Phosphorylation Targets of DNA-PK and Their Role in HIV-1 Replication


Abstract
:1. Introduction
2. Cellular Functions of DNA-PK
3. Phosphorylation Targets of DNA-PK
4. DNA-PK Targets in HIV Replication
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, J.F.; Knudsen, K.E. Beyond DNA Repair: DNA-PK Function in Cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.H. The role of DNA-PK in aging and energy metabolism. FEBS J. 2018, 285, 1959–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, J.F.; Kothari, V.; Drake, J.M.; Zhao, S.; Dylgjeri, E.; Dean, J.L.; Schiewer, M.J.; McNair, C.; Jones, J.K.; Aytes, A.; et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell 2015, 28, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Ting, N.S.Y.; Pohorelic, B.; Yu, Y.; Lees-Miller, S.P.; Beattie, T.L. The human telomerase RNA component, hTR, activates the DNA-dependent protein kinase to phosphorylate heterogeneous nuclear ribonucleoprotein A1. Nucleic Acids Res. 2009, 37, 6105–6115. [Google Scholar] [CrossRef] [Green Version]
- Sui, J.; Zhang, S.; Chen, B.P. DNA-dependent protein kinase in telomere maintenance and protection. Cell. Mol. Biol. Lett. 2020, 25, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.S.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef]
- Malu, S.; Malshetty, V.; Francis, D.; Cortes, P. Role of non-homologous end joining in V(D)J recombination. Immunol. Res. 2012, 54, 233–246. [Google Scholar] [CrossRef]
- Zhang, S.; Schlott, B.; Görlach, M.; Grosse, F. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res. 2004, 32, 1–10. [Google Scholar] [CrossRef]
- Knyazhanskaya, E.S.; Anisenko, A.; Shadrina, O.; Kalinina, A.; Zatsepin, T.; Zalevsky, A.; Mazurov, D.; Gottikh, M. NHEJ pathway is involved in post-integrational DNA repair due to Ku70 binding to HIV-1 integrase. Retrovirology 2019, 16, 30. [Google Scholar] [CrossRef]
- Skalka, A.M.; Katz, R.A. Retroviral DNA integration and the DNA damage response. Cell Death Differ. 2005, 12, 971–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Isaguliants, M.G.; Gottikh, M.B. A qPCR assay for measuring the post-integrational DNA repair in HIV-1 replication. J. Virol. Methods 2018, 262, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Bunch, H.; Zheng, X.; Burkholder, A.; Dillon, S.T.; Motola, S.; Birrane, G.; Ebmeier, C.C.; Levine, S.S.; Fargo, D.; Hu, G.; et al. TRIM28 regulates RNA polymerase II promoter-proximal pausing and pause release. Nat. Struct. Mol. Biol. 2014, 21, 876–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zicari, S.; Sharma, A.L.; Sahu, G.; Dubrovsky, L.; Sun, L.; Yue, H.; Jada, T.; Ochem, A.; Simon, G.; Bukrinsky, M.; et al. DNA dependent protein kinase (DNA-PK) enhances HIV transcription by promoting RNA polymerase II activity and recruitment of transcription machinery at HIV LTR. Oncotarget 2020, 11, 699–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dynan, W.S.; Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotula, E.; Faigle, W.; Berthault, N.; Dingli, F.; Loew, D.; Sun, J.-S.; Dutreix, M.; Quanz, M. DNA-PK Target Identification Reveals Novel Links between DNA Repair Signaling and Cytoskeletal Regulation. PLoS ONE 2013, 8, e80313. [Google Scholar] [CrossRef] [Green Version]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.A.; Dang, V.; Douglas, P.; Wold, M.S.; Lees-Miller, S.P.; Meek, K. Inhibition of Homologous Recombination by DNA-Dependent Protein Kinase Requires Kinase Activity, Is Titratable, and Is Modulated by Autophosphorylation. Mol. Cell. Biol. 2011, 31, 1719–1733. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lee, J.-H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Davis, A.J.; Fattah, K.R.; So, S.; Sun, J.; Lee, K.-J.; Harrison, L.; Yang, J.; Chen, D.J. Persistently bound Ku at DNA ends attenuates DNA end resection and homologous recombination. DNA Repair 2012, 11, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Britton, S.; Coates, J.; Jackson, S.P. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013, 202, 579–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, R.B.; Yaneva, M.; Lieber, M.R. Productive and Nonproductive Complexes of Ku and DNA-Dependent Protein Kinase at DNA Termini. Mol. Cell. Biol. 1998, 18, 5908–5920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [PubMed]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2013, 750, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q. Structural mechanism of DNA-end synapsis in the non-homologous end joining pathway for repairing double-strand breaks: Bridge over troubled ends. Biochem. Soc. Trans. 2019, 47, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 146–152. [Google Scholar] [CrossRef]
