TRPCs: Influential Mediators in Skeletal Muscle

{kind=link}
Abstract
1. Introduction
2. Store-Operated Ca2+ Entry (SOCE) as a Mechanism of Extracellular Ca2+ Entry into Skeletal Muscle
3. Orais and STIMs in Skeletal SOCE
4. General Aspects of TRPCs
5. TRPCs as SOCE Channels in Skeletal Muscle
5.1. TRPC1 and TRPC4 in Skeletal Muscle
5.2. TRPC3 and TRPC6 in Skeletal Muscle
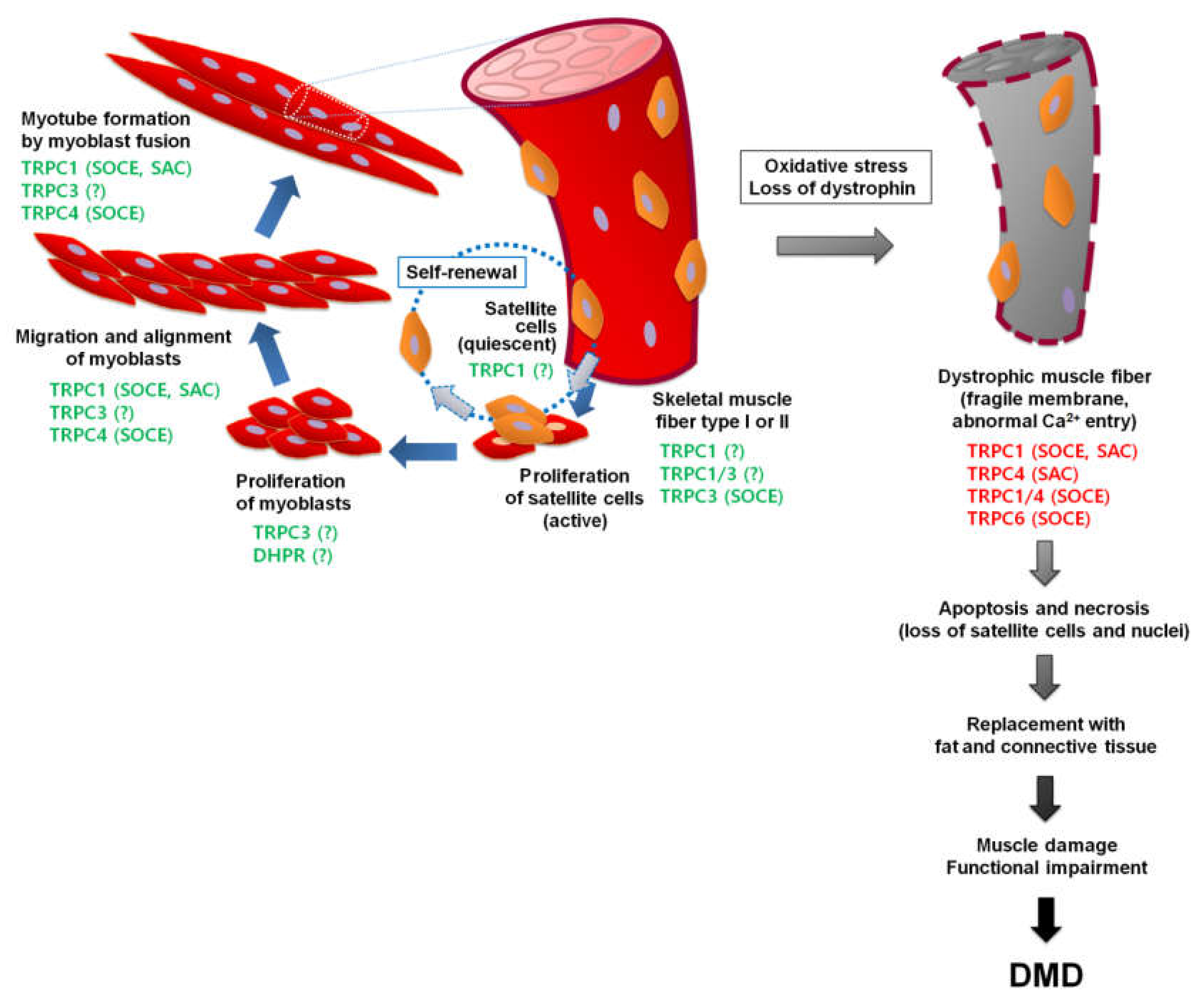
6. TRPCs in Muscular Dystrophy
6.1. Duchenne Muscular Dystrophy (DMD)
6.2. TRPCs in DMD
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Zucchi, R.; Ronca-Testoni, S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: Modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. 1997, 49, 1–51. [Google Scholar]
- Cho, C.H.; Lee, K.J.; Lee, E.H. With the greatest care, stromal interaction molecule (STIM) proteins verify what skeletal muscle is doing. BMB Rep. 2018, 51, 378–387. [Google Scholar] [CrossRef]
- Cho, C.H.; Woo, J.S.; Perez, C.F.; Lee, E.H. A focus on extracellular Ca(2+) entry into skeletal muscle. Exp. Mol. Med. 2017, 49, e378. [Google Scholar] [CrossRef]
- Lee, E.H. Ca2+ channels and skeletal muscle diseases. Prog. Biophys Mol. Biol. 2010, 103, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Kim, D.H.; Allen, P.D. Interplay between intra- and extracellular calcium ions. Mol. Cells 2006, 21, 315–329. [Google Scholar] [PubMed]
- Rios, E.; Pizarro, G.; Stefani, E. Charge movement and the nature of signal transduction in skeletal muscle excitation-contraction coupling. Annu. Rev. Physiol. 1992, 54, 109–133. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, N.; Ogawa, Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. 2001, 533, 185–199. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 2006, 443, 226–229. [Google Scholar] [CrossRef]
- Vig, M.; Beck, A.; Billingsley, J.M.; Lis, A.; Parvez, S.; Peinelt, C.; Koomoa, D.L.; Soboloff, J.; Gill, D.L.; Fleig, A.; et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 2006, 16, 2073–2079. [Google Scholar] [CrossRef]
- Launikonis, B.S.; Barnes, M.; Stephenson, D.G. Identification of the coupling between skeletal muscle store-operated Ca2+ entry and the inositol trisphosphate receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 2941–2944. [Google Scholar] [CrossRef] [PubMed]
- Ducret, T.; Vandebrouck, C.; Cao, M.L.; Lebacq, J.; Gailly, P. Functional role of store-operated and stretch-activated channels in murine adult skeletal muscle fibres. J. Physiol. 2006, 575, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Stiber, J.; Hawkins, A.; Zhang, Z.S.; Wang, S.; Burch, J.; Graham, V.; Ward, C.C.; Seth, M.; Finch, E.; Malouf, N.; et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Yarotskyy, V.; Dirksen, R.T. Temperature and RyR1 regulate the activation rate of store-operated Ca(2)+ entry current in myotubes. Biophys. J. 2012, 103, 202–211. [Google Scholar] [CrossRef][Green Version]
- Pan, Z.; Brotto, M.; Ma, J. Store-operated Ca2+ entry in muscle physiology and diseases. BMB Rep. 2014, 47, 69–79. [Google Scholar] [CrossRef]
- Pan, Z.; Yang, D.; Nagaraj, R.Y.; Nosek, T.A.; Nishi, M.; Takeshima, H.; Cheng, H.; Ma, J. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat. Cell Biol. 2002, 4, 379–383. [Google Scholar] [CrossRef]
- Dirksen, R.T. Checking your SOCCs and feet: The molecular mechanisms of Ca2+ entry in skeletal muscle. J. Physiol. 2009, 587, 3139–3147. [Google Scholar] [CrossRef]
- Stiber, J.A.; Rosenberg, P.B. The role of store-operated calcium influx in skeletal muscle signaling. Cell Calcium 2011, 49, 341–349. [Google Scholar] [CrossRef]
- Wei-Lapierre, L.; Carrell, E.M.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 2013, 4, 2805. [Google Scholar] [CrossRef]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Skeletal muscle fatigue: Cellular mechanisms. Physiol. Rev. 2008, 88, 287–332. [Google Scholar] [CrossRef]
- Sztretye, M.; Geyer, N.; Vincze, J.; Al-Gaadi, D.; Olah, T.; Szentesi, P.; Kis, G.; Antal, M.; Balatoni, I.; Csernoch, L.; et al. SOCE Is Important for Maintaining Sarcoplasmic Calcium Content and Release in Skeletal Muscle Fibers. Biophys. J. 2017, 113, 2496–2507. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef]
- Rosado, J.A.; Diez, R.; Smani, T.; Jardin, I. STIM and Orai1 Variants in Store-Operated Calcium Entry. Front. Pharmacol. 2015, 6, 325. [Google Scholar] [CrossRef]
- Wissenbach, U.; Philipp, S.E.; Gross, S.A.; Cavalie, A.; Flockerzi, V. Primary structure, chromosomal localization and expression in immune cells of the murine ORAI and STIM genes. Cell Calcium 2007, 42, 439–446. [Google Scholar] [CrossRef]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef]
