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Reuss, H.; Mojet, M.H.; Chyb, S.; Hardie, R.C. In vivo analysis of the drosophila light-sensitive channels, TRP and TRPL. Neuron 1997, 19, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Obukhov, A.G.; Harteneck, C.; Zobel, A.; Harhammer, R.; Kalkbrenner, F.; Leopoldt, D.; Luckhoff, A.; Nurnberg, B.; Schultz, G. Direct activation of trpl cation channels by G alpha11 subunits. EMBO J. 1996, 15, 5833–5838. [Google Scholar] [CrossRef]
- Obukhov, A.G.; Schultz, G.; Luckhoff, A. Regulation of heterologously expressed transient receptor potential-like channels by calcium ions. Neuroscience 1998, 85, 487–495. [Google Scholar] [CrossRef]
- Liu, C.H.; Wang, T.; Postma, M.; Obukhov, A.G.; Montell, C.; Hardie, R.C. In vivo identification and manipulation of the Ca2+ selectivity filter in the Drosophila transient receptor potential channel. J. Neurosci. 2007, 27, 604–615. [Google Scholar] [CrossRef]
- Hardie, R.C. A brief history of trp: Commentary and personal perspective. Pflug. Arch. 2011, 461, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Yoshioka, T.; Hotta, Y. A genetic study of inositol trisphosphate involvement in phototransduction using Drosophila mutants. Biochem. Biophys. Res. Commun. 1985, 132, 513–519. [Google Scholar] [CrossRef]
- Devary, O.; Heichal, O.; Blumenfeld, A.; Cassel, D.; Suss, E.; Barash, S.; Rubinstein, C.T.; Minke, B.; Selinger, Z. Coupling of photoexcited rhodopsin to inositol phospholipid hydrolysis in fly photoreceptors. Proc. Natl. Acad. Sci. USA 1987, 84, 6939–6943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Putney, J.W. Forms and functions of store-operated calcium entry mediators, STIM and Orai. Adv. Biol. Regul. 2018, 68, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Minke, B.; Selinger, Z. The inositol-lipid pathway is necessary for light excitation in fly photoreceptors. Soc. Gen. Physiol. Ser. 1992, 47, 201–217. [Google Scholar]
- Pollock, J.A.; Assaf, A.; Peretz, A.; Nichols, C.D.; Mojet, M.H.; Hardie, R.C.; Minke, B. TRP, a protein essential for inositide-mediated Ca2+ influx is localized adjacent to the calcium stores in Drosophila photoreceptors. J. Neurosci. 1995, 15, 3747–3760. [Google Scholar] [CrossRef] [PubMed]
- Hardie, R.C. Excitation of Drosophila photoreceptors by BAPTA and ionomycin: Evidence for capacitative Ca2+ entry? Cell Calcium 1996, 20, 315–327. [Google Scholar] [CrossRef]
- Scott, K.; Sun, Y.; Beckingham, K.; Zuker, C.S. Calmodulin regulation of Drosophila light-activated channels and receptor function mediates termination of the light response in vivo. Cell 1997, 91, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Vaca, L.; Zhu, X.; Birnbaumer, L.; Kunze, D.L.; Schilling, W.P. Appearance of a novel Ca2+ influx pathway in Sf9 insect cells following expression of the transient receptor potential-like (trpl) protein of Drosophila. Biochem. Biophys. Res. Commun. 1994, 201, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Vaca, L.; Sinkins, W.G.; Hu, Y.; Kunze, D.L.; Schilling, W.P. Activation of recombinant trp by thapsigargin in Sf9 insect cells. Am. J. Physiol. 1994, 267, C1501–C1505. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Schilling, W.P. Receptor-mediated activation of recombinant Trpl expressed in Sf9 insect cells. Biochem. J. 1995, 305, 605–611. [Google Scholar] [CrossRef] [Green Version]
- Harteneck, C.; Obukhov, A.G.; Zobel, A.; Kalkbrenner, F.; Schultz, G. The Drosophila cation channel trpl expressed in insect Sf9 cells is stimulated by agonists of G-protein-coupled receptors. FEBS Lett. 1995, 358, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Gillo, B.; Chorna, I.; Cohen, H.; Cook, B.; Manistersky, I.; Chorev, M.; Arnon, A.; Pollock, J.A.; Selinger, Z.; Minke, B. Coexpression of Drosophila TRP and TRP-like proteins in Xenopus oocytes reconstitutes capacitative Ca2+ entry. Proc. Natl. Acad. Sci. USA 1996, 93, 14146–14151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.Z.; Li, H.S.; Guggino, W.B.; Montell, C. Coassembly of TRP and TRPL produces a distinct store-operated conductance. Cell 1997, 89, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, G.; Montell, C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Chu, P.B.; Peyton, M.; Birnbaumer, L. Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Lett. 1995, 373, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 1996, 85, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Vannier, B.; Peyton, M.; Boulay, G.; Brown, D.; Qin, N.; Jiang, M.; Zhu, X.; Birnbaumer, L. Mouse trp2, the homologue of the human trpc2 pseudogene, encodes mTrp2, a store depletion-activated capacitative Ca2+ entry channel. Proc. Natl. Acad. Sci. USA 1999, 96, 2060–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philipp, S.; Cavalie, A.; Freichel, M.; Wissenbach, U.; Zimmer, S.; Trost, C.; Marquart, A.; Murakami, M.; Flockerzi, V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO J. 1996, 15, 6166–6171. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Hambrecht, J.; Braslavski, L.; Schroth, G.; Freichel, M.; Murakami, M.; Cavalie, A.; Flockerzi, V. A novel capacitative calcium entry channel expressed in excitable cells. EMBO J. 1998, 17, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Shimizu, S.; Wakamori, M.; Maeda, A.; Kurosaki, T.; Takada, N.; Imoto, K.; Mori, Y. Molecular cloning and functional characterization of a novel receptor-activated TRP Ca2+ channel from mouse brain. J. Biol. Chem. 1998, 273, 10279–10287. [Google Scholar] [CrossRef] [Green Version]
- Boulay, G.; Zhu, X.; Peyton, M.; Jiang, M.; Hurst, R.; Stefani, E.; Birnbaumer, L. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J. Biol. Chem. 1997, 272, 29672–29680. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Inoue, R.; Yamazaki, K.; Maeda, A.; Kurosaki, T.; Yamakuni, T.; Tanaka, I.; Shimizu, S.; Ikenaka, K.; Imoto, K.; et al. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca(2+)-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J. Biol. Chem. 1999, 274, 27359–27370. [Google Scholar] [CrossRef] [Green Version]
- Riccio, A.; Mattei, C.; Kelsell, R.E.; Medhurst, A.D.; Calver, A.R.; Randall, A.D.; Davis, J.B.; Benham, C.D.; Pangalos, M.N. Cloning and functional expression of human short TRP7, a candidate protein for store-operated Ca2+ influx. J. Biol. Chem. 2002, 277, 12302–12309. [Google Scholar] [CrossRef] [Green Version]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Zitt, C.; Obukhov, A.G.; Strubing, C.; Zobel, A.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J. Cell Biol. 1997, 138, 1333–1341. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Schaefer, M.; Plant, T.D.; Obukhov, A.G.; Hofmann, T.; Gudermann, T.; Schultz, G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J. Biol. Chem. 2000, 275, 17517–17526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lof, C.; Viitanen, T.; Sukumaran, P.; Tornquist, K. TRPC2: Of mice but not men. Adv. Exp. Med. Biol. 2011, 704, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, K.; Xu, X.; Mozhayeva, G.; Kuo, T.; Pessah, I.; Mignery, G.; Zhu, X.; Birnbaumer, L.; Muallem, S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature 1998, 396, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Lin, Y.; Zhang, Z.; Tikunova, S.; Birnbaumer, L.; Zhu, M.X. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J. Biol. Chem. 2001, 276, 21303–21310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tang, J.; Tikunova, S.; Johnson, J.D.; Chen, Z.; Qin, N.; Dietrich, A.; Stefani, E.; Birnbaumer, L.; Zhu, M.X. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc. Natl. Acad. Sci. USA 2001, 98, 3168–3173. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M.; St, J.B.G.; McKay, R.R.; Birnbaumer, L.; Putney, J.W., Jr. Signaling mechanism for receptor-activated canonical transient receptor potential 3 (TRPC3) channels. J. Biol. Chem. 2003, 278, 16244–16252. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Zheng, F.; Gill, D.L. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J. Biol. Chem. 2003, 278, 29031–29040. [Google Scholar] [CrossRef] [Green Version]
- Lemonnier, L.; Trebak, M.; Putney, J.W., Jr. Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4,5-bisphosphate. Cell Calcium 2008, 43, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M.; Lemonnier, L.; DeHaven, W.I.; Wedel, B.J.; Bird, G.S.; Putney, J.W., Jr. Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflug. Arch. 2009, 457, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Lichtenegger, M.; Tiapko, O.; Svobodova, B.; Stockner, T.; Glasnov, T.N.; Schreibmayer, W.; Platzer, D.; de la Cruz, G.G.; Krenn, S.; Schober, R.; et al. An optically controlled probe identifies lipid-gating fenestrations within the TRPC3 channel. Nat. Chem. Biol. 2018, 14, 396–404. [Google Scholar] [CrossRef]
- Tang, Y.; Tang, J.; Chen, Z.; Trost, C.; Flockerzi, V.; Li, M.; Ramesh, V.; Zhu, M.X. Association of mammalian trp4 and phospholipase C isozymes with a PDZ domain-containing protein, NHERF. J. Biol. Chem. 2000, 275, 37559–37564. [Google Scholar] [CrossRef] [Green Version]
- Obukhov, A.G.; Nowycky, M.C. TRPC5 activation kinetics are modulated by the scaffolding protein ezrin/radixin/moesin-binding phosphoprotein-50 (EBP50). J. Cell Physiol. 2004, 201, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Storch, U.; Forst, A.L.; Pardatscher, F.; Erdogmus, S.; Philipp, M.; Gregoritza, M.; Mederos, Y.S.M.; Gudermann, T. Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc. Natl. Acad. Sci. USA 2017, 114, E37–E46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Muhle, A.; Schaefer, M.; Strotmann, R.; Schultz, G.; Plant, T.D. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J. Biol. Chem. 2003, 278, 3562–3571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Berwick, Z.C.; Bartlett, P.J.; Kumar, S.; Thomas, A.P.; Sturek, M.; Tune, J.D.; Obukhov, A.G. Bromoenol lactone inhibits voltage-gated Ca2+ and transient receptor potential canonical channels. J. Pharmacol. Exp. Ther. 2011, 339, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, W.; Riley, A.M.; Soliman, M.; Chakraborty, S.; Stamatkin, C.W.; Obukhov, A.G. Molecular Determinants of the Sensitivity to Gq/11-Phospholipase C-dependent Gating, Gd3+ Potentiation, and Ca2+ Permeability in the Transient Receptor Potential Canonical Type 5 (TRPC5) Channel. J. Biol. Chem. 2017, 292, 898–911. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Xu, S.Z.; Jackson, P.K.; McHugh, D.; Kumar, B.; Fountain, S.J.; Beech, D.J. Human TRPC5 channel activated by a multiplicity of signals in a single cell. J. Physiol. 2004, 559, 739–750. [Google Scholar] [CrossRef]