- Scutts, S.R.; Ember, S.W.; Ren, H.; Ye, C.; Lovejoy, C.A.; Mazzon, M.; Veyer, D.L.; Sumner, R.P.; Smith, G.L. DNA-PK Is Targeted by Multiple Vaccinia Virus Proteins to Inhibit DNA Sensing. Cell Rep. 2018, 25, 1953–1965. [Google Scholar] [CrossRef] [Green Version]
- Burleigh, K.; Maltbaek, J.H.; Cambier, S.; Green, R.; Gale, M.; James, R.G.; Stetson, D.B. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 2020, 5, eaba4219. [Google Scholar] [CrossRef] [Green Version]
- Lamaa, A.; Le Bras, M.; Skuli, N.; Britton, S.; Frit, P.; Calsou, P.; Prats, H.; Cammas, A.; Millevoi, S. A novel cytoprotective function for the DNA repair protein Ku in regulating p53 mRNA translation and function. EMBO Rep. 2016, 17, 508–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.; Sapkota, G.P.; Morrice, N.; Yu, Y.; Goodarzi, A.A.; Merkle, D.; Meek, K.; Alessi, D.R.; Lees-Miller, S.P. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem. J. 2002, 368, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y.; Suzuki, N.; Namba, N.; Umeda, N.; Ma, X.J.; Morita, A.; Tomita, M.; Enomoto, A.; Serizawa, S.; Hirano, K.; et al. Cleavage and phosphorylation of XRCC4 protein induced by X-irradiation. FEBS Lett. 2000, 478, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.W.; Ye, R.; Veillette, C.J.; Lees-Miller, S.P. DNA-Dependent Protein Kinase Phosphorylation Sites in Ku 70/80 Heterodimer. Biochemistry 1999, 38, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Chan, D.W.; Kobayashi, J.; Burma, S.; Asaithamby, A.; Morotomi-Yano, K.; Botvinick, E.; Qin, J.; Chen, D.J. Cell Cycle Dependence of DNA-dependent Protein Kinase Phosphorylation in Response to DNA Double Strand Breaks. J. Biol. Chem. 2005, 280, 14709–14715. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Pannicke, U.; Lu, H.; Niewolik, D.; Schwarz, K.; Lieber, M.R. The DNA-dependent Protein Kinase Catalytic Subunit Phosphorylation Sites in Human Artemis. J. Biol. Chem. 2005, 280, 33839–33846. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, P.; Piotrowski, J.; Brosh, R.M.; Sommers, J.A.; Miller, S.P.L.; Cheng, W.H.; Snowden, C.M.; Ramsden, D.A.; Bohr, V.A. Werner Protein Is a Target of DNA-dependent Protein Kinase In Vivo and In Vitro, and Its Catalytic Activities Are Regulated by Phosphorylation. J. Biol. Chem. 2002, 277, 18291–18302. [Google Scholar] [CrossRef] [Green Version]
- Kusumoto-Matsuo, R.; Ghosh, D.; Karmakar, P.; May, A.; Ramsden, D.; Bohr, V.A. Serines 440 and 467 in the Werner syndrome protein are phosphorylated by DNA-PK and affects its dynamics in response to DNA double strand breaks. Aging 2014, 6, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wang, W.; Ding, Q.; Ye, R.; Chen, D.; Merkle, D.; Schriemer, D.; Meek, K.; Lees-Miller, S.P. DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair 2003, 2, 1239–1252. [Google Scholar] [CrossRef]
- Leber, R.; Wise, T.W.; Mizuta, R.; Meek, K. The XRCC4 Gene Product Is a Target for and Interacts with the DNA-dependent Protein Kinase. J. Biol. Chem. 1998, 273, 1794–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Critchlow, S.E.; Bowater, R.P.; Jackson, S.P. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol. 1997, 7, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Moghani, A.R.A.; Sharma, M.K.; Matsumoto, Y. In cellulo phosphorylation of DNA double-strand break repair protein XRCC4 on Ser260 by DNA-PK. J. Radiat. Res. 2018, 59, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Dragoi, A.M.; Fu, X.; Ivanov, S.S.; Zhang, P.; Sheng, L.; Wu, D.; Li, G.C.; Chu, W.M. DNA-PKcs, but not TLR9, is required for activation of Akt by CpG-DNA. EMBO J. 2005, 24, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Ji, C.; Yang, B.; Yang, Z.; Gu, H.; Lu, C.C.; Wang, R.; Su, Z.L.; Chen, B.; Sun, W.L.; et al. DNA-dependent protein kinase catalytic subunit (DNA-PKcs)-SIN1 association mediates ultraviolet B (UVB)-induced Akt Ser-473 phosphorylation and skin cell survival. Mol. Cancer 2013, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavuzer, U.; Smith, G.C.; Bliss, T.; Werner, D.; Jackson, S.P. DNA end-independent activation of DNA-PK mediated via association with the DNA-binding protein C1D. Genes Dev. 1998, 12, 2188–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, A.A.; Yu, Y.; Riballo, E.; Douglas, P.; Walker, S.A.; Ye, R.; Härer, C.; Marchetti, C.; Morrice, N.; Jeggo, P.A.; et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 2006, 25, 3880–3889. [Google Scholar] [CrossRef] [Green Version]
- Soubeyrand, S.; Pope, L.; De Chasseval, R.; Gosselin, D.; Dong, F.; De Villartay, J.-P.; Haché, R.J.G. Artemis Phosphorylated by DNA-dependent Protein Kinase Associates Preferentially with Discrete Regions of Chromatin. J. Mol. Biol. 2006, 358, 1200–1211. [Google Scholar] [CrossRef]