- Peinelt, C.; Vig, M.; Koomoa, D.L.; Beck, A.; Nadler, M.J.; Koblan-Huberson, M.; Lis, A.; Fleig, A.; Penner, R.; Kinet, J.P. Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell Biol. 2006, 8, 771–773. [Google Scholar] [CrossRef] [PubMed]
- Lyfenko, A.D.; Dirksen, R.T. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J. Physiol. 2008, 586, 4815–4824. [Google Scholar] [CrossRef] [PubMed]
- Kiviluoto, S.; Decuypere, J.P.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet Muscle 2011, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Sabourin, J.; Sauc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca(2+) release in human myotubes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Erxleben, C.; Yildirim, E.; Abramowitz, J.; Armstrong, D.L.; Birnbaumer, L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 4682–4687. [Google Scholar] [CrossRef]
- Lopez, J.J.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J. Biol. Chem. 2006, 281, 28254–28264. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef]
- Vandebrouck, C.; Martin, D.; Colson-Van Schoor, M.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef]
- McCarl, C.A.; Picard, C.; Khalil, S.; Kawasaki, T.; Rother, J.; Papolos, A.; Kutok, J.; Hivroz, C.; Ledeist, F.; Plogmann, K.; et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 2009, 124, 1311–1318. [Google Scholar] [CrossRef]
- Launikonis, B.S.; Stephenson, D.G.; Friedrich, O. Rapid Ca2+ flux through the transverse tubular membrane, activated by individual action potentials in mammalian skeletal muscle. J. Physiol. 2009, 587, 2299–2312. [Google Scholar] [CrossRef]
- Edwards, J.N.; Murphy, R.M.; Cully, T.R.; von Wegner, F.; Friedrich, O.; Launikonis, B.S. Ultra-rapid activation and deactivation of store-operated Ca(2+) entry in skeletal muscle. Cell Calcium 2010, 47, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Woo, J.S.; Hwang, J.H.; Hyun, C.; Cho, C.H.; Kim, D.H.; Lee, E.H. STIM1 negatively regulates Ca(2)(+) release from the sarcoplasmic reticulum in skeletal myotubes. Biochem. J. 2013, 453, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Roberts-Thomson, S.J.; Peters, A.A.; Grice, D.M.; Monteith, G.R. ORAI-mediated calcium entry: Mechanism and roles, diseases and pharmacology. Pharmacol. Ther. 2010, 127, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Carrell, E.M.; Coppola, A.R.; McBride, H.J.; Dirksen, R.T. Orai1 enhances muscle endurance by promoting fatigue-resistant type I fiber content but not through acute store-operated Ca2+ entry. FASEB J. 2016, 30, 4109–4119. [Google Scholar] [CrossRef] [PubMed]
- Bohm, J.; Bulla, M.; Urquhart, J.E.; Malfatti, E.; Williams, S.G.; O’Sullivan, J.; Szlauer, A.; Koch, C.; Baranello, G.; Mora, M.; et al. ORAI1 Mutations with Distinct Channel Gating Defects in Tubular Aggregate Myopathy. Hum. Mutat. 2017, 38, 426–438. [Google Scholar] [CrossRef]
- Zhao, X.; Moloughney, J.G.; Zhang, S.; Komazaki, S.; Weisleder, N. Orai1 mediates exacerbated Ca(2+) entry in dystrophic skeletal muscle. PLoS ONE 2012, 7, e49862. [Google Scholar] [CrossRef]
- Li, H.; Ding, X.; Lopez, J.R.; Takeshima, H.; Ma, J.; Allen, P.D.; Eltit, J.M. Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J. Biol. Chem. 2010, 285, 39171–39179. [Google Scholar] [CrossRef]
- Ahn, M.K.; Lee, K.J.; Cai, C.; Huang, M.; Cho, C.H.; Ma, J.; Lee, E.H. Mitsugumin 53 regulates extracellular Ca(2+) entry and intracellular Ca(2+) release via Orai1 and RyR1 in skeletal muscle. Sci. Rep. 2016, 6, 36909. [Google Scholar] [CrossRef]
- Zhao, X.; Weisleder, N.; Thornton, A.; Oppong, Y.; Campbell, R.; Ma, J.; Brotto, M. Compromised store-operated Ca2+ entry in aged skeletal muscle. Aging Cell 2008, 7, 561–568. [Google Scholar] [CrossRef]
- Darbellay, B.; Arnaudeau, S.; Ceroni, D.; Bader, C.R.; Konig, S.; Bernheim, L. Human muscle economy myoblast differentiation and excitation-contraction coupling use the same molecular partners, STIM1 and STIM2. J. Biol. Chem. 2010, 285, 22437–22447. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, T.; Higashi, T.; Higa, T.; Terada, K.; Mai, Y.; Aoyagi, H.; Hatate, C.; Nepal, P.; Horiguchi, M.; Harada, T.; et al. Different binding property of STIM1 and its novel splice variant STIM1L to Orai1, TRPC3, and TRPC6 channels. Biochem. Biophys. Res. Commun. 2012, 428, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Koenig, S.; Bernheim, L.; Frieden, M. During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca(2)(+) entry. J. Cell Sci. 2013, 126, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Darbellay, B.; Arnaudeau, S.; Konig, S.; Jousset, H.; Bader, C.; Demaurex, N.; Bernheim, L. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J. Biol. Chem. 2009, 284, 5370–5380. [Google Scholar] [CrossRef]
- Li, T.; Finch, E.A.; Graham, V.; Zhang, Z.S.; Ding, J.D.; Burch, J.; Oh-hora, M.; Rosenberg, P. STIM1-Ca(2+) signaling is required for the hypertrophic growth of skeletal muscle in mice. Mol. Cell Biol. 2012, 32, 3009–3017. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef]