- Subedi, K.P.; Ong, H.L.; Ambudkar, I.S. Assembly of ER-PM Junctions: A Critical Determinant in the Regulation of SOCE and TRPC1. Adv. Exp. Med. Biol. 2017, 981, 253–276. [Google Scholar] [CrossRef]
- Liao, Y.; Erxleben, C.; Yildirim, E.; Abramowitz, J.; Armstrong, D.L.; Birnbaumer, L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 4682–4687. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.H.; Li, Q.; Kim, M.S.; Shin, D.M.; Feske, S.; Birnbaumer, L.; Cheng, K.T.; Ambudkar, I.S.; Muallem, S. Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 2011, 12, 232–245. [Google Scholar] [CrossRef] [Green Version]
- So, I.; Chae, M.R.; Kim, S.J.; Lee, S.W. Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces the change of calcium mobilization via TRPC ion channels in cultured human corporal smooth muscle cells. Int. J. Impot. Res. 2005, 17, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemming, P.K.; Dedman, A.M.; Xu, S.Z.; Li, J.; Zeng, F.; Naylor, J.; Benham, C.D.; Bateson, A.N.; Muraki, K.; Beech, D.J. Sensing of lysophospholipids by TRPC5 calcium channel. J. Biol. Chem. 2006, 281, 4977–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.Z.; Muraki, K.; Zeng, F.; Li, J.; Sukumar, P.; Shah, S.; Dedman, A.M.; Flemming, P.K.; McHugh, D.; Naylor, J.; et al. A sphingosine-1-phosphate-activated calcium channel controlling vascular smooth muscle cell motility. Circ. Res. 2006, 98, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Gomis, A.; Soriano, S.; Belmonte, C.; Viana, F. Hypoosmotic- and pressure-induced membrane stretch activate TRPC5 channels. J. Physiol. 2008, 586, 5633–5649. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.; Ding, M.; Ding, Y.; Sours-Brothers, S.; Luchowski, R.; Gryczynski, Z.; Yorio, T.; Ma, H.; Ma, R. Canonical transient receptor potential 6 (TRPC6), a redox-regulated cation channel. J. Biol. Chem. 2010, 285, 23466–23476. [Google Scholar] [CrossRef] [Green Version]
- Spassova, M.A.; Hewavitharana, T.; Xu, W.; Soboloff, J.; Gill, D.L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. USA 2006, 103, 16586–16591. [Google Scholar] [CrossRef] [Green Version]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Hirama, T.; Lu, S.M.; Kay, J.G.; Maekawa, M.; Kozlov, M.M.; Grinstein, S.; Fairn, G.D. Membrane curvature induced by proximity of anionic phospholipids can initiate endocytosis. Nat. Commun. 2017, 8, 1393. [Google Scholar] [CrossRef] [Green Version]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185. [Google Scholar] [CrossRef]
- Dietrich, A.; Kalwa, H.; Storch, U.; Mederos y Schnitzler, M.; Salanova, B.; Pinkenburg, O.; Dubrovska, G.; Essin, K.; Gollasch, M.; Birnbaumer, L.; et al. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflug. Arch. 2007, 455, 465–477. [Google Scholar] [CrossRef]
- Gottlieb, P.; Folgering, J.; Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Bowman, C.; Bichet, D.; Patel, A.; Sachs, F.; et al. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflug. Arch. 2008, 455, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Staaf, S.; Maxvall, I.; Lind, U.; Husmark, J.; Mattsson, J.P.; Ernfors, P.; Pierrou, S. Down regulation of TRPC1 by shRNA reduces mechanosensitivity in mouse dorsal root ganglion neurons in vitro. Neurosci. Lett. 2009, 457, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, S.R.; Dietrich, A.; Stucky, C.L. TRPC1 contributes to light-touch sensation and mechanical responses in low-threshold cutaneous sensory neurons. J. Neurophysiol. 2012, 107, 913–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessandri-Haber, N.; Dina, O.A.; Chen, X.; Levine, J.D. TRPC1 and TRPC6 channels cooperate with TRPV4 to mediate mechanical hyperalgesia and nociceptor sensitization. J. Neurosci. 2009, 29, 6217–6228. [Google Scholar] [CrossRef] [Green Version]
- Semtner, M.; Schaefer, M.; Pinkenburg, O.; Plant, T.D. Potentiation of TRPC5 by protons. J. Biol. Chem. 2007, 282, 33868–33878. [Google Scholar] [CrossRef] [Green Version]
- Obukhov, A.G.; Nowycky, M.C. TRPC5 channels undergo changes in gating properties during the activation-deactivation cycle. J. Cell Physiol. 2008, 216, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 2001, 29, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Obukhov, A.G.; Nowycky, M.C. A cytosolic residue mediates Mg2+ block and regulates inward current amplitude of a transient receptor potential channel. J. Neurosci. 2005, 25, 1234–1239. [Google Scholar] [CrossRef] [Green Version]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar] [CrossRef] [Green Version]
- Yip, H.; Chan, W.Y.; Leung, P.C.; Kwan, H.Y.; Liu, C.; Huang, Y.; Michel, V.; Yew, D.T.; Yao, X. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochem. Cell Biol. 2004, 122, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Freichel, M.; Vennekens, R.; Olausson, J.; Stolz, S.; Philipp, S.E.; Weissgerber, P.; Flockerzi, V. Functional role of TRPC proteins in native systems: Implications from knockout and knock-down studies. J. Physiol. 2005, 567, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, C.Y.; Babich, L.; Word, R.A.; Zhong, M.; Ulloa, A.; Monga, M.; Sanborn, B.M. Expression of transient receptor channel proteins in human fundal myometrium in pregnancy. J. Soc. Gynecol. Investig. 2006, 13, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Kunert-Keil, C.; Bisping, F.; Kruger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elg, S.; Marmigere, F.; Mattsson, J.P.; Ernfors, P. Cellular subtype distribution and developmental regulation of TRPC channel members in the mouse dorsal root ganglion. J. Comp. Neurol. 2007, 503, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Wuensch, T.; Thilo, F.; Krueger, K.; Scholze, A.; Ristow, M.; Tepel, M. High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes 2010, 59, 844–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Xu, J.; Li, Z.; Yang, Z. Expression of TRPC6 in renal cortex and hippocampus of mouse during postnatal development. PLoS ONE 2012, 7, e38503. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Z.; Beech, D.J. TrpC1 is a membrane-spanning subunit of store-operated Ca(2+) channels in native vascular smooth muscle cells. Circ. Res. 2001, 88, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Startek, J.B.; Boonen, B.; Talavera, K.; Meseguer, V. TRP Channels as Sensors of Chemically-Induced Changes in Cell Membrane Mechanical Properties. Int. J. Mol. Sci. 2019, 20, 371. [Google Scholar] [CrossRef] [Green Version]
- Beech, D.J.; Muraki, K.; Flemming, R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J. Physiol. 2004, 559, 685–706. [Google Scholar] [CrossRef]
- Hu, G.; Oboukhova, E.A.; Kumar, S.; Sturek, M.; Obukhov, A.G. Canonical transient receptor potential channels expression is elevated in a porcine model of metabolic syndrome. Mol. Endocrinol. 2009, 23, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Chakraborty, S.; Barbosa, C.; Brustovetsky, T.; Brustovetsky, N.; Obukhov, A.G. Mechanisms controlling neurite outgrowth in a pheochromocytoma cell line: The role of TRPC channels. J. Cell Physiol. 2012, 227, 1408–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.; Yang, S.; Liu, Y.; Shao, W. TRPC Channels in Health and Disease. Adv. Exp. Med. Biol. 2017, 976, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiser, J.; Polu, K.R.; Moller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Choi, W.; Sun, W.; Du, J.; Lu, W. Structure of the human lipid-gated cation channel TRPC3. Elife 2018, 7. [Google Scholar] [CrossRef]
- Sierra-Valdez, F.; Azumaya, C.M.; Romero, L.O.; Nakagawa, T.; Cordero-Morales, J.F. Structure-function analyses of the ion channel TRPC3 reveal that its cytoplasmic domain allosterically modulates channel gating. J. Biol. Chem. 2018, 293, 16102–16114. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Guo, W.; Zheng, L.; Wu, J.X.; Liu, M.; Zhou, X.; Zhang, X.; Chen, L. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res. 2018, 28, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Azumaya, C.M.; Sierra-Valdez, F.; Cordero-Morales, J.F.; Nakagawa, T. Cryo-EM structure of the cytoplasmic domain of murine transient receptor potential cation channel subfamily C member 6 (TRPC6). J. Biol. Chem. 2018, 293, 10381–10391. [Google Scholar] [CrossRef] [Green Version]
- Vinayagam, D.; Mager, T.; Apelbaum, A.; Bothe, A.; Merino, F.; Hofnagel, O.; Gatsogiannis, C.; Raunser, S. Electron cryo-microscopy structure of the canonical TRPC4 ion channel. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Li, J.; Zeng, B.; Chen, G.L.; Peng, X.; Zhang, Y.; Wang, J.; Clapham, D.E.; Li, Z.; Zhang, J. Structure of the mouse TRPC4 ion channel. Nat. Commun. 2018, 9, 3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Li, J.; Chen, G.L.; Ge, Y.; Liu, J.; Xie, K.; Peng, X.; Zhou, W.; Zhong, J.; Zhang, Y.; et al. Cryo-EM structure of TRPC5 at 2.8-A resolution reveals unique and conserved structural elements essential for channel function. Sci. Adv. 2019, 5, eaaw7935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poteser, M.; Schleifer, H.; Lichtenegger, M.; Schernthaner, M.; Stockner, T.; Kappe, C.O.; Glasnov, T.N.; Romanin, C.; Groschner, K. PKC-dependent coupling of calcium permeation through transient receptor potential canonical 3 (TRPC3) to calcineurin signaling in HL-1 myocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10556–10561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenegger, M.; Stockner, T.; Poteser, M.; Schleifer, H.; Platzer, D.; Romanin, C.; Groschner, K. A novel homology model of TRPC3 reveals allosteric coupling between gate and selectivity filter. Cell Calcium 2013, 54, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.Z.; Sukumar, P.; Zeng, F.; Li, J.; Jairaman, A.; English, A.; Naylor, J.; Ciurtin, C.; Majeed, Y.; Milligan, C.J.; et al. TRPC channel activation by extracellular thioredoxin. Nature 2008, 451, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Kwak, M.; Myeong, J.; Ha, K.; Wie, J.; Jeon, J.H.; So, I. Extracellular disulfide bridges stabilize TRPC5 dimerization, trafficking, and activity. Pflug. Arch. 2015, 467, 703–712. [Google Scholar] [CrossRef]
- Xu, S.Z.; Zeng, F.; Lei, M.; Li, J.; Gao, B.; Xiong, C.; Sivaprasadarao, A.; Beech, D.J. Generation of functional ion-channel tools by E3 targeting. Nat. Biotechnol. 2005, 23, 1289–1293. [Google Scholar] [CrossRef]