- Ting, N.S.Y.; Kao, P.N.; Chan, D.W.; Lintott, L.G.; Lees-Miller, S.P. DNA-dependent protein kinase interacts with antigen receptor response element binding proteins NF90 and NF45. J. Biol. Chem. 1998, 273, 2136–2145. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is Phosphorylated by DNA-PK and Accumulates in the Cytoplasm after DNA Damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef] [Green Version]
- Farber-Katz, S.E.; Dippold, H.C.; Buschman, M.D.; Peterman, M.C.; Xing, M.; Noakes, C.J.; Tat, J.; Ng, M.M.; Rahajeng, J.; Cowan, D.M.; et al. DNA Damage Triggers Golgi Dispersal via DNA-PK and GOLPH3. Cell 2014, 156, 413–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chibazakura, T.; Watanabe, F.; Kitajima, S.; Tsukada, K.; Yasukochi, Y.; Teraoka, H. Phosphorylation of human general transcription factors TATA-binding protein and transcription factor IIB by DNA-dependent protein kinase—Synergistic stimulation of RNA polymerase II basal transcription in vitro. JBIC J. Boil. Inorg. Chem. 1997, 247, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Chan, D.W.; Park, J.-H.; Oettinger, M.A.; Kwon, J. DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res. 2003, 31, 6819–6827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Huang, Y.C.; Xu, Q.Z.; Zhou, L.J.; Shang, Z.F.; Huang, B.; Wang, Y.; Liu, X.D.; Wu, D.C.; Zhou, P.K. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol. Biol. 2010, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Koike, M.; Sugasawa, J.; Yasuda, M.; Koike, A. Tissue-specific DNA-PK-dependent H2AX phosphorylation and γ-H2AX elimination after X-irradiation in vivo. Biochem. Biophys. Res. Commun. 2008, 376, 52–55. [Google Scholar] [CrossRef]
- Wang, H.; Wang, M.; Wang, H.; Böcker, W.; Iliakis, G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J. Cell. Physiol. 2005, 202, 492–502. [Google Scholar] [CrossRef]
- Reitsema, T.; Klokov, D.; Banáth, J.P.; Olive, P.L. DNA-PK is responsible for enhanced phosphorylation of histone H2AX under hypertonic conditions. DNA Repair 2005, 4, 1172–1181. [Google Scholar] [CrossRef]
- Sui, J.; Lin, Y.-F.; Xu, K.; Lee, K.-J.; Wang, D.; Chen, B.P. DNA-PKcs phosphorylates hnRNP-A1 to facilitate the RPA-to-POT1 switch and telomere capping after replication. Nucleic Acids Res. 2015, 43, 5971–5983. [Google Scholar] [CrossRef] [Green Version]
- Berglund, F.M.; Clarke, P.R. hnRNP-U is a specific DNA-dependent protein kinase substrate phosphorylated in response to DNA double-strand breaks. Biochem. Biophys. Res. Commun. 2009, 381, 59–64. [Google Scholar] [CrossRef]
- Britton, S.; Froment, C.; Frit, P.; Monsarrat, B.; Salles, B.; Calsou, P. Cell nonhomologous end joining capacity controls SAF-A phosphorylation by DNA-PK in response to DNA double-strand breaks inducers. Cell Cycle 2009, 8, 3717–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.; Hunt, T.; Jackson, R.; Anderson, C. Double-stranded DNA induces the phosphorylation of several proteins including the 90 000 mol. wt. heat-shock protein in animal cell extracts. EMBO J. 1985, 4, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Quanz, M.; Herbette, A.; Sayarath, M.; De Koning, L.; Dubois, T.; Sun, J.-S.; Dutreix, M. Heat Shock Protein 90α (Hsp90α) Is Phosphorylated in Response to DNA Damage and Accumulates in Repair Foci. J. Biol. Chem. 2012, 287, 8803–8815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees-Miller, S.P.; Anderson, C.W. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J. Biol. Chem. 1989, 264, 17275–17280. [Google Scholar]
- Bannister, A.J.; Gottlieb, T.M.; Kouzarides, T.; Jackson, S.P. c-Jun is phosphorylated by the DNA-dependent protein kinase in vitro; definition of the minimal kinase recognition motif. Nucleic Acids Res. 1993, 21, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Mayo, L.D.; Turchi, J.J.; Berberich, S.J. Mdm-2 phosphorylation by DNA-dependent protein kinase prevents interaction with p53. Cancer Res. 1997, 57, 5013–5016. [Google Scholar]
- Iijima, S.; Teraoka, H.; Date, T.; Tsukada, K. DNA-activated protein kinase in Raji Burkitt’s lymphoma cells. Eur. J. Biochem. 1992, 206, 595–603. [Google Scholar] [CrossRef]
- Liu, L.; Kwak, Y.-T.; Bex, F.; Garcia-Martinez, L.F.; Li, X.-H.; Meek, K.; Lane, W.S.; Gaynor, R.B. DNA-Dependent Protein Kinase Phosphorylation of IκBα and IκBβ Regulates NF-κB DNA Binding Properties. Mol. Cell. Biol. 1998, 18, 4221–4234. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Mahaney, B.L.; Yano, K.-I.; Ye, R.; Fang, S.; Douglas, P.; Chen, D.J.; Lees-Miller, S.P. DNA-PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair 2008, 7, 1680–1692. [Google Scholar] [CrossRef] [Green Version]