- Edwards, J.N.; Friedrich, O.; Cully, T.R.; von Wegner, F.; Murphy, R.M.; Launikonis, B.S. Upregulation of store-operated Ca2+ entry in dystrophic mdx mouse muscle. Am. J. Physiol. Cell Physiol. 2010, 299, C42–C50. [Google Scholar] [CrossRef]
- Onopiuk, M.; Brutkowski, W.; Young, C.; Krasowska, E.; Rog, J.; Ritso, M.; Wojciechowska, S.; Arkle, S.; Zablocki, K.; Gorecki, D.C. Store-operated calcium entry contributes to abnormal Ca(2)(+) signalling in dystrophic mdx mouse myoblasts. Arch. Biochem. Biophys. 2015, 569, 1–9. [Google Scholar] [CrossRef]
- Choi, J.H.; Huang, M.; Hyun, C.; Oh, M.R.; Lee, K.J.; Cho, C.H.; Lee, E.H. A muscular hypotonia-associated STIM1 mutant at R429 induces abnormalities in intracellular Ca(2+) movement and extracellular Ca(2+) entry in skeletal muscle. Sci. Rep. 2019, 9, 19140. [Google Scholar] [CrossRef]
- Lee, K.J.; Hyun, C.; Woo, J.S.; Park, C.S.; Kim, D.H.; Lee, E.H. Stromal interaction molecule 1 (STIM1) regulates sarcoplasmic/endoplasmic reticulum Ca(2)(+)-ATPase 1a (SERCA1a) in skeletal muscle. Pflugers Arch. 2014, 466, 987–1001. [Google Scholar] [CrossRef]
- Sauc, S.; Frieden, M. Neurological and Motor Disorders: TRPC in the Skeletal Muscle. Adv. Exp. Med. Biol. 2017, 993, 557–575. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.R.; Lee, K.J.; Huang, M.; Kim, J.O.; Kim, D.H.; Cho, C.H.; Lee, E.H. STIM2 regulates both intracellular Ca(2+) distribution and Ca(2+) movement in skeletal myotubes. Sci. Rep. 2017, 7, 17936. [Google Scholar] [CrossRef] [PubMed]
- Phuong, T.T.T.; Kang, T.M. Stromal interaction molecule 2 regulates C2C12 myoblast differentiation. Integr. Med. Res. 2015, 4, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordonez, A.; Rosado, J.A.; Smani, T. The Complex Role of Store Operated Calcium Entry Pathways and Related Proteins in the Function of Cardiac, Skeletal and Vascular Smooth Muscle Cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Damann, N.; Voets, T.; Nilius, B. TRPs in our senses. Curr. Biol. 2008, 18, R880–R889. [Google Scholar] [CrossRef]
- Minke, B.; Cook, B. TRP channel proteins and signal transduction. Physiol. Rev. 2002, 82, 429–472. [Google Scholar] [CrossRef]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 2005, re3. [Google Scholar] [CrossRef]
- Montell, C. The venerable inveterate invertebrate TRP channels. Cell Calcium 2003, 33, 409–417. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Montell, C.; Birnbaumer, L.; Flockerzi, V. The TRP channels, a remarkably functional family. Cell 2002, 108, 595–598. [Google Scholar] [CrossRef]
- Minke, B. The history of the Drosophila TRP channel: The birth of a new channel superfamily. J. Neurogenet. 2010, 24, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP channels: An overview. Cell Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Owsianik, G.; D’Hoedt, D.; Voets, T.; Nilius, B. Structure-function relationship of the TRP channel superfamily. Rev. Physiol. Biochem. Pharmacol. 2006, 156, 61–90. [Google Scholar] [PubMed]
- Wissenbach, U.; Niemeyer, B.; Himmerkus, N.; Fixemer, T.; Bonkhoff, H.; Flockerzi, V. TRPV6 and prostate cancer: Cancer growth beyond the prostate correlates with increased TRPV6 Ca2+ channel expression. Biochem. Biophys. Res. Commun. 2004, 322, 1359–1363. [Google Scholar] [CrossRef]
- Abramowitz, J.; Birnbaumer, L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009, 23, 297–328. [Google Scholar] [CrossRef]
- Vazquez, G.; Wedel, B.J.; Aziz, O.; Trebak, M.; Putney, J.W., Jr. The mammalian TRPC cation channels. Biochim. Biophys. Acta 2004, 1742, 21–36. [Google Scholar] [CrossRef]
- Schindl, R.; Romanin, C. Assembly domains in TRP channels. Biochem. Soc. Trans. 2007, 35, 84–85. [Google Scholar] [CrossRef]
- Zhu, M.X. Multiple roles of calmodulin and other Ca(2+)-binding proteins in the functional regulation of TRP channels. Pflugers Arch. 2005, 451, 105–115. [Google Scholar] [CrossRef]
- Singh, B.B.; Liu, X.; Tang, J.; Zhu, M.X.; Ambudkar, I.S. Calmodulin regulates Ca(2+)-dependent feedback inhibition of store-operated Ca(2+) influx by interaction with a site in the C terminus of TrpC1. Mol. Cell 2002, 9, 739–750. [Google Scholar] [CrossRef]
- Beech, D.J. Characteristics of transient receptor potential canonical calcium-permeable channels and their relevance to vascular physiology and disease. Circ. J. 2013, 77, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Odell, A.F.; Scott, J.L.; Van Helden, D.F. Epidermal growth factor induces tyrosine phosphorylation, membrane insertion, and activation of transient receptor potential channel 4. J. Biol. Chem. 2005, 280, 37974–37987. [Google Scholar] [CrossRef] [PubMed]