- Vinayagam, D.Q.D.; Sitsel, O.; Merino, F.; Stabrin, M.; Hofnagel, O.; Yu, M.; Ledeboer, M.W.; Malojcic, G.; Raunser, S. Structural basis of TRPC4 regulation by calmodulin and pharmacological agents. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhu, M.X. Multiple roles of calmodulin and other Ca(2+)-binding proteins in the functional regulation of TRP channels. Pflug. Arch. 2005, 451, 105–115. [Google Scholar] [CrossRef]
- Liu, C.H.; Gong, Z.; Liang, Z.L.; Liu, Z.X.; Yang, F.; Sun, Y.J.; Ma, M.L.; Wang, Y.J.; Ji, C.R.; Wang, Y.H.; et al. Arrestin-biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Nat. Commun. 2017, 8, 14335. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Hao, Q.; Zheng, Y.M.; Liu, Q.H.; Wang, Y.X. Inositol 1,4,5-trisphosphate activates TRPC3 channels to cause extracellular Ca2+ influx in airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1455–L1466. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jeon, J.P.; Hong, C.; Kim, J.; Myeong, J.; Jeon, J.H.; So, I. An essential role of PI(4,5)P(2) for maintaining the activity of the transient receptor potential canonical (TRPC)4beta. Pflug. Arch. 2013, 465, 1011–1021. [Google Scholar] [CrossRef]
- Imai, Y.; Itsuki, K.; Okamura, Y.; Inoue, R.; Mori, M.X. A self-limiting regulation of vasoconstrictor-activated TRPC3/C6/C7 channels coupled to PI(4,5)P(2)-diacylglycerol signalling. J. Physiol. 2012, 590, 1101–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, S.N.; Albert, A.P.; Large, W.A. Activation of native TRPC1/C5/C6 channels by endothelin-1 is mediated by both PIP3 and PIP2 in rabbit coronary artery myocytes. J. Physiol. 2009, 587, 5361–5375. [Google Scholar] [CrossRef] [Green Version]
- Itsuki, K.; Imai, Y.; Hase, H.; Okamura, Y.; Inoue, R.; Mori, M.X. PLC-mediated PI(4,5)P2 hydrolysis regulates activation and inactivation of TRPC6/7 channels. J. Gen. Physiol. 2014, 143, 183–201. [Google Scholar] [CrossRef] [Green Version]
- Myeong, J.; Kwak, M.; Jeon, J.P.; Hong, C.; Jeon, J.H.; So, I. Close spatio-association of the transient receptor potential canonical 4 (TRPC4) channel with Galphai in TRPC4 activation process. Am. J. Physiol. Cell Physiol. 2015, 308, C879–C889. [Google Scholar] [CrossRef] [Green Version]
- Thakur, D.P.; Tian, J.B.; Jeon, J.; Xiong, J.; Huang, Y.; Flockerzi, V.; Zhu, M.X. Critical roles of Gi/o proteins and phospholipase C-delta1 in the activation of receptor-operated TRPC4 channels. Proc. Natl. Acad. Sci. USA 2016, 113, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, J.; Latta, L.; Beck, A.; Leidinger, P.; Fecher-Trost, C.; Schlenstedt, G.; Meese, E.; Wissenbach, U.; Flockerzi, V. Trans-activation response (TAR) RNA-binding protein 2 is a novel modulator of transient receptor potential canonical 4 (TRPC4) protein. J. Biol. Chem. 2014, 289, 9766–9780. [Google Scholar] [CrossRef] [Green Version]
- Bousquet, S.M.; Monet, M.; Boulay, G. Protein kinase C-dependent phosphorylation of transient receptor potential canonical 6 (TRPC6) on serine 448 causes channel inhibition. J. Biol. Chem. 2010, 285, 40534–40543. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Egly, C.; Riley, A.M.; Li, W.; Tewson, P.; Hughes, T.E.; Quinn, A.M.; Obukhov, A.G. PKC-dependent Phosphorylation of the H1 Histamine Receptor Modulates TRPC6 Activity. Cells 2014, 3, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagmann, H.; Mangold, N.; Rinschen, M.M.; Koenig, T.; Kunzelmann, K.; Schermer, B.; Benzing, T.; Brinkkoetter, P.T. Proline-dependent and basophilic kinases phosphorylate human TRPC6 at serine 14 to control channel activity through increased membrane expression. FASEB J. 2018, 32, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Trebak, M. Transient receptor potential canonical 7: A diacylglycerol-activated non-selective cation channel. Handb. Exp. Pharmacol. 2014, 222, 189–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeHaven, W.I.; Jones, B.F.; Petranka, J.G.; Smyth, J.T.; Tomita, T.; Bird, G.S.; Putney, J.W., Jr. TRPC channels function independently of STIM1 and Orai1. J. Physiol. 2009, 587, 2275–2298. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef]
- Broker-Lai, J.; Kollewe, A.; Schindeldecker, B.; Pohle, J.; Nguyen Chi, V.; Mathar, I.; Guzman, R.; Schwarz, Y.; Lai, A.; Weissgerber, P.; et al. Heteromeric channels formed by TRPC1, TRPC4 and TRPC5 define hippocampal synaptic transmission and working memory. EMBO J. 2017, 36, 2770–2789. [Google Scholar] [CrossRef] [PubMed]
- Neuner, S.M.; Wilmott, L.A.; Hope, K.A.; Hoffmann, B.; Chong, J.A.; Abramowitz, J.; Birnbaumer, L.; O’Connell, K.M.; Tryba, A.K.; Greene, A.S.; et al. TRPC3 channels critically regulate hippocampal excitability and contextual fear memory. Behav. Brain Res. 2015, 281, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014, 28, 4677–4685. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.S.; Xiao, J.H.; Wang, Y.; Xu, B.M.; Gao, L.; Wang, J.L. Upregulation of TRPC1 contributes to contractile function in isoproterenol-induced hypertrophic myocardium of rat. Cell Physiol. Biochem. 2013, 32, 951–959. [Google Scholar] [CrossRef]
- Seo, K.; Rainer, P.P.; Shalkey Hahn, V.; Lee, D.I.; Jo, S.H.; Andersen, A.; Liu, T.; Xu, X.; Willette, R.N.; Lepore, J.J.; et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 1551–1556. [Google Scholar] [CrossRef] [Green Version]
- Han, J.W.; Lee, Y.H.; Yoen, S.I.; Abramowitz, J.; Birnbaumer, L.; Lee, M.G.; Kim, J.Y. Resistance to pathologic cardiac hypertrophy and reduced expression of CaV1.2 in Trpc3-depleted mice. Mol. Cell Biochem. 2016, 421, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Sunggip, C.; Shimoda, K.; Oda, S.; Tanaka, T.; Nishiyama, K.; Mangmool, S.; Nishimura, A.; Numaga-Tomita, T.; Nishida, M. TRPC5-eNOS Axis Negatively Regulates ATP-Induced Cardiomyocyte Hypertrophy. Front. Pharmacol. 2018, 9, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Li, J.; Wu, Q.; Zheng, X.; Gao, Y.; Yang, Q.; Sun, N.; He, M.; Zhou, Y. Effect of SKF96365 on cardiomyocyte hypertrophy induced by angiotensin II. Mol. Med. Rep. 2020, 21, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Yao, F.; Wang, H.; Wang, X.; Shen, J.; Dai, B.; Wu, H.; Zhou, D.; Guo, F.; Wang, J.; et al. Inhibition of TRPC1 prevents cardiac hypertrophy via NF-kappaB signaling pathway in human pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell Cardiol 2019, 126, 143–154. [Google Scholar] [CrossRef]
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.M.; Benitah, J.P.; Perros, F.; Humbert, M.; Antigny, F. Ca(2+) handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell Cardiol 2018, 118, 208–224. [Google Scholar] [CrossRef]
- Doleschal, B.; Primessnig, U.; Wolkart, G.; Wolf, S.; Schernthaner, M.; Lichtenegger, M.; Glasnov, T.N.; Kappe, C.O.; Mayer, B.; Antoons, G.; et al. TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc. Res. 2015, 106, 163–173. [Google Scholar] [CrossRef]
- Ju, Y.K.; Lee, B.H.; Trajanovska, S.; Hao, G.; Allen, D.G.; Lei, M.; Cannell, M.B. The involvement of TRPC3 channels in sinoatrial arrhythmias. Front. Physiol. 2015, 6, 86. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Iribe, G.; Kaneko, T.; Takahashi, K.; Numaga-Tomita, T.; Nishida, M.; Birnbaumer, L.; Naruse, K. TRPC3 participates in angiotensin II type 1 receptor-dependent stress-induced slow increase in intracellular Ca(2+) concentration in mouse cardiomyocytes. J. Physiol. Sci. 2018, 68, 153–164. [Google Scholar] [CrossRef]
- Eder, P.; Probst, D.; Rosker, C.; Poteser, M.; Wolinski, H.; Kohlwein, S.D.; Romanin, C.; Groschner, K. Phospholipase C-dependent control of cardiac calcium homeostasis involves a TRPC3-NCX1 signaling complex. Cardiovasc. Res. 2007, 73, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Camacho Londono, J.E.; Tian, Q.; Hammer, K.; Schroder, L.; Camacho Londono, J.; Reil, J.C.; He, T.; Oberhofer, M.; Mannebach, S.; Mathar, I.; et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur. Heart J. 2015, 36, 2257–2266. [Google Scholar] [CrossRef] [Green Version]
- Washburn, D.G.; Holt, D.A.; Dodson, J.; McAtee, J.J.; Terrell, L.R.; Barton, L.; Manns, S.; Waszkiewicz, A.; Pritchard, C.; Gillie, D.J.; et al. The discovery of potent blockers of the canonical transient receptor channels, TRPC3 and TRPC6, based on an anilino-thiazole pharmacophore. Bioorg. Med. Chem. Lett. 2013, 23, 4979–4984. [Google Scholar] [CrossRef] [PubMed]
- Welsh, D.G.; Morielli, A.D.; Nelson, M.T.; Brayden, J.E. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ. Res. 2002, 90, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.L.; Yang, Y.; Sullivan, M.N.; Sanders, L.; Dabertrand, F.; Hill-Eubanks, D.C.; Nelson, M.T.; Earley, S. A PLCgamma1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci. Signal. 2014, 7, ra49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Miguel, I.; Cidad, P.; Perez-Garcia, M.T.; Lopez-Lopez, J.R. Differences in TRPC3 and TRPC6 channels assembly in mesenteric vascular smooth muscle cells in essential hypertension. J. Physiol. 2017, 595, 1497–1513. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, A.; Mederos, Y.S.M.; Gollasch, M.; Gross, V.; Storch, U.; Dubrovska, G.; Obst, M.; Yildirim, E.; Salanova, B.; Kalwa, H.; et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol. Cell Biol. 2005, 25, 6980–6989. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Miralles, F.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Store depletion induces Galphaq-mediated PLCbeta1 activity to stimulate TRPC1 channels in vascular smooth muscle cells. FASEB J. 2016, 30, 702–715. [Google Scholar] [CrossRef]
- Shi, J.; Miralles, F.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Store-operated interactions between plasmalemmal STIM1 and TRPC1 proteins stimulate PLCbeta1 to induce TRPC1 channel activation in vascular smooth muscle cells. J. Physiol. 2017, 595, 1039–1058. [Google Scholar] [CrossRef]
- Avila-Medina, J.; Calderon-Sanchez, E.; Gonzalez-Rodriguez, P.; Monje-Quiroga, F.; Rosado, J.A.; Castellano, A.; Ordonez, A.; Smani, T. Orai1 and TRPC1 Proteins Co-localize with CaV1.2 Channels to Form a Signal Complex in Vascular Smooth Muscle Cells. J. Biol. Chem. 2016, 291, 21148–21159. [Google Scholar] [CrossRef] [Green Version]