- Malewicz, M.; Kadkhodaei, B.; Kee, N.; Volakakis, N.; Hellman, U.; Viktorsson, K.; Leung, C.Y.; Chen, B.; Lewensohn, R.; Van Gent, D.C.; et al. Essential role for DNA-PK-mediated phosphorylation of NR4A nuclear orphan receptors in DNA double-strand break repair. Genes Dev. 2011, 25, 2031–2040. [Google Scholar] [CrossRef] [Green Version]
- Ariumi, Y.; Masutani, M.; Copeland, T.D.; Mimori, T.; Sugimura, T.; Shimotohno, K.; Ueda, K.; Hatanaka, M.; Noda, M. Suppression of the poly(ADP-ribose) polymerase activity by DNA-dependent protein kinase in vitro. Oncogene 1999, 18, 4616–4625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolner, A.E.; Abdou, I.; Ye, R.; Mani, R.S.; Fanta, M.; Yu, Y.; Douglas, P.; Tahbaz, N.; Fang, S.; Dobbs, T.; et al. Phosphorylation of polynucleotide kinase/ phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage. Nucleic Acids Res. 2011, 39, 9224–9237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre-Moreno, G.; Pryor, J.M.; Moreno-Oñate, M.; Herrero-Ruiz, A.M.; Cortés-Ledesma, F.; Blanco, L.; Ramsden, D.A.; Ruiz, J.F. Regulation of human polλ by ATM-mediated phosphorylation during non-homologous end joining. DNA Repair 2017, 51, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.R.; Dvir, A.; Anderson, C.W.; Dynan, W.S. DNA binding provides a signal for phosphorylation of the RNA polymerase II heptapeptide repeats. Genes Dev. 1992, 6, 426–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schild-Poulter, C.; Shih, A.; Yarymowich, N.C.; Haché, R.J.G. Down-regulation of histone H2B by DNA-dependent protein kinase in response to DNA damage through modulation of octamer transcription factor 1. Cancer Res. 2003, 63, 7197–7205. [Google Scholar] [PubMed]
- Block, W.D.; Yu, Y.; Lees-Miller, S.P. Phosphatidyl inositol 3-kinase-like serine/threonine protein kinases (PIKKs) are required for DNA damage-induced phosphorylation of the 32 kDa subunit of replication protein A at threonine 21. Nucleic Acids Res. 2004, 32, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Liaw, H.J.; Lee, D.; Myung, K. DNA-PK-Dependent RPA2 Hyperphosphorylation Facilitates DNA Repair and Suppresses Sister Chromatid Exchange. PLoS ONE 2011, 6, e21424. [Google Scholar] [CrossRef]
- Shao, R.; Cao, C.; Zhang, H.; Kohn, K.W.; Wold, M.S.; Pommier, Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999, 18, 1397–1406. [Google Scholar] [CrossRef]
- Jackson, S.P.; Macdonald, J.J.; Lees-Miller, S.; Tjian, R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell 1990, 63, 155–165. [Google Scholar] [CrossRef]
- Liu, S.H.; Ma, J.T.; Yueh, A.Y.; Lees-Miller, S.P.; Anderson, C.W.; Ng, S.Y. The carboxyl-terminal transactivation domain of human serum response factor contains DNA-activated protein kinase phosphorylation sites. J. Biol. Chem. 1993, 268, 21147–21154. [Google Scholar] [PubMed]
- Komiyama, S.; Taniguchi, S.; Matsumoto, Y.; Tsunoda, E.; Ohto, T.; Suzuki, Y.; Yin, H.L.; Tomita, M.; Enomoto, A.; Morita, A.; et al. Potentiality of DNA-dependent protein kinase to phosphorylate Ser46 of human p53. Biochem. Biophys. Res. Commun. 2004, 323, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Lees-Miller, S.P.; Sakaguchi, K.; Ullrich, S.J.; Appella, E.; Anderson, C.W. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol. Cell. Biol. 1992, 12, 5041–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; He, Y.; Hu, Y.; Lu, H.; Cao, Z.; Yi, X.; Wang, J. Real-time surface plasmon resonance monitoring of site-specific phosphorylation of p53 protein and its interaction with MDM2 protein. Analyst 2019, 144, 6033–6040. [Google Scholar] [CrossRef]
- Tomimatsu, N.; Mukherjee, B.; Burma, S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009, 10, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Bunch, H.; Lawney, B.P.; Lin, Y.-F.; Asaithamby, A.; Murshid, A.; Wang, Y.E.; Chen, B.P.C.; Calderwood, S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yannone, S.M.; Roy, S.; Chan, D.W.; Murphy, M.B.; Huang, S.; Campisi, J.; Chen, D.J. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J. Biol. Chem. 2001, 276, 38242–38248. [Google Scholar] [CrossRef]
- Douglas, P.; Gupta, S.; Morrice, N.; Meek, K.; Lees-Miller, S.P. DNA-PK-dependent phosphorylation of Ku70/80 is not required for non-homologous end joining. DNA Repair 2005, 4, 1006–1018. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 2010, 920161. [Google Scholar] [CrossRef] [Green Version]
- DAVID Functional Annotation Bioinformatics Microarray Analysis. Available online: https://david.ncifcrf.gov/ (accessed on 28 March 2020).