- Lockwich, T.P.; Liu, X.; Singh, B.B.; Jadlowiec, J.; Weiland, S.; Ambudkar, I.S. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J. Biol. Chem. 2000, 275, 11934–11942. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.; Ding, M.; Ding, Y.; Sours-Brothers, S.; Luchowski, R.; Gryczynski, Z.; Yorio, T.; Ma, H.; Ma, R. Canonical transient receptor potential 6 (TRPC6), a redox-regulated cation channel. J. Biol. Chem. 2010, 285, 23466–23476. [Google Scholar] [CrossRef] [PubMed]
- Bon, R.S.; Beech, D.J. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels -- mirage or pot of gold? Br. J. Pharmacol. 2013, 170, 459–474. [Google Scholar] [CrossRef]
- Harteneck, C.; Plant, T.D.; Schultz, G. From worm to man: Three subfamilies of TRP channels. Trends Neurosci. 2000, 23, 159–166. [Google Scholar] [CrossRef]
- Singh, B.B.; Lockwich, T.P.; Bandyopadhyay, B.C.; Liu, X.; Bollimuntha, S.; Brazer, S.C.; Combs, C.; Das, S.; Leenders, A.G.; Sheng, Z.H.; et al. VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol. Cell 2004, 15, 635–646. [Google Scholar] [CrossRef]
- Van Rossum, D.B.; Patterson, R.L.; Sharma, S.; Barrow, R.K.; Kornberg, M.; Gill, D.L.; Snyder, S.H. Phospholipase Cgamma1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature 2005, 434, 99–104. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Ramsey, I.S.; Kotecha, S.; Greka, A.; Clapham, D.E. Rapid vesicular translocation and insertion of TRP channels. Nat. Cell Biol. 2004, 6, 709–720. [Google Scholar] [CrossRef]
- Dietrich, A.; Mederos y Schnitzler, M.; Emmel, J.; Kalwa, H.; Hofmann, T.; Gudermann, T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J. Biol. Chem. 2003, 278, 47842–47852. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, P.; Sedo, A.; Li, J.; Wilson, L.A.; O’Regan, D.; Lippiat, J.D.; Porter, K.E.; Kearney, M.T.; Ainscough, J.F.; Beech, D.J. Constitutively active TRPC channels of adipocytes confer a mechanism for sensing dietary fatty acids and regulating adiponectin. Circ. Res. 2012, 111, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Cavalie, A.; Freichel, M.; Wissenbach, U.; Zimmer, S.; Trost, C.; Marquart, A.; Murakami, M.; Flockerzi, V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO J. 1996, 15, 6166–6171. [Google Scholar] [CrossRef] [PubMed]
- Warnat, J.; Philipp, S.; Zimmer, S.; Flockerzi, V.; Cavalie, A. Phenotype of a recombinant store-operated channel: Highly selective permeation of Ca2+. J. Physiol. 1999, 518(Pt. 3), 631–638. [Google Scholar] [CrossRef]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weissgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef]
- Vazquez, G.; Lievremont, J.P.; St, J.B.G.; Putney, J.W., Jr. Human Trp3 forms both inositol trisphosphate receptor-dependent and receptor-independent store-operated cation channels in DT40 avian B lymphocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 11777–11782. [Google Scholar] [CrossRef]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [CrossRef]
- Franco, A., Jr.; Lansman, J.B. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 1990, 344, 670–673. [Google Scholar] [CrossRef]
- Vandebrouck, C.; Duport, G.; Cognard, C.; Raymond, G. Cationic channels in normal and dystrophic human myotubes. Neuromuscul. Disord. 2001, 11, 72–79. [Google Scholar] [CrossRef]
- Tutdibi, O.; Brinkmeier, H.; Rudel, R.; Fohr, K.J. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J. Physiol. 1999, 515(Pt. 3), 859–868. [Google Scholar] [CrossRef]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Dryer, S.E. A mutation in TRPC6 channels abolishes their activation by hypoosmotic stretch but does not affect activation by diacylglycerol or G protein signaling cascades. Am. J. Physiol. Renal Physiol. 2014, 306, F1018–F1025. [Google Scholar] [CrossRef] [PubMed]
- Fels, B.; Nielsen, N.; Schwab, A. Role of TRPC1 channels in pressure-mediated activation of murine pancreatic stellate cells. Eur. Biophys. J. 2016, 45, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; He, Y.; Yang, G.; Yu, Q.; Li, M. Role of TRPC1 channels in pressure-mediated activation of airway remodeling. Respir Res. 2019, 20, 91. [Google Scholar] [CrossRef] [PubMed]
- Molnar, T.; Yarishkin, O.; Iuso, A.; Barabas, P.; Jones, B.; Marc, R.E.; Phuong, T.T.; Krizaj, D. Store-Operated Calcium Entry in Muller Glia Is Controlled by Synergistic Activation of TRPC and Orai Channels. J. Neurosci. 2016, 36, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Staaf, S.; Maxvall, I.; Lind, U.; Husmark, J.; Mattsson, J.P.; Ernfors, P.; Pierrou, S. Down regulation of TRPC1 by shRNA reduces mechanosensitivity in mouse dorsal root ganglion neurons in vitro. Neurosci. Lett. 2009, 457, 3–7. [Google Scholar] [CrossRef]