- Lemos, V.S.; Poburko, D.; Liao, C.H.; Cole, W.C.; van Breemen, C. Na+ entry via TRPC6 causes Ca2+ entry via NCX reversal in ATP stimulated smooth muscle cells. Biochem. Biophys. Res. Commun. 2007, 352, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Zulian, A.; Baryshnikov, S.G.; Linde, C.I.; Hamlyn, J.M.; Ferrari, P.; Golovina, V.A. Upregulation of Na+/Ca2+ exchanger and TRPC6 contributes to abnormal Ca2+ homeostasis in arterial smooth muscle cells from Milan hypertensive rats. Am. J. Physiol. Heart Circ. Physiol 2010, 299, H624–H633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosker, C.; Graziani, A.; Lukas, M.; Eder, P.; Zhu, M.X.; Romanin, C.; Groschner, K. Ca(2+) signaling by TRPC3 involves Na(+) entry and local coupling to the Na(+)/Ca(2+) exchanger. J. Biol. Chem. 2004, 279, 13696–13704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, R. The Na+/Ca2+ exchange inhibitor KB-R7943 potently blocks TRPC channels. Biochem. Biophys. Res. Commun. 2007, 361, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, X.; Riley, A.M.; Hiett, S.C.; Temm, C.J.; Beli, E.; Long, X.; Chakraborty, S.; Alloosh, M.; White, F.A.; et al. Long-term spironolactone treatment reduces coronary TRPC expression, vasoconstriction, and atherosclerosis in metabolic syndrome pigs. Basic Res. Cardiol. 2017, 112, 54. [Google Scholar] [CrossRef] [PubMed]
- Numaga-Tomita, T.; Shimauchi, T.; Oda, S.; Tanaka, T.; Nishiyama, K.; Nishimura, A.; Birnbaumer, L.; Mori, Y.; Nishida, M. TRPC6 regulates phenotypic switching of vascular smooth muscle cells through plasma membrane potential-dependent coupling with PTEN. FASEB J. 2019, 33, 9785–9796. [Google Scholar] [CrossRef] [Green Version]
- Koenig, S.; Schernthaner, M.; Maechler, H.; Kappe, C.O.; Glasnov, T.N.; Hoefler, G.; Braune, M.; Wittchow, E.; Groschner, K. A TRPC3 blocker, ethyl-1-(4-(2,3,3-trichloroacrylamide)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-c arboxylate (Pyr3), prevents stent-induced arterial remodeling. J. Pharmacol. Exp. Ther. 2013, 344, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Dreja, K.; Shah, S.S.; Cheong, A.; Xu, S.Z.; Sukumar, P.; Naylor, J.; Forte, A.; Cipollaro, M.; McHugh, D.; et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ. Res. 2006, 98, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Yang, D.; He, H.; Chen, X.; Cao, T.; Feng, X.; Ma, L.; Luo, Z.; Wang, L.; Yan, Z.; et al. Increased transient receptor potential canonical type 3 channels in vasculature from hypertensive rats. Hypertension 2009, 53, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Lin, S.; Wang, B.; Wang, Q.; Xia, W.; Zhang, H.; Cui, Y.; He, C.; Wu, H.; Sun, F.; et al. TRPC3 deficiency attenuates high salt-induced cardiac hypertrophy by alleviating cardiac mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2019, 519, 674–681. [Google Scholar] [CrossRef]
- Wang, B.; Xiong, S.; Lin, S.; Xia, W.; Li, Q.; Zhao, Z.; Wei, X.; Lu, Z.; Wei, X.; Gao, P.; et al. Enhanced Mitochondrial Transient Receptor Potential Channel, Canonical Type 3-Mediated Calcium Handling in the Vasculature From Hypertensive Rats. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.N.; Kappe, C.O.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca(2+) entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Weissmann, N.; Dietrich, A.; Fuchs, B.; Kalwa, H.; Ay, M.; Dumitrascu, R.; Olschewski, A.; Storch, U.; Mederos y Schnitzler, M.; Ghofrani, H.A.; et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. USA 2006, 103, 19093–19098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malczyk, M.; Erb, A.; Veith, C.; Ghofrani, H.A.; Schermuly, R.T.; Gudermann, T.; Dietrich, A.; Weissmann, N.; Sydykov, A. The Role of Transient Receptor Potential Channel 6 Channels in the Pulmonary Vasculature. Front. Immunol. 2017, 8, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malczyk, M.; Veith, C.; Fuchs, B.; Hofmann, K.; Storch, U.; Schermuly, R.T.; Witzenrath, M.; Ahlbrecht, K.; Fecher-Trost, C.; Flockerzi, V.; et al. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yang, X.R.; Fu, Z.; Paudel, O.; Abramowitz, J.; Birnbaumer, L.; Sham, J.S. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension 2014, 63, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weissgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Gene, G.G.; Tomas, M.; Plata, C.; Selent, J.; Pastor, M.; Fandos, C.; Senti, M.; Lucas, G.; Elosua, R.; et al. A gain-of-function SNP in TRPC4 cation channel protects against myocardial infarction. Cardiovasc. Res. 2011, 91, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.L.; Shen, Z.; Li, Z.; Ye, X.; Wu, M.; Hong, L.; Zhao, Y. TRPC1 Deficiency Impairs the Endothelial Progenitor Cell Function via Inhibition of Calmodulin/eNOS Pathway. J. Cardiovasc. Transl. Res. 2018, 11, 339–345. [Google Scholar] [CrossRef]
- Greenberg, H.Z.E.; Carlton-Carew, S.R.E.; Khan, D.M.; Zargaran, A.K.; Jahan, K.S.; Vanessa Ho, W.S.; Albert, A.P. Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vascul. Pharmacol. 2017, 96–98, 53–62. [Google Scholar] [CrossRef]
- Kuang, C.Y.; Yu, Y.; Wang, K.; Qian, D.H.; Den, M.Y.; Huang, L. Knockdown of transient receptor potential canonical-1 reduces the proliferation and migration of endothelial progenitor cells. Stem Cells Dev. 2012, 21, 487–496. [Google Scholar] [CrossRef]
- Yeon, S.I.; Kim, J.Y.; Yeon, D.S.; Abramowitz, J.; Birnbaumer, L.; Muallem, S.; Lee, Y.H. Transient receptor potential canonical type 3 channels control the vascular contractility of mouse mesenteric arteries. PLoS ONE 2014, 9, e110413. [Google Scholar] [CrossRef]
- Tauseef, M.; Knezevic, N.; Chava, K.R.; Smith, M.; Sukriti, S.; Gianaris, N.; Obukhov, A.G.; Vogel, S.M.; Schraufnagel, D.E.; Dietrich, A.; et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 2012, 209, 1953–1968. [Google Scholar] [CrossRef] [PubMed]
- Strielkov, I.; Krause, N.C.; Sommer, N.; Schermuly, R.T.; Ghofrani, H.A.; Grimminger, F.; Gudermann, T.; Dietrich, A.; Weissmann, N. Hypoxic pulmonary vasoconstriction in isolated mouse pulmonary arterial vessels. Exp. Physiol. 2018, 103, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Ballejo, G. Pharmacological characterization of the calcium influx pathways involved in nitric oxide production by endothelial cells. Einstein (Sao Paulo) 2019, 17, eAO4600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.; Shi, J.; Zhu, Y.; Kustov, M.; Tian, J.B.; Stevens, A.; Wu, M.; Xu, J.; Long, S.; Yang, P.; et al. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J. Biol. Chem. 2011, 286, 33436–33446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalba, N.; Sackheim, A.M.; Nunez, I.A.; Hill-Eubanks, D.C.; Nelson, M.T.; Wellman, G.C.; Freeman, K. Traumatic Brain Injury Causes Endothelial Dysfunction in the Systemic Microcirculation through Arginase-1-Dependent Uncoupling of Endothelial Nitric Oxide Synthase. J. Neurotrauma 2017, 34, 192–203. [Google Scholar] [CrossRef]
- Chen, X.; Taylor-Nguyen, N.N.; Riley, A.M.; Herring, B.P.; White, F.A.; Obukhov, A.G. The TRPC6 inhibitor, larixyl acetate, is effective in protecting against traumatic brain injury-induced systemic endothelial dysfunction. J. Neuroinflamm. 2019, 16, 21. [Google Scholar] [CrossRef] [Green Version]
- Urban, N.; Wang, L.; Kwiek, S.; Rademann, J.; Kuebler, W.M.; Schaefer, M. Identification and Validation of Larixyl Acetate as a Potent TRPC6 Inhibitor. Mol. Pharmacol. 2016, 89, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Tano, J.Y.; Solanki, S.; Lee, R.H.; Smedlund, K.; Birnbaumer, L.; Vazquez, G. Bone marrow deficiency of TRPC3 channel reduces early lesion burden and necrotic core of advanced plaques in a mouse model of atherosclerosis. Cardiovasc. Res. 2014, 101, 138–144. [Google Scholar] [CrossRef]
- Solanki, S.; Dube, P.R.; Birnbaumer, L.; Vazquez, G. Reduced Necrosis and Content of Apoptotic M1 Macrophages in Advanced Atherosclerotic Plaques of Mice with Macrophage-Specific Loss of Trpc3. Sci. Rep. 2017, 7, 42526. [Google Scholar] [CrossRef]
- Smedlund, K.B.; Birnbaumer, L.; Vazquez, G. Increased size and cellularity of advanced atherosclerotic lesions in mice with endothelial overexpression of the human TRPC3 channel. Proc. Natl. Acad. Sci. USA 2015, 112, E2201–E2206. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qin, W.; Zhang, L.; Wu, X.; Du, N.; Hu, Y.; Li, X.; Shen, N.; Xiao, D.; Zhang, H.; et al. MicroRNA-26a prevents endothelial cell apoptosis by directly targeting TRPC6 in the setting of atherosclerosis. Sci. Rep. 2015, 5, 9401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, P.; Rosenbaum, M.A.; Sinharoy, P.; Damron, D.S.; Birnbaumer, L.; Graham, L.M. Membrane translocation of TRPC6 channels and endothelial migration are regulated by calmodulin and PI3 kinase activation. Proc. Natl. Acad. Sci. USA 2016, 113, 2110–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, M.A.; Chaudhuri, P.; Graham, L.M. Hypercholesterolemia inhibits re-endothelialization of arterial injuries by TRPC channel activation. J. Vasc. Surg. 2015, 62, 1040–1047.e1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Cui, J.; Mi, B.; Yan, X.; Xu, W.; Ma, H.; Zhang, Q.; Xu, F. Isoliquiritigenin Inhibits Atherosclerosis by Blocking TRPC5 Channel Expression. Cardiovasc. Ther. 2020, 2020, 1926249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Zhang, Y.; Zhuo, D.; Lo, C.Y.; Yu, L.; Lau, C.W.; Kwan, Y.W.; Tse, G.; Huang, Y.; Yao, X. Endothelial cell transient receptor potential channel C5 (TRPC5) is essential for endothelium-dependent contraction in mouse carotid arteries. Biochem. Pharmacol. 2019, 159, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, H.J.; Vasudev, N.S.; Beech, D.J. Transient receptor potential canonical 4 and 5 proteins as targets in cancer therapeutics. Eur. Biophys. J. 2016, 45, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Asghar, M.Y.; Magnusson, M.; Kemppainen, K.; Sukumaran, P.; Lof, C.; Pulli, I.; Kalhori, V.; Tornquist, K. Transient Receptor Potential Canonical 1 (TRPC1) Channels as Regulators of Sphingolipid and VEGF Receptor Expression: IMPLICATIONS FOR THYROID CANCER CELL MIGRATION AND PROLIFERATION. J. Biol. Chem. 2015, 290, 16116–16131. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Cai, Y.; He, D.; Zou, C.; Zhang, P.; Lo, C.Y.; Xu, Z.; Chan, F.L.; Yu, S.; Chen, Y.; et al. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16282–16287. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Chen, Z.; Hua, D.; He, D.; Wang, L.; Zhang, P.; Wang, J.; Cai, Y.; Gao, C.; Zhang, X.; et al. Essential role for TrpC5-containing extracellular vesicles in breast cancer with chemotherapeutic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 6389–6394. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Chen, M.; Zhang, S.; Miao, Z.; Wang, J.; Lu, X.; Zhao, X. TRPC5 induced autophagy promotes the TMZ resistance of glioma cells via the CAMMKbeta/AMPKalpha/mTOR pathway. Oncol. Rep. 2019, 41, 3413–3423. [Google Scholar] [CrossRef]