- STRING: Functional Protein Association Networks. Available online: https://string-db.org/ (accessed on 28 March 2020).
- Ju, B.-G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A Topoisomerase IIβ-Mediated dsDNA Break Required for Regulated Transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Bunch, H. Role of genome guardian proteins in transcriptional elongation. FEBS Lett. 2016, 590, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc Regulates Transcriptional Pause Release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffner, M.C.; De Marzo, A.M.; Meeker, A.K.; Nelson, W.G.; Yegnasubramanian, S. Transcription-induced DNA double strand breaks: Both oncogenic force and potential therapeutic target? Clin. Cancer Res. 2011, 17, 3858–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankotai, T.; Bonhomme, C.; Chen, D.; Soutoglou, E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 2012, 19, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Iannelli, F.; Galbiati, A.; Capozzo, I.; Nguyen, Q.; Magnuson, B.; Michelini, F.; D’Alessandro, G.; Cabrini, M.; Roncador, M.; Francia, S.; et al. A damaged genome’s transcriptional landscape through multilayered expression profiling around in situ-mapped DNA double-strand breaks. Nat. Commun. 2017, 8, 15656. [Google Scholar] [CrossRef]
- Janssens, S.; Tschopp, J. Signals from within: The DNA-damage-induced NF-kappaB response. Cell Death Differ. 2006, 13, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Sutton, J.; Dotson, D.; Dong, X. Molecular Events in Late Stages of HIV-1 Replication. JSM Microbiol. 2013, 1, 1004. [Google Scholar]
- Daniel, R.; Katz, R.A.; Merkel, G.; Hittle, J.C.; Yen, T.J.; Skalka, A.M. Wortmannin Potentiates Integrase-Mediated Killing of Lymphocytes and Reduces the Efficiency of Stable Transduction by Retroviruses. Mol. Cell. Biol. 2001, 21, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Daniel, R.; Katz, R.A.; Skalka, A.M. A Role for DNA-PK in Retroviral DNA Integration. Science 1999, 284, 644–647. [Google Scholar] [CrossRef]
- Daniel, R.; Greger, J.G.; Katz, R.A.; Taganov, K.D.; Wu, X.; Kappes, J.C.; Skalka, A.M. Evidence that Stable Retroviral Transduction and Cell Survival following DNA Integration Depend on Components of the Nonhomologous End Joining Repair Pathway. J. Virol. 2004, 78, 8573–8581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeanson, L.; Subra, F.; Vaganay, S.; Hervy, M.; Marangoni, E.; Bourhis, J.; Mouscadet, J.F. Effect of Ku80 Depletion on the Preintegrative Steps of HIV-1 Replication in Human Cells. Virology 2002, 300, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waninger, S.; Kuhen, K.; Hu, X.; Chatterton, J.E.; Wong-Staal, F.; Tang, H.; Yang, X.; Tomov, V.; Kurteva, S.; Wang, L.; et al. Identification of Cellular Cofactors for Human Immunodeficiency Virus Replication via a Ribozyme-Based Genomics Approach. J. Virol. 2004, 78, 12829–12837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, Y.; Komatsu, K.; Agematsu, K.; Matsuoka, M. DNA double strand break repair enzymes function at multiple steps in retroviral infection. Retrovirology 2009, 6, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manic, G.; Maurin-Marlin, A.; Laurent, F.; Vitale, I.; Thierry, S.; Delelis, O.; Dessen, P.; Vincendeau, M.; Leib-Mösch, C.; Hazan, U.; et al. Impact of the Ku Complex on HIV-1 Expression and Latency. PLoS ONE 2013, 8, e69691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Swinbank, K.M.; Ahmed, P.S.; Taylor, D.L.; Jackson, S.P.; Smith, G.C.M.; O’Connor, M.J. Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat. Cell Biol. 2005, 7, 493–500. [Google Scholar] [CrossRef]