- Formigli, L.; Sassoli, C.; Squecco, R.; Bini, F.; Martinesi, M.; Chellini, F.; Luciani, G.; Sbrana, F.; Zecchi-Orlandini, S.; Francini, F.; et al. Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J. Cell Sci. 2009, 122, 1322–1333. [Google Scholar] [CrossRef]
- Gottlieb, P.; Folgering, J.; Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Bowman, C.; Bichet, D.; Patel, A.; Sachs, F.; et al. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch. 2008, 455, 1097–1103. [Google Scholar] [CrossRef]
- Mederos y Schnitzler, M.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008, 27, 3092–3103. [Google Scholar] [CrossRef]
- Kruger, J.; Kunert-Keil, C.; Bisping, F.; Brinkmeier, H. Transient receptor potential cation channels in normal and dystrophic mdx muscle. Neuromuscul. Disord. 2008, 18, 501–513. [Google Scholar] [CrossRef]
- Brinkmeier, H. TRP channels in skeletal muscle: Gene expression, function and implications for disease. Adv. Exp. Med. Biol. 2011, 704, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Gailly, P. TRP channels in normal and dystrophic skeletal muscle. Curr. Opin. Pharmacol. 2012, 12, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Zanou, N.; Shapovalov, G.; Louis, M.; Tajeddine, N.; Gallo, C.; Van Schoor, M.; Anguish, I.; Cao, M.L.; Schakman, O.; Dietrich, A.; et al. Role of TRPC1 channel in skeletal muscle function. Am. J. Physiol. Cell Physiol. 2010, 298, C149–C162. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.A.; Streiff, M.; Hunter, C.; Hu, Q.; Sachse, F.B. Physiological and pathophysiological role of transient receptor potential canonical channels in cardiac myocytes. Prog. Biophys. Mol. Biol. 2017, 130, 254–263. [Google Scholar] [CrossRef]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Galeano-Otero, I.; Redondo, P.C.; Rosado, J.A.; Smani, T. Pathophysiological Significance of Store-Operated Calcium Entry in Cardiovascular and Skeletal Muscle Disorders and Angiogenesis. Adv. Exp. Med. Biol. 2020, 1131, 489–504. [Google Scholar] [CrossRef]
- Jansen, K.M.; Pavlath, G.K. Molecular control of mammalian myoblast fusion. Methods Mol. Biol. 2008, 475, 115–133. [Google Scholar] [CrossRef]
- Schollmeyer, J.E. Role of Ca2+ and Ca2+-activated protease in myoblast fusion. Exp. Cell Res. 1986, 162, 411–422. [Google Scholar] [CrossRef]
- Wakelam, M.J. The fusion of myoblasts. Biochem. J. 1985, 228, 1–12. [Google Scholar] [CrossRef]
- Louis, M.; Zanou, N.; Van Schoor, M.; Gailly, P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. 2008, 121, 3951–3959. [Google Scholar] [CrossRef]
- Olah, T.; Fodor, J.; Ruzsnavszky, O.; Vincze, J.; Berbey, C.; Allard, B.; Csernoch, L. Overexpression of transient receptor potential canonical type 1 (TRPC1) alters both store operated calcium entry and depolarization-evoked calcium signals in C2C12 cells. Cell Calcium 2011, 49, 415–425. [Google Scholar] [CrossRef][Green Version]
- Ma, R.; Rundle, D.; Jacks, J.; Koch, M.; Downs, T.; Tsiokas, L. Inhibitor of myogenic family, a novel suppressor of store-operated currents through an interaction with TRPC1. J. Biol. Chem. 2003, 278, 52763–52772. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Cheung, K.K.; Yeung, S.S.; Yeung, E.W. The involvement of transient receptor potential canonical type 1 in skeletal muscle regrowth after unloading-induced atrophy. J. Physiol. 2016, 594, 3111–3126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.T.; Yeung, S.S.; Cheung, K.K.; Chai, Z.Y.; Yeung, E.W. Adaptive responses of TRPC1 and TRPC3 during skeletal muscle atrophy and regrowth. Muscle Nerve 2014, 49, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Zanou, N.; Schakman, O.; Louis, P.; Ruegg, U.T.; Dietrich, A.; Birnbaumer, L.; Gailly, P. Trpc1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J. Biol. Chem. 2012, 287, 14524–14534. [Google Scholar] [CrossRef]
- Liu, Y.; Schneider, M.F. FGF2 activates TRPC and Ca(2+) signaling leading to satellite cell activation. Front. Physiol. 2014, 5, 38. [Google Scholar] [CrossRef]
- Imaoka, Y.; Kawai, M.; Mori, F.; Miyata, H. Effect of eccentric contraction on satellite cell activation in human vastus lateralis muscle. J. Physiol Sci 2015, 65, 461–469. [Google Scholar] [CrossRef]
- Woo, J.S.; Kim, D.H.; Allen, P.D.; Lee, E.H. TRPC3-interacting triadic proteins in skeletal muscle. Biochem. J. 2008, 411, 399–405. [Google Scholar] [CrossRef]
- Sabourin, J.; Lamiche, C.; Vandebrouck, A.; Magaud, C.; Rivet, J.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J. Biol. Chem. 2009, 284, 36248–36261. [Google Scholar] [CrossRef]
- Woo, J.S.; Cho, C.H.; Kim, D.H.; Lee, E.H. TRPC3 cation channel plays an important role in proliferation and differentiation of skeletal muscle myoblasts. Exp. Mol. Med. 2010, 42, 614–627. [Google Scholar] [CrossRef]