- Sobradillo, D.; Hernandez-Morales, M.; Ubierna, D.; Moyer, M.P.; Nunez, L.; Villalobos, C. A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmons, M.F.; Anreddy, N.; Cuevas, J.; Steinberger, K.; Yang, S.; McLaughlin, M.; Silva, A.; Hazlehurst, L.A. MTI-101 treatment inducing activation of Stim1 and TRPC1 expression is a determinant of response in multiple myeloma. Sci. Rep. 2017, 7, 2685. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Zhao, N.; Jin, H.; Zhang, Z.; Liu, Y.; Wu, J.; Bast, R.C., Jr.; Yu, Y.; Feng, Y. FSH enhances the proliferation of ovarian cancer cells by activating transient receptor potential channel C3. Endocr. Relat. Cancer 2013, 20, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Bernichtein, S.; Pigat, N.; Barry Delongchamps, N.; Boutillon, F.; Verkarre, V.; Camparo, P.; Reyes-Gomez, E.; Mejean, A.; Oudard, S.M.; Lepicard, E.M.; et al. Vitamin D3 Prevents Calcium-Induced Progression of Early-Stage Prostate Tumors by Counteracting TRPC6 and Calcium Sensing Receptor Upregulation. Cancer Res. 2017, 77, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Wang, H.; Qu, C.; Xu, F.; Zhu, Y.; Lv, G.; Lu, Y.; Zhou, Q.; Zhou, H.; Zeng, X.; et al. Pyrazolo[1,5-a]pyrimidine TRPC6 antagonists for the treatment of gastric cancer. Cancer Lett. 2018, 432, 47–55. [Google Scholar] [CrossRef]
- Nielsen, N.; Kondratska, K.; Ruck, T.; Hild, B.; Kovalenko, I.; Schimmelpfennig, S.; Welzig, J.; Sargin, S.; Lindemann, O.; Christian, S.; et al. TRPC6 channels modulate the response of pancreatic stellate cells to hypoxia. Pflug. Arch. 2017, 469, 1567–1577. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, W.; Cui, W.; Shi, B.; Wang, H. PKCalpha promotes insulin secretion via TRPC1 phosphorylation in INS-1E cells. Biosci. Biotechnol. Biochem. 2019, 83, 1676–1682. [Google Scholar] [CrossRef]
- Krout, D.; Schaar, A.; Sun, Y.; Sukumaran, P.; Roemmich, J.N.; Singh, B.B.; Claycombe-Larson, K.J. The TRPC1 Ca(2+)-permeable channel inhibits exercise-induced protection against high-fat diet-induced obesity and type II diabetes. J. Biol. Chem. 2017, 292, 20799–20807. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Jin, X.; Li, Q.; Wang, W.; Wang, Y.; Zhang, J. Association of TRPC1 gene polymorphisms with type 2 diabetes and diabetic nephropathy in Han Chinese population. Endocr. Res. 2013, 38, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chang, J.H.; Buckley, A.F.; Spurney, R.F. Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney Int. 2019, 95, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Lanner, J.T.; Bruton, J.D.; Assefaw-Redda, Y.; Andronache, Z.; Zhang, S.J.; Severa, D.; Zhang, Z.B.; Melzer, W.; Zhang, S.L.; Katz, A.; et al. Knockdown of TRPC3 with siRNA coupled to carbon nanotubes results in decreased insulin-mediated glucose uptake in adult skeletal muscle cells. FASEB J. 2009, 23, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Yoshida, M.; Ito, K.; Dezaki, K.; Yada, T.; Ishikawa, S.E.; Kakei, M. Potentiation of Glucose-stimulated Insulin Secretion by the GPR40-PLC-TRPC Pathway in Pancreatic beta-Cells. Sci. Rep. 2016, 6, 25912. [Google Scholar] [CrossRef] [PubMed]
- Spires, D.; Ilatovskaya, D.V.; Levchenko, V.; North, P.E.; Geurts, A.M.; Palygin, O.; Staruschenko, A. Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am. J. Physiol. Renal. Physiol. 2018, 315, F1091–F1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilatovskaya, D.V.; Blass, G.; Palygin, O.; Levchenko, V.; Pavlov, T.S.; Grzybowski, M.N.; Winsor, K.; Shuyskiy, L.S.; Geurts, A.M.; Cowley, A.W., Jr.; et al. A NOX4/TRPC6 Pathway in Podocyte Calcium Regulation and Renal Damage in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1917–1927. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, R.; Schlotterer, A.; Schumacher, D.; Matka, C.; Mathar, I.; Dietrich, N.; Medert, R.; Kriebs, U.; Lin, J.; Nawroth, P.; et al. TRPC proteins contribute to development of diabetic retinopathy and regulate glyoxalase 1 activity and methylglyoxal accumulation. Mol. Metab. 2018, 9, 156–167. [Google Scholar] [CrossRef]
- Marabita, F.; Islam, M.S. Expression of Transient Receptor Potential Channels in the Purified Human Pancreatic beta-Cells. Pancreas 2017, 46, 97–101. [Google Scholar] [CrossRef]
- Islam, M.S. Molecular Regulations and Functions of the Transient Receptor Potential Channels of the Islets of Langerhans and Insulinoma Cells. Cells 2020, 9, 685. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Liu, E.; Zhou, Q.; Li, S.; Wang, X.; Liu, Y.; Wang, L.; Sun, D.; Ye, J.; Gao, Y.; et al. TRPC1 Null Exacerbates Memory Deficit and Apoptosis Induced by Amyloid-beta. J. Alzheimers Dis. 2018, 63, 761–772. [Google Scholar] [CrossRef]
- Martinez-Galan, J.R.; Verdejo, A.; Caminos, E. TRPC1 Channels Are Expressed in Pyramidal Neurons and in a Subset of Somatostatin Interneurons in the Rat Neocortex. Front. Neuroanat. 2018, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yu, H.; Xu, B.; Xu, H.; Zhang, Z.; Ren, X.; Yuan, J.; Liu, J.; Guo, Y.; Spencer, P.S.; et al. TRPC1 Deletion Causes Striatal Neuronal Cell Apoptosis and Proteomic Alterations in Mice. Front. Aging Neurosci. 2018, 10, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Jia, Y. TRPC Channels and Programmed Cell Death. Adv. Exp. Med. Biol. 2017, 976, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ma, Q.; Deng, P.; Yang, J.; Yang, L.; Lin, M.; Yu, Z.; Zhou, Z. Critical role of TRPC1 in thyroid hormone-dependent dopaminergic neuron development. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1900–1912. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.B.; Webb, S.E.; Yue, J.; Moreau, M.; Leclerc, C.; Miller, A.L. TRPC3 is required for the survival, pluripotency and neural differentiation of mouse embryonic stem cells (mESCs). Sci. China Life Sci. 2018, 61, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Stroh, O.; Freichel, M.; Kretz, O.; Birnbaumer, L.; Hartmann, J.; Egger, V. NMDA receptor-dependent synaptic activation of TRPC channels in olfactory bulb granule cells. J. Neurosci. 2012, 32, 5737–5746. [Google Scholar] [CrossRef] [Green Version]
- Greka, A.; Navarro, B.; Oancea, E.; Duggan, A.; Clapham, D.E. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat. Neurosci. 2003, 6, 837–845. [Google Scholar] [CrossRef]
- Riccio, A.; Li, Y.; Moon, J.; Kim, K.S.; Smith, K.S.; Rudolph, U.; Gapon, S.; Yao, G.L.; Tsvetkov, E.; Rodig, S.J.; et al. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell 2009, 137, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Phelan, K.D.; Shwe, U.T.; Abramowitz, J.; Wu, H.; Rhee, S.W.; Howell, M.D.; Gottschall, P.E.; Freichel, M.; Flockerzi, V.; Birnbaumer, L.; et al. Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol. Pharmacol. 2013, 83, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Just, S.; Chenard, B.L.; Ceci, A.; Strassmaier, T.; Chong, J.A.; Blair, N.T.; Gallaschun, R.J.; Del Camino, D.; Cantin, S.; D’Amours, M.; et al. Treatment with HC-070, a potent inhibitor of TRPC4 and TRPC5, leads to anxiolytic and antidepressant effects in mice. PLoS ONE 2018, 13, e0191225. [Google Scholar] [CrossRef]
- Ko, A.R.; Kang, T.C. TRPC6-mediated ERK1/2 phosphorylation prevents dentate granule cell degeneration via inhibiting mitochondrial elongation. Neuropharmacology 2017, 121, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ryskamp, D.; Birnbaumer, L.; Bezprozvanny, I. Inhibition of TRPC1-Dependent Store-Operated Calcium Entry Improves Synaptic Stability and Motor Performance in a Mouse Model of Huntington’s Disease. J. Huntingtons Dis. 2018, 7, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Lepannetier, S.; Gualdani, R.; Tempesta, S.; Schakman, O.; Seghers, F.; Kreis, A.; Yerna, X.; Slimi, A.; de Clippele, M.; Tajeddine, N.; et al. Activation of TRPC1 Channel by Metabotropic Glutamate Receptor mGluR5 Modulates Synaptic Plasticity and Spatial Working Memory. Front. Cell Neurosci. 2018, 12, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerna, X.; Schakman, O.; Ratbi, I.; Kreis, A.; Lepannetier, S.; de Clippele, M.; Achouri, Y.; Tajeddine, N.; Tissir, F.; Gualdani, R.; et al. Role of the TRPC1 Channel in Hippocampal Long-Term Depression and in Spatial Memory Extinction. Int. J. Mol. Sci. 2020, 21, 1712. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, Y.; Oleinikov, K.; Schindeldecker, B.; Wyatt, A.; Weissgerber, P.; Flockerzi, V.; Boehm, U.; Freichel, M.; Bruns, D. TRPC channels regulate Ca2+-signaling and short-term plasticity of fast glutamatergic synapses. PLoS Biol. 2019, 17, e3000445. [Google Scholar] [CrossRef] [Green Version]
- Klipec, W.D.; Burrow, K.R.; O’Neill, C.; Cao, J.L.; Lawyer, C.R.; Ostertag, E.; Fowler, M.; Bachtell, R.K.; Illig, K.R.; Cooper, D.C. Loss of the trpc4 gene is associated with a reduction in cocaine self-administration and reduced spontaneous ventral tegmental area dopamine neuronal activity, without deficits in learning for natural rewards. Behav. Brain Res. 2016, 306, 117–127. [Google Scholar] [CrossRef]
- Egorov, A.V.; Schumacher, D.; Medert, R.; Birnbaumer, L.; Freichel, M.; Draguhn, A. TRPC channels are not required for graded persistent activity in entorhinal cortex neurons. Hippocampus 2019, 29, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lu, M.; He, X.; Ma, L.; Birnbaumer, L.; Liao, Y. TRPC3/6/7 Knockdown Protects the Brain from Cerebral Ischemia Injury via Astrocyte Apoptosis Inhibition and Effects on NF-small ka, CyrillicB Translocation. Mol. Neurobiol. 2017, 54, 7555–7566. [Google Scholar] [CrossRef]
- Guo, J.; Li, J.; Xia, L.; Wang, Y.; Zhu, J.; Du, J.; Lu, Y.; Liu, G.; Yao, X.; Shen, B. Transient Receptor Potential Canonical 5-Scramblase Signaling Complex Mediates Neuronal Phosphatidylserine Externalization and Apoptosis. Cells 2020, 9, 547. [Google Scholar] [CrossRef] [Green Version]
- Park, S.E.; Song, J.H.; Hong, C.; Kim, D.E.; Sul, J.W.; Kim, T.Y.; Seo, B.R.; So, I.; Kim, S.Y.; Bae, D.J.; et al. Contribution of Zinc-Dependent Delayed Calcium Influx via TRPC5 in Oxidative Neuronal Death and its Prevention by Novel TRPC Antagonist. Mol. Neurobiol. 2019, 56, 2822–2835. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Meng, H.; Liu, T.; Feng, Y.; Qi, Y.; Wang, H. TRPC1 Deficiency Exacerbates Cerebral Ischemia/Reperfusion-Induced Neurological Injury by Potentiating Nox4-Derived Reactive Oxygen Species Generation. Cell Physiol. Biochem. 2018, 51, 1723–1738. [Google Scholar] [CrossRef] [PubMed]
- Griesi-Oliveira, K.; Acab, A.; Gupta, A.R.; Sunaga, D.Y.; Chailangkarn, T.; Nicol, X.; Nunez, Y.; Walker, M.F.; Murdoch, J.D.; Sanders, S.J.; et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatr. 2015, 20, 1350–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.; Konnerth, A. TRPC3-dependent synaptic transmission in central mammalian neurons. J. Mol. Med. 2015, 93, 983–989. [Google Scholar] [CrossRef]