- Yang, J.; Yu, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J. ATM, ATR and DNA-PK: Initiators of the cellular genotoxic stress responses. Carcinogenesis 2003, 24, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Wang, F.X.; Zhang, H.; Wu, K.J.; Williams, K.J.; Daniel, R. Evidence that the Nijmegen breakage syndrome protein, an early sensor of double-strand DNA breaks (DSB), is involved in HIV-1 post-integration repair by recruiting the ataxia telangiectasia-mutated kinase in a process similar to, but distinct from, cellular DSB repair. Virol. J. 2008, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Borgatti, P.; Zauli, G.; Colamussi, M.L.; Gibellini, D.; Previati, M.; Cantley, L.C.; Capitani, S. Extracellular HIV-1 Tat protein activates phosphatidylinositol 3- and Akt/PKB kinases in CD4+ T lymphoblastoid Jurkat cells. Eur. J. Immunol. 1997, 27, 2805–2811. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-Scale RNAi Screen for Host Factors Required for HIV Replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Dehart, J.L.; Andersen, J.L.; Zimmerman, E.S.; Ardon, O.; An, D.S.; Blackett, J.; Kim, B.; Planelles, V. The Ataxia Telangiectasia-Mutated and Rad3-Related Protein Is Dispensable for Retroviral Integration. J. Virol. 2005, 79, 1389–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espeseth, A.S.; Fishel, R.; Hazuda, D.; Huang, Q.; Xu, M.; Yoder, K.; Zhou, H. siRNA Screening of a Targeted Library of DNA Repair Factors in HIV Infection Reveals a Role for Base Excision Repair in HIV Integration. PLoS ONE 2011, 6, e17612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, S.; Singh, G.; Bolinger, C.; Song, Z.; Boeras, I.; Weng, K.; Trent, B.; Brown, W.C.; Singh, K.; Boris-Lawrie, K.; et al. Virion-associated, host-derived DHX9/RNA helicase A enhances the processivity of HIV-1 reverse transcriptase on genomic RNA. J. Biol. Chem. 2019, 294, 11473–11485. [Google Scholar] [CrossRef] [PubMed]
- Boeras, I.; Song, Z.; Moran, A.; Franklin, J.; Brown, W.C.; Johnson, M.; Boris-Lawrie, K.; Heng, X. DHX9/RHA Binding to the PBS-Segment of the Genomic RNA during HIV-1 Assembly Bolsters Virion Infectivity. J. Mol. Biol. 2016, 428, 2418–2429. [Google Scholar] [CrossRef] [Green Version]
- Ndzinu, J.K.; Takeuchi, H.; Saito, H.; Yoshida, T.; Yamaoka, S. eIF4A2 is a host factor required for efficient HIV-1 replication. Microbes Infect. 2018, 20, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Krasnopolsky, S.; Marom, L.; Victor, R.A.; Kuzmina, A.; Schwartz, J.C.; Fujinaga, K.; Taube, R. Fused in sarcoma silences HIV gene transcription and maintains viral latency through suppressing AFF4 gene activation. Retrovirology 2019, 16, 16. [Google Scholar] [CrossRef] [Green Version]
- Daniel, R.; Ramcharan, J.; Rogakou, E.; Taganov, K.D.; Greger, J.G.; Bonner, W.; Nussenzweig, A.; Katz, R.A.; Skalka, A.M. Histone H2AX Is Phosphorylated at Sites of Retroviral DNA Integration but Is Dispensable for Postintegration Repair. J. Biol. Chem. 2004, 279, 45810–45814. [Google Scholar] [CrossRef] [Green Version]
- Monette, A.; Ajamian, L.; López-Lastra, M.; Mouland, A.J. Human Immunodeficiency Virus Type 1 (HIV-1) Induces the Cytoplasmic Retention of Heterogeneous Nuclear Ribonucleoprotein A1 by Disrupting Nuclear Import: Implications for HIV-1 gene expression. J. Biol. Chem. 2009, 284, 31350–31362. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, T.; Ishizaka, A.; Suzuki, Y.; Iba, H. 7SK small nuclear ribonucleoprotein complex is recruited to the HIV-1 promoter via short viral transcripts. FEBS Lett. 2014, 588, 1630–1636. [Google Scholar] [CrossRef]
- Low, J.S.; Fassati, A. Hsp90: A chaperone for HIV-1. Parasitology 2014, 141, 1192–1202. [Google Scholar] [CrossRef]
- Van Der Sluis, R.M.; Derking, R.; Breidel, S.; Speijer, D.; Berkhout, B.; Jeeninga, R.E. Interplay between viral Tat protein and c-Jun transcription factor in controlling LTR promoter activity in different human immunodeficiency virus type I subtypes. J. Gen. Virol. 2014, 95, 968–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, Y.; Desrosiers, V.; Ouellet, M.; Deshiere, A.; Torresilla, C.; Cohen, É.A.; Tremblay, M.J. Expression of MDM2 in Macrophages Promotes the Early Postentry Steps of HIV-1 Infection through Inhibition of p53. J. Virol. 2019, 93, e01871-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, T.; Takaori-Kondo, A.; Shirakawa, K.; Higashitsuji, H.; Itoh, K.; Io, K.; Matsui, M.; Iwai, K.; Kondoh, H.; Sato, T.; et al. MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology 2009, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brès, V.; Kiernan, R.E.; Linares, L.K.; Chable-Bessia, C.; Plechakova, O.; Treand, C.; Emiliani, S.; Peloponese, J.-M.; Jeang, K.-T.; Coux, O.; et al. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 2003, 5, 754–761. [Google Scholar] [CrossRef]