- Woo, J.S.; Lee, K.J.; Huang, M.; Cho, C.H.; Lee, E.H. Heteromeric TRPC3 with TRPC1 formed via its ankyrin repeats regulates the resting cytosolic Ca2+ levels in skeletal muscle. Biochem. Biophys. Res. Commun. 2014, 446, 454–459. [Google Scholar] [CrossRef]
- Rosenberg, P.; Hawkins, A.; Stiber, J.; Shelton, J.M.; Hutcheson, K.; Bassel-Duby, R.; Shin, D.M.; Yan, Z.; Williams, R.S. TRPC3 channels confer cellular memory of recent neuromuscular activity. Proc. Natl. Acad. Sci. USA 2004, 101, 9387–9392. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, M.W.; Houweling, P.J.; Thomas, K.C.; Gordish-Dressman, H.; Bello, L.; Cooperative International Neuromuscular Research, G.; Pegoraro, E.; Hoffman, E.P.; Head, S.I.; North, K.N. Evidence for ACTN3 as a genetic modifier of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14143. [Google Scholar] [CrossRef] [PubMed]
- Lanner, J.T.; Bruton, J.D.; Assefaw-Redda, Y.; Andronache, Z.; Zhang, S.J.; Severa, D.; Zhang, Z.B.; Melzer, W.; Zhang, S.L.; Katz, A.; et al. Knockdown of TRPC3 with siRNA coupled to carbon nanotubes results in decreased insulin-mediated glucose uptake in adult skeletal muscle cells. FASEB J. 2009, 23, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Cherednichenko, G.; Pessah, I.N.; Allen, P.D. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J. Biol. Chem. 2006, 281, 10042–10048. [Google Scholar] [CrossRef]
- Sampieri, A.; Diaz-Munoz, M.; Antaramian, A.; Vaca, L. The foot structure from the type 1 ryanodine receptor is required for functional coupling to store-operated channels. J. Biol. Chem. 2005, 280, 24804–24815. [Google Scholar] [CrossRef]
- Dietrich, A.; Gudermann, T. Trpc6. Handb Exp. Pharmacol. 2007. [Google Scholar] [CrossRef]
- Spassova, M.A.; Hewavitharana, T.; Xu, W.; Soboloff, J.; Gill, D.L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. USA 2006, 103, 16586–16591. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Nilius, B. Transient receptor potential channels in mechanosensing and cell volume regulation. Methods Enzymol. 2007, 428, 183–207. [Google Scholar] [CrossRef]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef]
- Reiser, J.; Polu, K.R.; Moller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P. Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell 1995, 80, 675–679. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Sonnemann, K.J. Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol. 2008, 265, 191–225. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Moens, P.; Baatsen, P.H.; Marechal, G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil. 1993, 14, 446–451. [Google Scholar] [CrossRef]
- Allard, B. Sarcolemmal ion channels in dystrophin-deficient skeletal muscle fibres. J. Muscle Res. Cell Motil. 2006, 27, 367–373. [Google Scholar] [CrossRef]
- Ruegg, U.T.; Gillis, J.M. Calcium homeostasis in dystrophic muscle. Trends Pharmacol. Sci. 1999, 20, 351–352. [Google Scholar] [CrossRef]
- Gailly, P. New aspects of calcium signaling in skeletal muscle cells: Implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta 2002, 1600, 38–44. [Google Scholar] [CrossRef]
- Gillis, J.M. Understanding dystrophinopathies: An inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J. Muscle Res. Cell Motil. 1999, 20, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Lansman, J.B.; Franco, A., Jr. What does dystrophin do in normal muscle? J. Muscle Res. Cell Motil. 1991, 12, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Proske, U.; Morgan, D.L. Muscle damage from eccentric exercise: Mechanism, mechanical signs, adaptation and clinical applications. J. Physiol. 2001, 537, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Oberc, M.A.; Engel, W.K. Ultrastructural localization of calcium in normal and abnormal skeletal muscle. Lab. Invest. 1977, 36, 566–577. [Google Scholar] [PubMed]
- Robert, V.; Massimino, M.L.; Tosello, V.; Marsault, R.; Cantini, M.; Sorrentino, V.; Pozzan, T. Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 2001, 276, 4647–4651. [Google Scholar] [CrossRef] [PubMed]
- Mallouk, N.; Jacquemond, V.; Allard, B. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibers detected with Ca2+-activated K+ channels. Proc. Natl. Acad. Sci. USA 2000, 97, 4950–4955. [Google Scholar] [CrossRef]
- Mongini, T.; Ghigo, D.; Doriguzzi, C.; Bussolino, F.; Pescarmona, G.; Pollo, B.; Schiffer, D.; Bosia, A. Free cytoplasmic Ca++ at rest and after cholinergic stimulus is increased in cultured muscle cells from Duchenne muscular dystrophy patients. Neurology 1988, 38, 476–480. [Google Scholar] [CrossRef]
- Turner, P.R.; Westwood, T.; Regen, C.M.; Steinhardt, R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 1988, 335, 735–738. [Google Scholar] [CrossRef]
- Williams, D.A.; Head, S.I.; Bakker, A.J.; Stephenson, D.G. Resting calcium concentrations in isolated skeletal muscle fibres of dystrophic mice. J. Physiol. 1990, 428, 243–256. [Google Scholar] [CrossRef]