- Tian, J.; Zhu, M.X. GABAB Receptors Augment TRPC3-Mediated Slow Excitatory Postsynaptic Current to Regulate Cerebellar Purkinje Neuron Response to Type-1 Metabotropic Glutamate Receptor Activation. Cells 2018, 7, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, H.; John, T.T.; Chia, J.X.; Tariq, M.F.; Phillips, R.S.; Mosher, B.; Chen, Y.; Thompson, R.; Zhang, R.; Koshiya, N.; et al. Transient Receptor Potential Channels TRPM4 and TRPC3 Critically Contribute to Respiratory Motor Pattern Formation but not Rhythmogenesis in Rodent Brainstem Circuits. eNeuro 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Phelan, K.D.; Shwe, U.T.; Abramowitz, J.; Birnbaumer, L.; Zheng, F. Critical role of canonical transient receptor potential channel 7 in initiation of seizures. Proc. Natl. Acad. Sci. USA 2014, 111, 11533–11538. [Google Scholar] [CrossRef] [Green Version]
- Phelan, K.D.; Shwe, U.T.; Cozart, M.A.; Wu, H.; Mock, M.M.; Abramowitz, J.; Birnbaumer, L.; Zheng, F. TRPC3 channels play a critical role in the theta component of pilocarpine-induced status epilepticus in mice. Epilepsia 2017, 58, 247–254. [Google Scholar] [CrossRef]
- Phelan, K.D.; Mock, M.M.; Kretz, O.; Shwe, U.T.; Kozhemyakin, M.; Greenfield, L.J.; Dietrich, A.; Birnbaumer, L.; Freichel, M.; Flockerzi, V.; et al. Heteromeric canonical transient receptor potential 1 and 4 channels play a critical role in epileptiform burst firing and seizure-induced neurodegeneration. Mol. Pharmacol. 2012, 81, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.H.; Cui, A.F.; McDonald, M.F.; Grandl, J. Transduction of Repetitive Mechanical Stimuli by Piezo1 and Piezo2 Ion Channels. Cell Rep. 2017, 19, 2572–2585. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, Y.; Geng, J.; Zhou, S.; Xiao, B. Mechanically Activated Piezo Channels Mediate Touch and Suppress Acute Mechanical Pain Response in Mice. Cell Rep. 2019, 26, 1419–1431.e1414. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Beckel, J.M.; Daugherty, S.L.; Wang, T.; Woodcock, S.R.; Freeman, B.A.; de Groat, W.C. Activation of TRPC channels contributes to OA-NO2-induced responses in guinea-pig dorsal root ganglion neurons. J. Physiol. 2014, 592, 4297–4312. [Google Scholar] [CrossRef] [PubMed]
- Alkhani, H.; Ase, A.R.; Grant, R.; O’Donnell, D.; Groschner, K.; Seguela, P. Contribution of TRPC3 to store-operated calcium entry and inflammatory transductions in primary nociceptors. Mol. Pain 2014, 10, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alawi, K.M.; Russell, F.A.; Aubdool, A.A.; Srivastava, S.; Riffo-Vasquez, Y.; Baldissera, L., Jr.; Thakore, P.; Saleque, N.; Fernandes, E.S.; Walsh, D.A.; et al. Transient receptor potential canonical 5 (TRPC5) protects against pain and vascular inflammation in arthritis and joint inflammation. Ann. Rheum. Dis. 2017, 76, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewauw, I.; De Clercq, K.; Mulier, M.; Held, K.; Pinto, S.; Van Ranst, N.; Segal, A.; Voet, T.; Vennekens, R.; Zimmermann, K.; et al. A TRP channel trio mediates acute noxious heat sensing. Nature 2018, 555, 662–666. [Google Scholar] [CrossRef]
- Sexton, J.E.; Desmonds, T.; Quick, K.; Taylor, R.; Abramowitz, J.; Forge, A.; Kros, C.J.; Birnbaumer, L.; Wood, J.N. The contribution of TRPC1, TRPC3, TRPC5 and TRPC6 to touch and hearing. Neurosci. Lett. 2016, 610, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Dalrymple, A.; Slater, D.M.; Beech, D.; Poston, L.; Tribe, R.M. Molecular identification and localization of Trp homologues, putative calcium channels, in pregnant human uterus. Mol. Hum. Reprod. 2002, 8, 946–951. [Google Scholar] [CrossRef] [Green Version]
- Persoons, E.; Hennes, A.; De Clercq, K.; Van Bree, R.; Vriens, G.; O, D.F.; Peterse, D.; Vanhie, A.; Meuleman, C.; Voets, T.; et al. Functional Expression of TRP Ion Channels in Endometrial Stromal Cells of Endometriosis Patients. Int. J. Mol. Sci. 2018, 19, 2467. [Google Scholar] [CrossRef] [Green Version]
- Kawarabayashi, Y.; Hai, L.; Honda, A.; Horiuchi, S.; Tsujioka, H.; Ichikawa, J.; Inoue, R. Critical role of TRPC1-mediated Ca(2)(+) entry in decidualization of human endometrial stromal cells. Mol. Endocrinol. 2012, 26, 846–858. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, K.; Held, K.; Van Bree, R.; Meuleman, C.; Peeraer, K.; Tomassetti, C.; Voets, T.; D’Hooghe, T.; Vriens, J. Functional expression of transient receptor potential channels in human endometrial stromal cells during the luteal phase of the menstrual cycle. Hum. Reprod. 2015, 30, 1421–1436. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Nakade, U.P.; Choudhury, S.; Garg, S.K. Functional involvement of protein kinase C, Rho-kinase and TRPC3 decreases while PLC increases with advancement of pregnancy in mediating oxytocin-induced myometrial contractions in water buffaloes (Bubalus bubalis). Theriogenology 2017, 92, 176–189. [Google Scholar] [CrossRef]
- Jing, C.; Dongming, Z.; Hong, C.; Quan, N.; Sishi, L.; Caixia, L. TRPC3 Overexpression Promotes the Progression of Inflammation-Induced Preterm Labor and Inhibits T Cell Activation. Cell Physiol. Biochem. 2018, 45, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Hasna, J.; Abi Nahed, R.; Sergent, F.; Alfaidy, N.; Bouron, A. The Deletion of TRPC6 Channels Perturbs Iron and Zinc Homeostasis and Pregnancy Outcome in Mice. Cell Physiol. Biochem. 2019, 52, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Cobos, J.C.; Trebak, M. TRPC channels in smooth muscle cells. Front. Biosci. 2010, 15, 1023–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, L.; Rhee, P.L.; Lowe, V.; Zheng, H.; Peri, L.; Ro, S.; Sanders, K.M.; Koh, S.D. Basally activated nonselective cation currents regulate the resting membrane potential in human and monkey colonic smooth muscle. Am. J. Physiol. Gastrointest Liver Physiol. 2011, 301, G287–G296. [Google Scholar] [CrossRef]
- Tsvilovskyy, V.V.; Zholos, A.V.; Aberle, T.; Philipp, S.E.; Dietrich, A.; Zhu, M.X.; Birnbaumer, L.; Freichel, M.; Flockerzi, V. Deletion of TRPC4 and TRPC6 in mice impairs smooth muscle contraction and intestinal motility in vivo. Gastroenterology 2009, 137, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Dryn, D.O.; Melnyk, M.I.; Al Kury, L.T.; Prylutskyy, Y.I.; Ritter, U.; Zholos, A.V. C60 fullerenes disrupt cellular signalling leading to TRPC4 and TRPC6 channels opening by the activation of muscarinic receptors and G-proteins in small intestinal smooth muscles. Cell Signal. 2018, 43, 40–46. [Google Scholar] [CrossRef]
- Dryn, D.; Luo, J.; Melnyk, M.; Zholos, A.; Hu, H. Inhalation anaesthetic isoflurane inhibits the muscarinic cation current and carbachol-induced gastrointestinal smooth muscle contractions. Eur. J. Pharmacol. 2018, 820, 39–44. [Google Scholar] [CrossRef]
- Schlondorff, J. TRPC6 and kidney disease: Sclerosing more than just glomeruli? Kidney Int. 2017, 91, 773–775. [Google Scholar] [CrossRef]
- Heeringa, S.F.; Moller, C.C.; Du, J.; Yue, L.; Hinkes, B.; Chernin, G.; Vlangos, C.N.; Hoyer, P.F.; Reiser, J.; Hildebrandt, F. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE 2009, 4, e7771. [Google Scholar] [CrossRef]
- Mukerji, N.; Damodaran, T.V.; Winn, M.P. TRPC6 and FSGS: The latest TRP channelopathy. Biochim. Biophys. Acta 2007, 1772, 859–868. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Dryer, S.E. A mutation in TRPC6 channels abolishes their activation by hypoosmotic stretch but does not affect activation by diacylglycerol or G protein signaling cascades. Am. J. Physiol. Renal. Physiol. 2014, 306, F1018–F1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheissari, A.; Meamar, R.; Kheirollahi, M.; Rouigari, M.; Dehbashi, M.; Dehghani, L.; Abedini, A. TRPC6 Mutational Analysis in Iranian Children With Focal Segmental Glomerulosclerosis. Iran. J. Kidney Dis. 2018, 12, 341–349. [Google Scholar] [PubMed]
- Riehle, M.; Buscher, A.K.; Gohlke, B.O.; Kassmann, M.; Kolatsi-Joannou, M.; Brasen, J.H.; Nagel, M.; Becker, J.U.; Winyard, P.; Hoyer, P.F.; et al. TRPC6 G757D Loss-of-Function Mutation Associates with FSGS. J. Am. Soc. Nephrol. 2016, 27, 2771–2783. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.; Wang, L.; Spurney, R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells 2019, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Castonguay, P.; Sidhom, E.H.; Clark, A.R.; Dvela-Levitt, M.; Kim, S.; Sieber, J.; Wieder, N.; Jung, J.Y.; Andreeva, S.; et al. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 2017, 358, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.C.; Birnbaumer, L.; Heesom, K.J.; Xu, S.Z.; Saleem, M.A.; et al. TRPC6 Binds to and Activates Calpain, Independent of Its Channel Activity, and Regulates Podocyte Cytoskeleton, Cell Adhesion, and Motility. J. Am. Soc. Nephrol. 2019, 30, 1910–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheijden, K.A.T.; Sonneveld, R.; Bakker-van Bebber, M.; Wetzels, J.F.M.; van der Vlag, J.; Nijenhuis, T. The Calcium-Dependent Protease Calpain-1 Links TRPC6 Activity to Podocyte Injury. J. Am. Soc. Nephrol. 2018, 29, 2099–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlondorff, J.; Del Camino, D.; Carrasquillo, R.; Lacey, V.; Pollak, M.R. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am. J. Physiol. Cell Physiol. 2009, 296, C558–C569. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, D.; Shibata, S.; Ji, T.; Zhang, L.; Zhang, R.; Yang, H.; Ma, L.; Jiao, J. Group I metabotropic glutamate receptor activation induces TRPC6-dependent calcium influx and RhoA activation in cultured human kidney podocytes. Biochem. Biophys. Res. Commun. 2019, 511, 374–380. [Google Scholar] [CrossRef]
- Sun, X.; Chu, Y.; Zhang, C.; Du, X.; He, F.; Chen, S.; Gao, P.; Liu, J.; Zhu, Z.; Meng, X. Effect of TRPC6 knockdown on puromycin aminonucleoside-induced podocyte injury. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 340–345. [Google Scholar] [CrossRef]
- Kong, W.; Haschler, T.N.; Nurnberg, B.; Kramer, S.; Gollasch, M.; Marko, L. Renal Fibrosis, Immune Cell Infiltration and Changes of TRPC Channel Expression after Unilateral Ureteral Obstruction in Trpc6−/− Mice. Cell Physiol. Biochem. 2019, 52, 1484–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Xiao, H.; Zhang, Y.; Zeng, X.; Huang, M.; Chen, X.; Birnbaumer, L.; Liao, Y. Transient receptor potential channel 6 knockdown prevents apoptosis of renal tubular epithelial cells upon oxidative stress via autophagy activation. Cell Death Dis. 2018, 9, 1015. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Y.; Sun, M.R.; Wu, C.L.; Li, Y.; Du, J.J.; Zeng, J.Y.; Bi, H.L.; Sun, Y.H. Activation of calcium-sensing receptor increases TRPC3/6 expression in T lymphocyte in sepsis. Mol. Immunol. 2015, 64, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Riazanski, V.; Gabdoulkhakova, A.G.; Boynton, L.S.; Eguchi, R.R.; Deriy, L.V.; Hogarth, D.K.; Loaec, N.; Oumata, N.; Galons, H.; Brown, M.E.; et al. TRPC6 channel translocation into phagosomal membrane augments phagosomal function. Proc. Natl. Acad. Sci. USA 2015, 112, E6486–E6495. [Google Scholar] [CrossRef] [Green Version]