- Sun, Y.; Clark, E.C. Expression of the c-myc Proto-oncogene Is Essential for HIV-1 Infection in Activated T Cells. J. Exp. Med. 1999, 189, 1391–1398. [Google Scholar] [CrossRef]
- Jiang, G.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. c-Myc and Sp1 Contribute to Proviral Latency by Recruiting Histone Deacetylase 1 to the Human Immunodeficiency Virus Type 1 Promoter. J. Virol. 2007, 81, 10914–10923. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, G.; Zaikos, T.D.; Khan, S.Z.; Jacobi, A.M.; Behlke, M.A.; Zeichner, S.L. Targeting IκB Proteins for HIV Latency Activation: The Role of Individual IκB and NF-κB Proteins. J. Virol. 2013, 87, 3966–3978. [Google Scholar] [CrossRef] [Green Version]
- Kameoka, M.; Nukuzuma, S.; Itaya, A.; Tanaka, Y.; Ota, K.; Ikuta, K.; Yoshihara, K. RNA Interference Directed against Poly(ADP-Ribose) Polymerase 1 Efficiently Suppresses Human Immunodeficiency Virus Type 1 Replication in Human Cells. J. Virol. 2004, 78, 8931–8934. [Google Scholar] [CrossRef] [Green Version]
- Kameoka, M.; Nukuzuma, S.; Itaya, A.; Tanaka, Y.; Ota, K.; Inada, Y.; Ikuta, K.; Yoshihara, K. Poly(ADP-ribose)polymerase-1 is required for integration of the human immunodeficiency virus type 1 genome near centromeric alphoid DNA in human and murine cells. Biochem. Biophys. Res. Commun. 2005, 334, 412–417. [Google Scholar] [CrossRef]
- Gäken, J.A.; Tavassoli, M.; Gan, S.U.; Vallian, S.; Giddings, I.; Darling, D.C.; Galea-Lauri, J.; Thomas, M.G.; Abedi, H.; Schreiber, V.; et al. Efficient retroviral infection of mammalian cells is blocked by inhibition of poly(ADP-ribose) polymerase activity. J. Virol. 1996, 70, 3992–4000. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.C.; Juluri, K.; Zhou, Y.; Leung, S.; Hermankova, M.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proc. Natl. Acad. Sci. USA 2001, 98, 3364–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siva, A.C.; Bushman, F.D. Poly(ADP-Ribose) Polymerase 1 Is Not Strictly Required for Infection of Murine Cells by Retroviruses. J. Virol. 2002, 76, 11904–11910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariumi, Y.; Turelli, P.; Masutani, M.; Trono, D. DNA Damage Sensors ATM, ATR, DNA-PKcs, and PARP-1 Are Dispensable for Human Immunodeficiency Virus Type 1 Integration. J. Virol. 2005, 79, 2973–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baekelandt, V.; Claeys, A.; Cherepanov, P.; De Clercq, E.; De Strooper, B.; Nuttin, B.; Debyser, Z.; Brown, A.J.L.; Precious, H.M.; Whitcomb, J.M.; et al. DNA-Dependent Protein Kinase Is Not Required for Efficient Lentivirus Integration. J. Virol. 2000, 74, 11278–11285. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Liu, R.; Yang, G.; Zhou, Q. The PARP1-Siah1 Axis Controls HIV-1 Transcription and Expression of Siah1 Substrates. Cell Rep. 2018, 23, 3741–3749. [Google Scholar] [CrossRef]
- Bueno, M.T.D.; Reyes, D.; Valdes, L.; Saheba, A.; Urias, E.; Mendoza, C.; Fregoso, O.I.; Llano, M. Poly(ADP-Ribose) Polymerase 1 Promotes Transcriptional Repression of Integrated Retroviruses. J. Virol. 2013, 87, 2496–2507. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-Z.; Latchman, D.S. The octamer-binding proteins Oct-1 and Oct-2 repress the HIV long terminal repeat promoter and its transactivation by Tat. Biochem. J. 1997, 322, 155–158. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Iqbal, M.; Tariq, M.; Baig, S.M.; Abbas, W. Epigenetic regulation of HIV-1 latency: Focus on polycomb group (PcG) proteins. Clin. Epigenetics 2018, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Colin, L.; Verdin, E.; Van Lint, C. HIV-1 Chromatin, Transcription, and the Regulatory Protein Tat. Methods Mol. Biol. 2014, 1087, 85–101. [Google Scholar] [CrossRef]
- Majello, B.; Napolitano, G.; Lania, L. Recruitment of the TATA-binding protein to the HIV-1 promoter is a limiting step for Tat transactivation. AIDS 1998, 12, 1957–1964. [Google Scholar] [CrossRef]