- Sandri, M.; El Meslemani, A.H.; Sandri, C.; Schjerling, P.; Vissing, K.; Andersen, J.L.; Rossini, K.; Carraro, U.; Angelini, C. Caspase 3 expression correlates with skeletal muscle apoptosis in Duchenne and facioscapulo human muscular dystrophy. A potential target for pharmacological treatment? J. Neuropathol. Exp. Neurol. 2001, 60, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, V.M.; Kropotov, A.V.; Zelenin, A.V.; Krutilina, R.I.; Kolesnikov, V.A.; Zelenina, I.A.; Baranov, A.N.; Shtein, G.I.; Ostapenko, O.V.; Tomilin, N.V.; et al. The BCL-xL and ACR-1 genes promote differentiation and reduce apoptosis in muscle fibers of mdx mice. Genetika 2002, 38, 1445–1450. [Google Scholar]
- Sandri, M.; Carraro, U. Apoptosis of skeletal muscles during development and disease. Int. J. Biochem. Cell Biol. 1999, 31, 1373–1390. [Google Scholar] [CrossRef]
- Collins, C.A.; Morgan, J.E. Duchenne’s muscular dystrophy: Animal models used to investigate pathogenesis and develop therapeutic strategies. Int. J. Exp. Pathol. 2003, 84, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Imbert, N.; Vandebrouck, C.; Constantin, B.; Duport, G.; Guillou, C.; Cognard, C.; Raymond, G. Hypoosmotic shocks induce elevation of resting calcium level in Duchenne muscular dystrophy myotubes contracting in vitro. Neuromuscul. Disord. 1996, 6, 351–360. [Google Scholar] [CrossRef]
- Gailly, P.; Boland, B.; Himpens, B.; Casteels, R.; Gillis, J.M. Critical evaluation of cytosolic calcium determination in resting muscle fibres from normal and dystrophic (mdx) mice. Cell Calcium 1993, 14, 473–483. [Google Scholar] [CrossRef]
- Mallouk, N.; Allard, B. Stretch-induced activation of Ca(2+)-activated K(+) channels in mouse skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 2000, 278, C473–C479. [Google Scholar] [CrossRef]
- Gailly, P.; De Backer, F.; Van Schoor, M.; Gillis, J.M. In situ measurements of calpain activity in isolated muscle fibres from normal and dystrophin-lacking mdx mice. J. Physiol. 2007, 582, 1261–1275. [Google Scholar] [CrossRef]
- Vandebrouck, A.; Ducret, T.; Basset, O.; Sebille, S.; Raymond, G.; Ruegg, U.; Gailly, P.; Cognard, C.; Constantin, B. Regulation of store-operated calcium entries and mitochondrial uptake by minidystrophin expression in cultured myotubes. FASEB J. 2006, 20, 136–138. [Google Scholar] [CrossRef]
- Gervasio, O.L.; Whitehead, N.P.; Yeung, E.W.; Phillips, W.D.; Allen, D.G. TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J. Cell Sci. 2008, 121, 2246–2255. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P. Duchenne muscular dystrophy--what causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 2011, 43, 290–294. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Yeung, E.W. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: Role of ionic changes. J. Physiol. 2005, 567, 723–735. [Google Scholar] [CrossRef]
- Gailly, P.; Hermans, E.; Octave, J.N.; Gillis, J.M. Specific increase of genetic expression of parvalbumin in fast skeletal muscles of mdx mice. FEBS Lett 1993, 326, 272–274. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef]
- Alderton, J.M.; Steinhardt, R.A. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J. Biol. Chem. 2000, 275, 9452–9460. [Google Scholar] [CrossRef]
- Fong, P.Y.; Turner, P.R.; Denetclaw, W.F.; Steinhardt, R.A. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science 1990, 250, 673–676. [Google Scholar] [CrossRef]
- Hopf, F.W.; Reddy, P.; Hong, J.; Steinhardt, R.A. A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. J. Biol. Chem. 1996, 271, 22358–22367. [Google Scholar] [CrossRef]
- Wang, X.; Weisleder, N.; Collet, C.; Zhou, J.; Chu, Y.; Hirata, Y.; Zhao, X.; Pan, Z.; Brotto, M.; Cheng, H.; et al. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat. Cell Biol. 2005, 7, 525–530. [Google Scholar] [CrossRef]
- Franco-Obregon, A., Jr.; Lansman, J.B. Mechanosensitive ion channels in skeletal muscle from normal and dystrophic mice. J. Physiol. 1994, 481(Pt. 2), 299–309. [Google Scholar] [CrossRef]
- Rolland, J.F.; De Luca, A.; Burdi, R.; Andreetta, F.; Confalonieri, P.; Conte Camerino, D. Overactivity of exercise-sensitive cation channels and their impaired modulation by IGF-1 in mdx native muscle fibers: Beneficial effect of pentoxifylline. Neurobiol. Dis. 2006, 24, 466–474. [Google Scholar] [CrossRef]
- Watkins, S.C.; Cullen, M.J. A qualitative and quantitative study of the ultrastructure of regenerating muscle fibres in Duchenne muscular dystrophy and polymyositis. J. Neurol. Sci. 1987, 82, 181–192. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.R.; von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Zulian, A.; Schiavone, M.; Giorgio, V.; Bernardi, P. Forty years later: Mitochondria as therapeutic targets in muscle diseases. Pharmacol. Res. 2016, 113, 563–573. [Google Scholar] [CrossRef]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Yeung, E.W.; Froehner, S.C.; Allen, D.G. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS ONE 2010, 5, e15354. [Google Scholar] [CrossRef]