- Pereira, D.M.S.; Mendes, S.J.F.; Alawi, K.; Thakore, P.; Aubdool, A.; Sousa, N.C.F.; da Silva, J.F.R.; Castro, J.A., Jr.; IC, P.P.; Silva, L.C.N.; et al. Transient Receptor Potential Canonical Channels 4 and 5 Mediate Escherichia coli-Derived Thioredoxin Effects in Lipopolysaccharide-Injected Mice. Oxid Med. Cell Longev. 2018, 2018, 4904696. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.; Varga-Szabo, D.; Kleinschnitz, C.; Pleines, I.; Bender, M.; Austinat, M.; Bosl, M.; Stoll, G.; Nieswandt, B. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood 2009, 113, 2056–2063. [Google Scholar] [CrossRef]
- Pulcinelli, F.M.; Trifiro, E.; Massimi, I.; Di Renzo, L. A functional interaction between TRPC/NCKX induced by DAG plays a role in determining calcium influx independently from PKC activation. Platelets 2013, 24, 554–559. [Google Scholar] [CrossRef]
- Mahaut-Smith, M.P. A role for platelet TRPC channels in the Ca2+ response that induces procoagulant activity. Sci. Signal. 2013, 6, pe23. [Google Scholar] [CrossRef]
- Albarran, L.; Berna-Erro, A.; Dionisio, N.; Redondo, P.C.; Lopez, E.; Lopez, J.J.; Salido, G.M.; Brull Sabate, J.M.; Rosado, J.A. TRPC6 participates in the regulation of cytosolic basal calcium concentration in murine resting platelets. Biochim. Biophys. Acta 2014, 1843, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yao, T.; Deng, Z.; Sohn, J.W.; Sun, J.; Huang, Y.; Kong, X.; Yu, K.J.; Wang, R.T.; Chen, H.; et al. TrpC5 Mediates Acute Leptin and Serotonin Effects via Pomc Neurons. Cell Rep. 2017, 18, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Rode, B.; Yuldasheva, N.Y.; Baxter, P.D.; Sedo, A.; Ainscough, J.F.; Shires, M.; Kearney, M.T.; Bailey, M.A.; Wheatcroft, S.B.; Beech, D.J. TRPC5 ion channel permeation promotes weight gain in hypercholesterolaemic mice. Sci. Rep. 2019, 9, 773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfrum, C.; Kiehlmann, E.; Pelczar, P. TRPC1 regulates brown adipose tissue activity in a PPARgamma-dependent manner. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E825–E832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alawi, K.M.; Tandio, D.; Xu, J.; Thakore, P.; Papacleovoulou, G.; Fernandes, E.S.; Legido-Quigley, C.; Williamson, C.; Brain, S.D. Transient receptor potential canonical 5 channels plays an essential role in hepatic dyslipidemia associated with cholestasis. Sci. Rep. 2017, 7, 2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sel, S.; Rost, B.R.; Yildirim, A.O.; Sel, B.; Kalwa, H.; Fehrenbach, H.; Renz, H.; Gudermann, T.; Dietrich, A. Loss of classical transient receptor potential 6 channel reduces allergic airway response. Clin. Exp. Allergy 2008, 38, 1548–1558. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Z.; Ma, L.; Guo, Y.; Li, X.; Zhao, L.; Tian, C.; Tang, X.; Cheng, D.; Chen, Z.; et al. The effects of transient receptor potential channel (TRPC) on airway smooth muscle cell isolated from asthma model mice. J. Cell Biochem. 2018, 119, 6033–6044. [Google Scholar] [CrossRef]
- Pu, Q.; Zhao, Y.; Sun, Y.; Huang, T.; Lin, P.; Zhou, C.; Qin, S.; Singh, B.B.; Wu, M. TRPC1 intensifies house dust mite-induced airway remodeling by facilitating epithelial-to-mesenchymal transition and STAT3/NF-kappaB signaling. FASEB J. 2019, 33, 1074–1085. [Google Scholar] [CrossRef]
- Kepura, F.; Braun, E.; Dietrich, A.; Plant, T.D. TRPC1 Regulates the Activity of a Voltage-Dependent Nonselective Cation Current in Hippocampal CA1 Neurons. Cells 2020, 9, 459. [Google Scholar] [CrossRef] [Green Version]
- Oda, S.; Numaga-Tomita, T.; Kitajima, N.; Toyama, T.; Harada, E.; Shimauchi, T.; Nishimura, A.; Ishikawa, T.; Kumagai, Y.; Birnbaumer, L.; et al. TRPC6 counteracts TRPC3-Nox2 protein complex leading to attenuation of hyperglycemia-induced heart failure in mice. Sci. Rep. 2017, 7, 7511. [Google Scholar] [CrossRef] [Green Version]
- Nunez, L.; Bird, G.S.; Hernando-Perez, E.; Perez-Riesgo, E.; Putney, J.W., Jr.; Villalobos, C. Store-operated Ca(2+) entry and Ca(2+) responses to hypothalamic releasing hormones in anterior pituitary cells from Orai1−/− and heptaTRPC knockout mice. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1124–1136. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef]
- Rubaiy, H.N. Treasure troves of pharmacological tools to study transient receptor potential canonical 1/4/5 channels. Br. J. Pharmacol. 2019, 176, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Minard, A.; Bauer, C.C.; Wright, D.J.; Rubaiy, H.N.; Muraki, K.; Beech, D.J.; Bon, R.S. Remarkable Progress with Small-Molecule Modulation of TRPC1/4/5 Channels: Implications for Understanding the Channels in Health and Disease. Cells 2018, 7, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bon, R.S.; Beech, D.J. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels -- mirage or pot of gold? Br. J. Pharmacol. 2013, 170, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Kiyonaka, S.; Kato, K.; Nishida, M.; Mio, K.; Numaga, T.; Sawaguchi, Y.; Yoshida, T.; Wakamori, M.; Mori, E.; Numata, T.; et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. USA 2009, 106, 5400–5405. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.R.; Shi, J.; Wu, M.; Engers, J.; Hopkins, C.R.; Lindsley, C.W.; Salovich, J.M.; Zhu, Y.; Tian, J.B.; Zhu, M.X.; et al. Novel Chemical Inhibitor of TRPC4 Channels. In Probe Reports from the NIH Molecular Libraries Program; Bethesda: Rockvill, MD, USA, 2010. [Google Scholar]
- Akbulut, Y.; Gaunt, H.J.; Muraki, K.; Ludlow, M.J.; Amer, M.S.; Bruns, A.; Vasudev, N.S.; Radtke, L.; Willot, M.; Hahn, S.; et al. (-)-Englerin A is a potent and selective activator of TRPC4 and TRPC5 calcium channels. Angew. Chem. Int. Ed. Engl. 2015, 54, 3787–3791. [Google Scholar] [CrossRef]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Potentiation and inhibition of Ca(2+) release-activated Ca(2+) channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3) receptors. J. Physiol. 2001, 536, 3–19. [Google Scholar] [CrossRef]
- Jairaman, A.; Prakriya, M. Molecular pharmacology of store-operated CRAC channels. Channels (Austin) 2013, 7, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.Z.; Gu, Q.; Wang, C.; Colton, C.K.; Tang, J.; Kinoshita-Kawada, M.; Lee, L.Y.; Wood, J.D.; Zhu, M.X. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 2004, 279, 35741–35748. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Z.; Zeng, F.; Boulay, G.; Grimm, C.; Harteneck, C.; Beech, D.J. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: A differential, extracellular and voltage-dependent effect. Br. J. Pharmacol. 2005, 145, 405–414. [Google Scholar] [CrossRef]
- Merritt, J.E.; Armstrong, W.P.; Benham, C.D.; Hallam, T.J.; Jacob, R.; Jaxa-Chamiec, A.; Leigh, B.K.; McCarthy, S.A.; Moores, K.E.; Rink, T.J. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 1990, 271, 515–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Hildebrand, M.E.; Garcia, E.; Snutch, T.P. The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharmacol. 2010, 160, 1464–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Yu, X.; Chen, H.; Horne, D.; White, R.; Wu, X.; Lee, P.; Gu, Y.; Ghimire-Rijal, S.; Lin, D.C.; et al. Structural basis for pharmacological modulation of the TRPC6 channel. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz, G.G.; Svobodova, B.; Lichtenegger, M.; Tiapko, O.; Groschner, K.; Glasnov, T. Intensified Microwave-Assisted N-Acylation Procedure—Synthesis and Activity Evaluation of TRPC3 Channel Agonists with a 1,3-Dihydro-2H-benzo[d]imidazol-2-one Core. Synlett 2017, 28, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Ding, M.; Zhu, Y.; Lu, Y.; Du, J.; Miller, M.; Tian, J.; Zhu, J.; Xu, J.; Wen, M.; et al. Pyrazolopyrimidines as Potent Stimulators for Transient Receptor Potential Canonical 3/6/7 Channels. J. Med. Chem. 2017, 60, 4680–4692. [Google Scholar] [CrossRef]
- Tiapko, O.; Shrestha, N.; Lindinger, S.; Guedes de la Cruz, G.; Graziani, A.; Klec, C.; Butorac, C.; Graier, W.F.; Kubista, H.; Freichel, M.; et al. Lipid-independent control of endothelial and neuronal TRPC3 channels by light. Chem. Sci. 2019, 10, 2837–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinders-Zufall, T.; Storch, U.; Bleymehl, K.; Mederos, Y.S.M.; Frank, J.A.; Konrad, D.B.; Trauner, D.; Gudermann, T.; Zufall, F. PhoDAGs Enable Optical Control of Diacylglycerol-Sensitive Transient Receptor Potential Channels. Cell Chem. Biol. 2018, 25, 215–223.e3. [Google Scholar] [CrossRef] [Green Version]
- Urban, N.; Schaefer, M. Direct Activation of TRPC3 Channels by the Antimalarial Agent Artemisinin. Cells 2020, 9, 202. [Google Scholar] [CrossRef] [Green Version]
- Motoyama, K.; Nagata, T.; Kobayashi, J.; Nakamura, A.; Miyoshi, N.; Kazui, M.; Sakurai, K.; Sakakura, T. Discovery of a bicyclo[4.3.0]nonane derivative DS88790512 as a potent, selective, and orally bioavailable blocker of transient receptor potential canonical 6 (TRPC6). Bioorg. Med. Chem. Lett. 2018, 28, 2222–2227. [Google Scholar] [CrossRef]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef] [Green Version]
- Rubaiy, H.N.; Ludlow, M.J.; Bon, R.S.; Beech, D.J. Pico145—Powerful new tool for TRPC1/4/5 channels. Channels (Austin) 2017, 11, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Naylor, J.; Minard, A.; Gaunt, H.J.; Amer, M.S.; Wilson, L.A.; Migliore, M.; Cheung, S.Y.; Rubaiy, H.N.; Blythe, N.M.; Musialowski, K.E.; et al. Natural and synthetic flavonoid modulation of TRPC5 channels. Br. J. Pharmacol. 2016, 173, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Follmann, M.; Hessler, G.; Kleemann, H.W.; Hachtel, S.; Fuchs, B.; Weissmann, N.; Linz, W.; Schmidt, T.; Lohn, M.; et al. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br. J. Pharmacol. 2015, 172, 3650–3660. [Google Scholar] [CrossRef] [PubMed]
- Hafner, S.; Burg, F.; Kannler, M.; Urban, N.; Mayer, P.; Dietrich, A.; Trauner, D.; Broichhagen, J.; Schaefer, M. A (+)-Larixol Congener with High Affinity and Subtype Selectivity toward TRPC6. ChemMedChem 2018, 13, 1028–1035. [Google Scholar] [CrossRef]

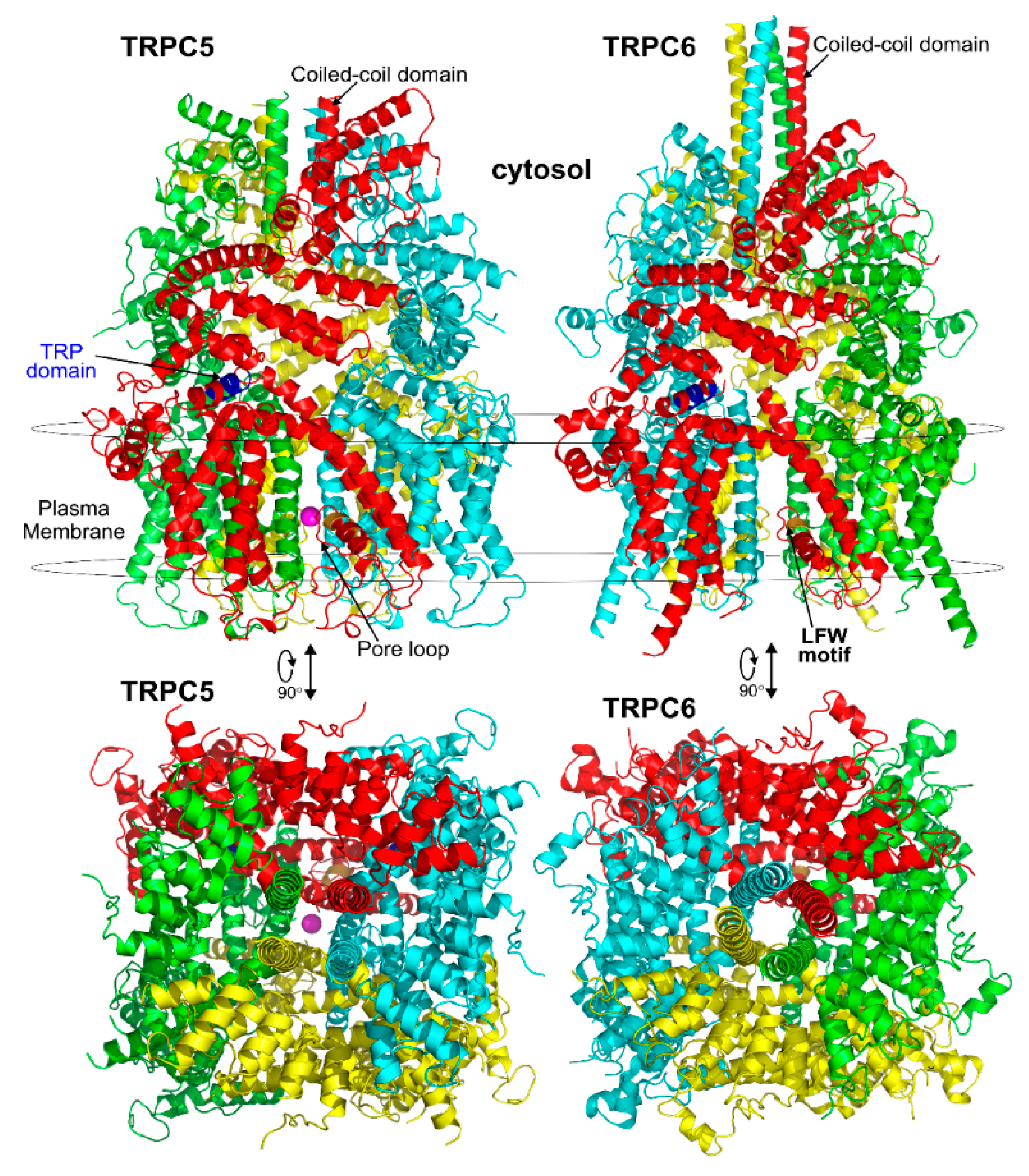
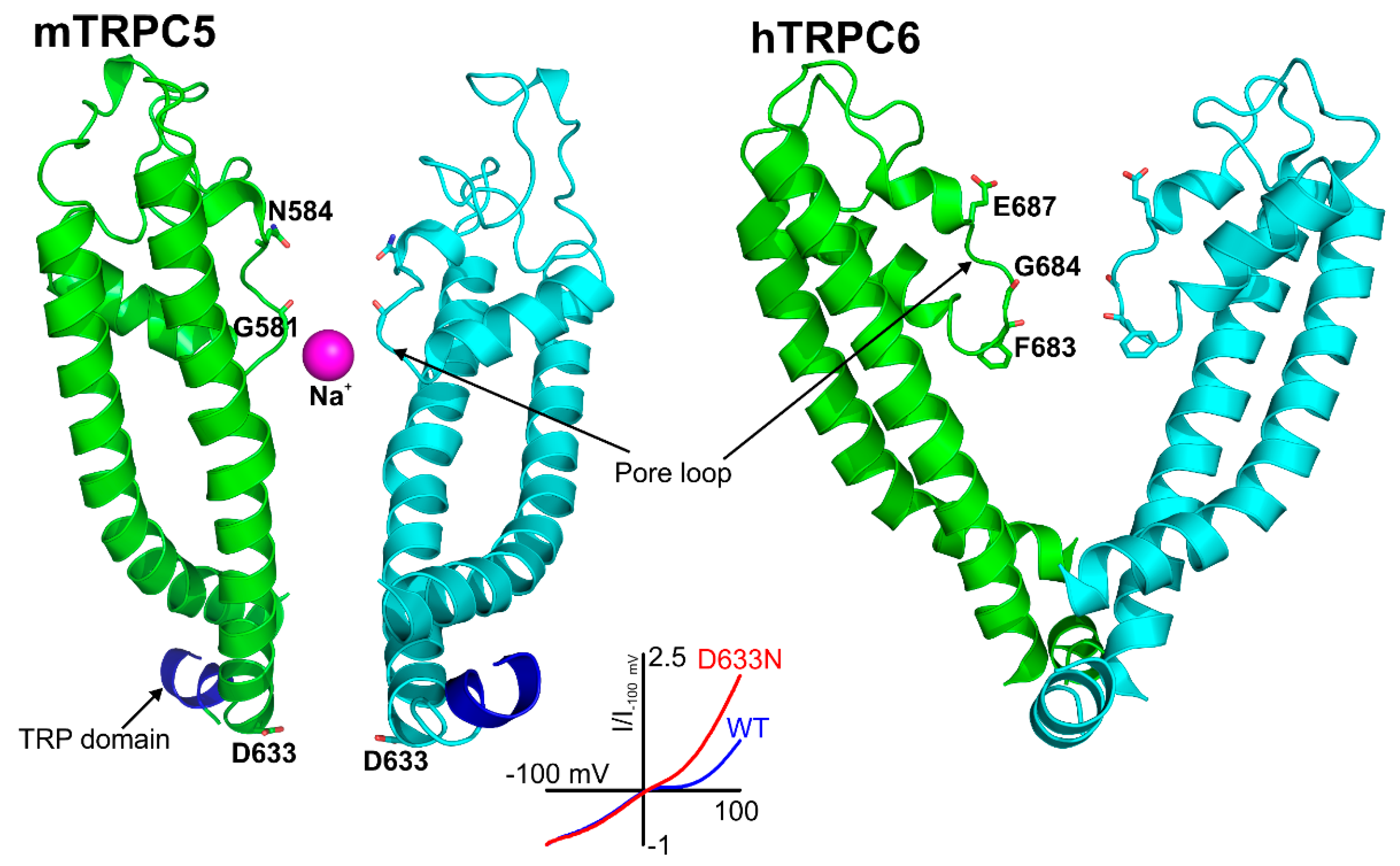
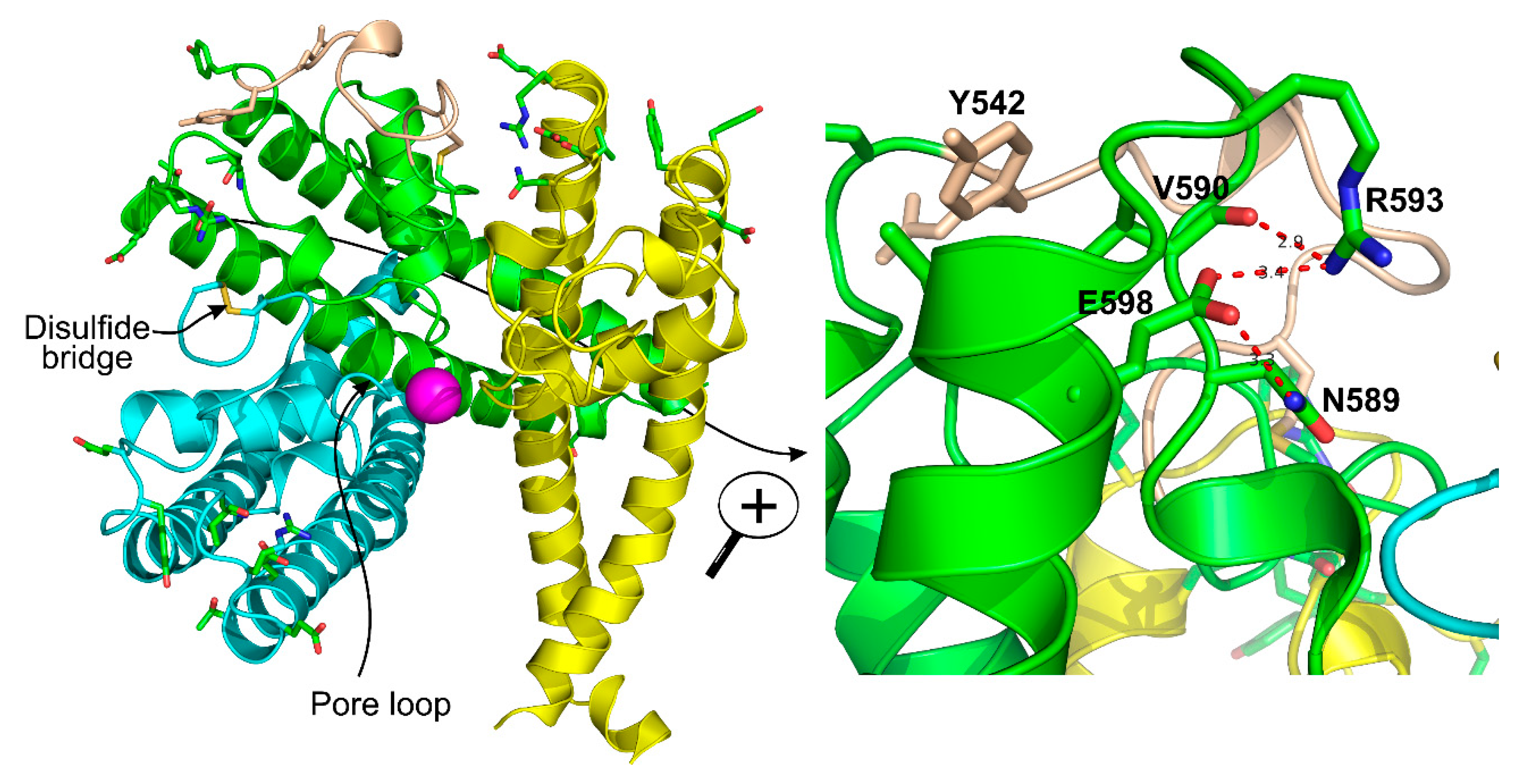
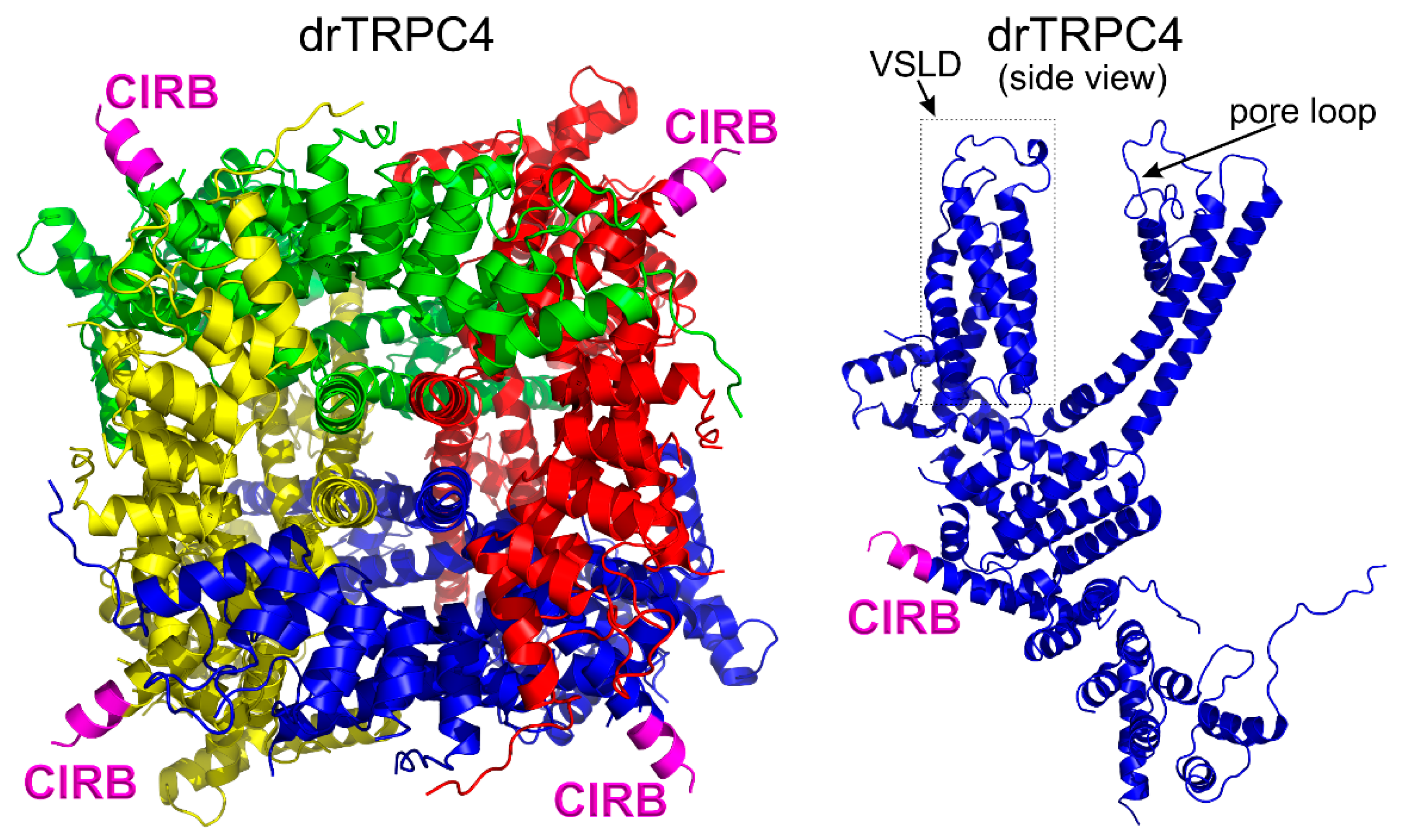
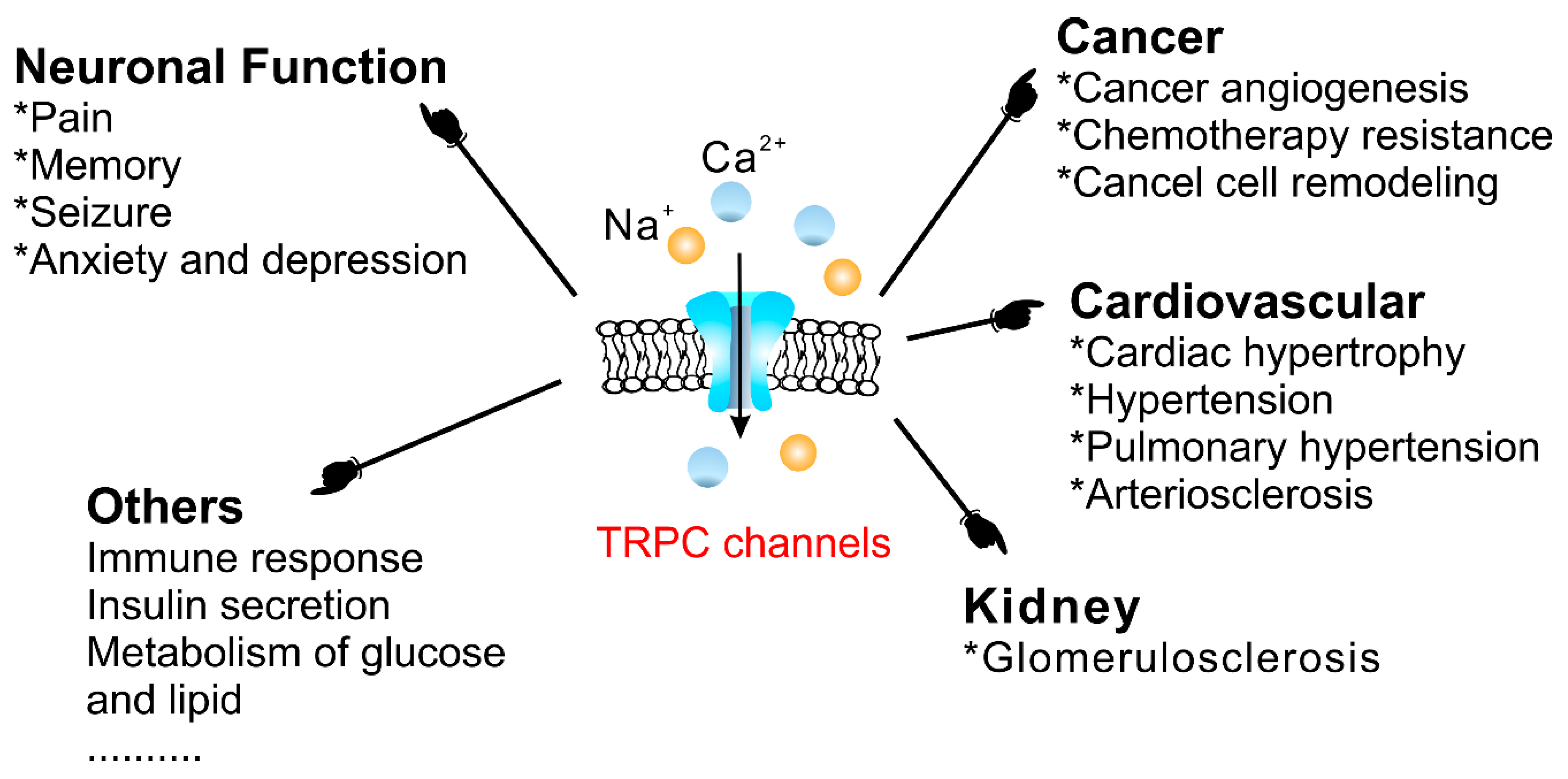
| Type | Effects | Authors | Reference |
|---|---|---|---|
| TRPC1 KO | TRPC1 KO mice exhibited increased mGluR agonist-induced non-selective inward currents in the CA1 neurons of the hippocampus | Kepura et al., 2020 | [287] |
| TRPC1 KO mice did not develop pulmonary hypertension under chronic hypoxia and had reduced vascular remodeling; however, right ventricular hypertrophy was similar to that in WT mice | Malczyk et al., 2013 | [163] | |
| TRPC1 KO mice showed impairment in spatial working memory and fear memory formation but innate fear behavior was unaffected; activation of mGluR increased inward currents in hippocampal neurons from WT but not from TRPC KO mice | Lepannetier et al., 2018 | [223] | |
| TRPC1 KO mice fed a high fat diet exhibited reduced fat mass, fasting glucose concentrations, and autophagy markers, whereas apoptosis markers were increased | Krout et al., 2017 | [200] | |
| Tamoxifen-inducible TRPC1 genetic ablation in the hippocampus impaired mGluR-induced ERK1/2 activation, diminished mGluR-LTD, and decreased memory extinction | Yerna et al., 2020 | [224] | |
| TRPC1 KO mice exhibited increased cerebral ischemia/reperfusion-induced infarction, neurological severity score, memory impairment, and oxidative stress. | Xu et al., 2018 | [231] | |
| TRPC1 KO mice exhibited a reduced airway remodeling following house dust mite challenge with decreases in mucus production, cytokine secretion, and collagen deposition. | Pu et al., 2019 | [286] | |
| TRPC1 KO mice exhibited no deficit of learning and memory under physiological conditions, but exacerbated learning and memory deficits induced by amyloid-β (Aβ), associated with Alzheimer’s disease | Li et al., 2018 | [210] | |
| TRPC1 KO mice exhibited a reduced endothelial progenitor cell function secondary to calmodulin/endothelial nitric oxide synthase downregulation | Du et al., 2018 | [167] | |
| Improved motor performance and an increased density of striatal medium spiny neuron spines in a mouse model of Huntington disease | Wu et al., 2018 | [222] | |
| TRPC3 KO | TRPC3 KO mice exhibited ameliorated hypertension through reduction of angiotensin II-induced mitochondrial ROS generation | Wang et al., 2017 | [159] |
| TRPC3 KO impaired the pluripotency of undifferentiated mouse embryonic stem cells and repressed their neural differentiation | Hao et al., 2018 | [215] | |
| TRPC3 KO mice exhibited reduced pilocarpine-induced seizures and theta activity involved in status epilepticus | Phelan et al., 2017 | [237] | |
| Phenylephrine-induced vasoconstriction and endothelium-dependent acetylcholine-induced vasodilation were reduced in TRPC3 KO mouse mesenteric arteries, whereas KCl- or pressure-induced vasoconstriction was unaltered. | Yeon et al., 2014 | [170] | |
| Increase of mGluR1-induced slow EPSC by GABABR activation was absent in TRPC3 KO mouse cerebellar Purkinje neurons but unaltered in TRPC1, TRPC4 and TRPC1/4/5/6 KO Purkinje neurons | Tian & Zhu, 2018 | [234] | |
| TRPC3 KO mice exhibited a delayed occurrence of inflammation-induced preterm labor | Jing et al., 2018 | [251] | |
| TRPC3 KD | TRPC3 KD resulted in a decreased insulin-mediated glucose uptake in adult skeletal muscle cells | Lanner et al., 2009 | [203] |
| TRPC3 KD resulted in a reduced angiotensin II-induced Ca2+ influx in VSMC from spontaneously hypertensive rats | Liu et al., 2009 | [157] | |
| TRPC4 KO | TRPC4 KO mice exhibited a reduced self-administration of cocaine, but normal learning for natural reward | Klipec et al., 2016 | [226] |
| TRPC4 KO mice lack store-operated Ca2+ currents and agonist-dependent vasorelaxation | Freichel et al., 2001 | [165] | |
| TRPC5 KO | Compared with WT mice, TRPC5 KO mice had a reduced phosphatidylserine externalization and less apoptosis of cerebral neurons following ischemia-reperfusion | Guo et al., 2020 | [229] |
| Cortical neurons from TRPC5 KO mice were resistant to H2O2 toxicity as compared to WT neurons | Park et al., 2019 | [230] | |
| Acetylchoine-induced endothelium-dependent contractions were smaller in TRPC5 KO carotid arteries compared to WT mice; TRPC5 contributed by stimulating COX-2-mediatedprostanoid production from carotid artery endothelial cells | Liang et al., 2019 | [185] | |
| TRPC5 KO mice fed supplemental cholic acid had less liver enlargement than WT; hepatic bile acid was lower in TRPC5 KO mice | Alawi et al., 2017 | [283] | |
| TRPC5 KO mice demonstrated reduced nociceptive thresholds (thermal and mechanical) in a complete Freund’s adjuvant-induced unilateral arthritis model | Alawi et al., 2017 | [243] | |
| TRPC5 KO did not alter body temperature but was associated with increased accumulation of peritoneal leukocytes secreting inflammatory mediator in thioredoxin-treated LPS- injected mice. | Pereira et al., 2018 | [275] | |
| Isoliquiritigenin from Glycyrrhiza glabra (Licorice) inhibits atherosclerosis by blocking TRPC5 channel expression and lipid serum levels in an apolipoprotein E (ApoE) knockout mouse model and angiotensin II-stimulated vascular smooth muscle cells (VSMCs) | Qi et al., 2020 | [184] | |
| TRPC5 KO mice exhibited diminished innate fear levels in response to aversive stimuli and reduced response to metabotropic glutamate receptor activation in amygdala | Riccio et al., 2009 | [218] | |
| TRPC5 KD | Knockdown of TRPC5 increased chemosensitivity to temozolomide in glioblastoma | Zou et al., 2019 | [190] |
| TRPC6 KO | Compared to Akita mice, TRPC6 KO Akita mice exhibited increased insulin resistance, mesangial expansion, and glomerular injury but reduced albuminuria and tubular injury | Wang et al., 2019 | [202] |
| In the unilateral ureter obstruction model, TRPC6 knockout was linked to decreased expression of pro-fibrotic, with pro-inflammatory genes being unaffected | Kong et al., 2019 | [271] | |
| Reduces allergic airway response | Sel et al., 2008 | [284] | |
| Induction of hypoventilation resulted in severe arterial hypoxemia not found in wild type | Weissmann et al., 2006 | [161] | |
| Displayed reduced litter sizes and structural changes of the placenta | Hasna et al., 2019 | [252] | |
| Protects mice from mTBI-induced aortic endothelial dysfunction; | Chen et al., 2019 | [176] | |
| TRPC6 knockout has reno-protective effects in diabetic kidney disease, protecting the podocytes but not the glomerulus as a whole | Spires et al., 2018 | [205] | |
| In TRPC6 KO mice, pancreatic stellate cells (PSCs) exhibited decrease migration as compared to WT PSCs under hypoxic conditions | Nielsen et al., 2017 | [198] | |
| TRPC6 KO mice exhibited an elevated blood pressure and enhanced agonist-induced aortic contractility due to upregulation of constitutively active TRPC3-type channels | Dietrich et al., 2005 | [145] | |
| TRPC6 KD | Attenuated Ca2+ influx and the stress fiber formation induced by DHPG | Wang et al., 2019 | [269] |
| Prevented podocyte permeability increase induced by puromucin aminonucleaside | Sun et al., 2012 | [270] | |
| Increased autophagic flux and mitigated oxidative stress-induced apoptosis of proximal tubular cells | Hou et al., 2018 | [272] | |
| Decreased hypoxic increases of Ca2+ influx in PASMCs | Malczyk et al., 2017 | [162] | |
| Increased TGF-β1-induced Akt phosphorylation at Ser473 in VSMCs | Numaga-Tomita et al., 2019 | [154] | |
| Mitochondrial elongation, reduced phosphorylation of dynamin-related protein 1, and extracellular-signal-regulated kinase 1/2 | Ko et al., 2017 | [221] | |
| Unaltered ROS production, but increased inflammatory cytokines production compared to WT mice | Oda et al., 2017 | [288] | |
| TRPC6 KD | Increased density of striatal medium spiny neuron spines in a mouse model of Huntington disease | Wu et al., 2018 | [222] |
| TRPC1 or TRPC3 KD | Asthmatic mouse airway smooth muscle cells exhibited upregulated TRPC1 and TRPC3 and increased proliferation. Knockdown either of the channels reduced proliferation. | Zhang et al., 2018 | [285] |
| TRPC6 KO and TRPC5/6 DKO | TRPC6 KO mice and TRPC5/6 double KO mice were protected from H2O2-induced damage in a model of diabetic kidney disease | Ilatovskaya et al., 2018 | [206] |
| TRPC1/4 KO TRPC5 KO TRPC1 KO | LTP and pilocarpine-induced seizures are reduced in TRPC5 KO mice, but are unaltered in TRPC1/4 KO mice; mGluR-mediated epileptiform bursting in the hippocampal CA1 area is not affected in TRPC5 KO mice, but is abolished in TRPC1 KO and TRPC1/4 DKO mice; seizure-induced neural cell death in the hippocampus was reduced in both TRPC5 KO and TRPC1/4 DKO mice | Phelan et al., 2013 | [219] |
| TRPC1/4/5 KO | TRPC1/4/5 KO mice exhibited deficits in spatial working memory and relearning task, while spatial reference memory was not affected | Bröker-Lai et al., 2017 | [126] |
| TRPC1/4/5 KO mice exhibited decreased basal-evoked secretion, reduces readily releasable vesicles pool size, and accelerated synaptic depression during high-frequency stimulation, whereas TRPC5 KI showed short-term enhancement of synaptic strength during high frequency stimulation | Schwarz et al., 2019 | [225] | |
| TRPC3/6/7 KO | Compared to WT mice, triple TRPC3/6/7 KO mice had smaller infarct size when subjected to cerebral ischemia-reperfusion | Chen et al., 2017 | [228] |
| TRPC1/4/5/6 KO | Tetra-TRPC KO mice are protected from hyperglycemia-evoked vasoregression. In addition, the KO mice are resistant to the STZ-induced reduction in retinal layer thickness | Sachdeva et al., 2018 | [207] |
| TRPC1/4/5 or hepta-TRPC-KO | No change in persistent activity in entorhinal cortex neurons of layer V | Egorov et al., 2019 | [227] |
| Hepta- TRPC KO | Thapsigargin-induced store-operated Ca2+ entry was absent in Orai1 KO anterior pituitary cells, but was unaffected in hepta TRPC KO; conversely, spontaneous intracellular Ca2+ oscillations related to electrical activity was reduced in hepta TRPC1-7 KO mouse cells and unaffected in Orai1 KO mouse cells | Núñez et al., 2019 | [289] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. https://doi.org/10.3390/cells9091983
Chen X, Sooch G, Demaree IS, White FA, Obukhov AG. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells. 2020; 9(9):1983. https://doi.org/10.3390/cells9091983
Chicago/Turabian StyleChen, Xingjuan, Gagandeep Sooch, Isaac S. Demaree, Fletcher A. White, and Alexander G. Obukhov. 2020. "Transient Receptor Potential Canonical (TRPC) Channels: Then and Now" Cells 9, no. 9: 1983. https://doi.org/10.3390/cells9091983
APA StyleChen, X., Sooch, G., Demaree, I. S., White, F. A., & Obukhov, A. G. (2020). Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells, 9(9), 1983. https://doi.org/10.3390/cells9091983