- Vandebrouck, A.; Sabourin, J.; Rivet, J.; Balghi, H.; Sebille, S.; Kitzis, A.; Raymond, G.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of capacitative calcium entries by alpha1-syntrophin: Association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. 2007, 21, 608–617. [Google Scholar] [CrossRef]
- Adams, M.E.; Butler, M.H.; Dwyer, T.M.; Peters, M.F.; Murnane, A.A.; Froehner, S.C. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron 1993, 11, 531–540. [Google Scholar] [CrossRef]
- Boittin, F.X.; Petermann, O.; Hirn, C.; Mittaud, P.; Dorchies, O.M.; Roulet, E.; Ruegg, U.T. Ca2+-independent phospholipase A2 enhances store-operated Ca2+ entry in dystrophic skeletal muscle fibers. J. Cell Sci. 2006, 119, 3733–3742. [Google Scholar] [CrossRef]
- Vaghy, P.L.; Fang, J.; Wu, W.; Vaghy, L.P. Increased caveolin-3 levels in mdx mouse muscles. FEBS Lett. 1998, 431, 125–127. [Google Scholar] [CrossRef]
- Venema, V.J.; Ju, H.; Zou, R.; Venema, R.C. Interaction of neuronal nitric-oxide synthase with caveolin-3 in skeletal muscle. Identification of a novel caveolin scaffolding/inhibitory domain. J. Biol Chem 1997, 272, 28187–28190. [Google Scholar] [CrossRef]
- Shibuya, S.; Wakayama, Y.; Inoue, M.; Oniki, H.; Kominami, E. Changes in the distribution and density of caveolin 3 molecules at the plasma membrane of mdx mouse skeletal muscles: A fracture-label electron microscopic study. Neurosci. Lett. 2002, 325, 171–174. [Google Scholar] [CrossRef]
- Galbiati, F.; Razani, B.; Lisanti, M.P. Caveolae and caveolin-3 in muscular dystrophy. Trends Mol. Med. 2001, 7, 435–441. [Google Scholar] [CrossRef]
- Allen, D.G.; Gervasio, O.L.; Yeung, E.W.; Whitehead, N.P. Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 2010, 88, 83–91. [Google Scholar] [CrossRef]
- Sotgia, F.; Lee, J.K.; Das, K.; Bedford, M.; Petrucci, T.C.; Macioce, P.; Sargiacomo, M.; Bricarelli, F.D.; Minetti, C.; Sudol, M.; et al. Caveolin-3 directly interacts with the C-terminal tail of beta -dystroglycan. Identification of a central WW-like domain within caveolin family members. J. Biol. Chem. 2000, 275, 38048–38058. [Google Scholar] [CrossRef]
- Stiber, J.A.; Tabatabaei, N.; Hawkins, A.F.; Hawke, T.; Worley, P.F.; Williams, R.S.; Rosenberg, P. Homer modulates NFAT-dependent signaling during muscle differentiation. Dev. Biol. 2005, 287, 213–224. [Google Scholar] [CrossRef]
- Stiber, J.A.; Zhang, Z.S.; Burch, J.; Eu, J.P.; Zhang, S.; Truskey, G.A.; Seth, M.; Yamaguchi, N.; Meissner, G.; Shah, R.; et al. Mice lacking Homer 1 exhibit a skeletal myopathy characterized by abnormal transient receptor potential channel activity. Mol. Cell Biol. 2008, 28, 2637–2647. [Google Scholar] [CrossRef]
- Bidou, L.; Allamand, V.; Rousset, J.P.; Namy, O. Sense from nonsense: Therapies for premature stop codon diseases. Trends Mol. Med. 2012, 18, 679–688. [Google Scholar] [CrossRef]
- Wagner, K.R.; Hamed, S.; Hadley, D.W.; Gropman, A.L.; Burstein, A.H.; Escolar, D.M.; Hoffman, E.P.; Fischbeck, K.H. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol. 2001, 49, 706–711. [Google Scholar] [CrossRef]
- Finkel, R.S. Read-through strategies for suppression of nonsense mutations in Duchenne/ Becker muscular dystrophy: Aminoglycosides and ataluren (PTC124). J. Child. Neurol. 2010, 25, 1158–1164. [Google Scholar] [CrossRef]
- Marchand, E.; Constantin, B.; Balghi, H.; Claudepierre, M.C.; Cantereau, A.; Magaud, C.; Mouzou, A.; Raymond, G.; Braun, S.; Cognard, C. Improvement of calcium handling and changes in calcium-release properties after mini- or full-length dystrophin forced expression in cultured skeletal myotubes. Exp. Cell Res. 2004, 297, 363–379. [Google Scholar] [CrossRef]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.H.; Jeong, S.Y.; Oh, M.R.; Allen, P.D.; Lee, E.H. TRPCs: Influential Mediators in Skeletal Muscle. Cells 2020, 9, 850. https://doi.org/10.3390/cells9040850
Choi JH, Jeong SY, Oh MR, Allen PD, Lee EH. TRPCs: Influential Mediators in Skeletal Muscle. Cells. 2020; 9(4):850. https://doi.org/10.3390/cells9040850
Chicago/Turabian StyleChoi, Jun Hee, Seung Yeon Jeong, Mi Ri Oh, Paul D. Allen, and Eun Hui Lee. 2020. "TRPCs: Influential Mediators in Skeletal Muscle" Cells 9, no. 4: 850. https://doi.org/10.3390/cells9040850
APA StyleChoi, J. H., Jeong, S. Y., Oh, M. R., Allen, P. D., & Lee, E. H. (2020). TRPCs: Influential Mediators in Skeletal Muscle. Cells, 9(4), 850. https://doi.org/10.3390/cells9040850