- Raha, T.; Cheng, S.W.G.; Green, M.R. HIV-1 Tat Stimulates Transcription Complex Assembly through Recruitment of TBP in the Absence of TAFs. PLoS Biol. 2005, 3, e44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, B.; Sharifi, H.J.; DiGrigoli, S.; Kinnetz, M.; Mellon, K.; Hu, W.; De Noronha, C.M. Inhibition of HIV early replication by the p53 and its downstream gene p21. Virol. J. 2018, 15, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnetz, M.; Alghamdi, F.; Racz, M.; Hu, W.; Shi, B. The impact of p53 on the early stage replication of retrovirus. Virol. J. 2017, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, C.-H.; Kim, S.-Y.; Byeon, S.E.; Jeong, Y.; Lee, J.; Kim, K.P.; Park, J.; Bae, Y.S. p53-Derived Host Restriction of HIV-1 Replication by Protein Kinase R-Mediated Tat Phosphorylation and Inactivation. J. Virol. 2015, 89, 4262–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargonetti, J.; Chicas, A.; White, D.; Prives, C. p53 represses Sp1 DNA binding and HIV-LTR directed transcription. Cell. Mol. Biol. 1997, 43, 935–949. [Google Scholar]
- Chicas, A.; Molina, P.; Bargonetti, J. Mutant p53 Forms a Complex with Sp1 on HIV-LTR DNA. Biochem. Biophys. Res. Commun. 2000, 279, 383–390. [Google Scholar] [CrossRef]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef]
- Fernández-Ortega, C.; Ramírez, A.; Casillas, D.; Paneque, T.; Ubieta, R.; Dubed, M.; Navea, L.; Castellanos-Serra, L.; Duarte, C.A.; Falcón-Cama, V.; et al. Identification of Vimentin as a Potential Therapeutic Target against HIV Infection. Viruses 2016, 8, 98. [Google Scholar] [CrossRef]
- Sharma, A.; Awasthi, S.; Harrod, C.K.; Matlock, E.F.; Khan, S.; Xu, L.; Chan, S.; Yang, H.; Thammavaram, C.K.; Rasor, R.A.; et al. The Werner Syndrome Helicase Is a Cofactor for HIV-1 Long Terminal Repeat Transactivation and Retroviral Replication. J. Biol. Chem. 2007, 282, 12048–12057. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, T.; Ishizaka, A.; Furuichi, Y. The Werner Protein Acts as a Coactivator of Nuclear Factor κB (NF-κB) on HIV-1 and Interleukin-8 (IL-8) Promoters. J. Biol. Chem. 2015, 290, 18391–18399. [Google Scholar] [CrossRef] [Green Version]
- Anisenko, A.N.; Gottikh, M.B. Role of Cellular DNA Repair Systems in HIV-1 Replication. Mol. Biol. 2019, 53, 313–322. [Google Scholar] [CrossRef]
- Gutiérrez, D.A.; Valdes, L.; Serguera, C.; Llano, M. Poly(ADP-ribose) polymerase-1 silences retroviruses independently of viral DNA integration or heterochromatin formation. J. Gen. Virol. 2016, 97, 1686–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rom, S.; Reichenbach, N.L.; Dykstra, H.; Persidsky, Y. The dual action of poly(ADP-ribose) polymerase -1 (PARP-1) inhibition in HIV-1 infection: HIV-1 LTR inhibition and diminution in Rho GTPase activity. Front. Microbiol. 2015, 6, 878. [Google Scholar] [CrossRef] [PubMed]
- Parent, M.; Yung, T.M.C.; Rancourt, A.; Ho, E.L.Y.; Vispé, S.; Suzuki-Matsuda, F.; Uehara, A.; Wada, T.; Handa, H.; Satoh, M.S.; et al. Poly(ADP-ribose) Polymerase-1 Is a Negative Regulator of HIV-1 Transcription through Competitive Binding to TAR RNA with Tat Positive Transcription Elongation Factor b (p-TEFb) Complex. J. Biol. Chem. 2005, 280, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Delling, U.; Chen, C.H.; Rosen, C.A.; Sonenberg, N. A bulge structure in HIV-1 TAR RNA is required for Tat binding and Tat-mediated trans-activation. Genes Dev. 1990, 4, 1365–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, H.; Lis, J.T. Control of Transcriptional Elongation. Annu. Rev. Genet. 2013, 47, 483–508. [Google Scholar] [CrossRef] [Green Version]
- Shadrina, O.A.; Knyazhanskaya, E.S.; Korolev, S.P.; Gottikh, M. Host Proteins Ku and HMGA1 As Participants of HIV-1 Transcription. Acta Nat. 2016, 8, 34–47. [Google Scholar] [CrossRef]
- Maldonado, E.; Shiekhattar, R.; Sheldon, M.; Cho, H.; Drapkin, R.; Rickert, P.; Lees, E.; Anderson, C.W.; Linn, S.; Reinberg, D. A human RNA polymerase II complex associated with SRB and DNA-repair proteins. Nature 1996, 381, 86–89. [Google Scholar] [CrossRef]
- Kaczmarski, W.; Khan, S.A. Lupus Autoantigen Ku Protein Binds HIV-1 TAR RNA in Vitro. Biochem. Biophys. Res. Commun. 1993, 196, 935–942. [Google Scholar] [CrossRef]
- Shadrina, O.; Garanina, I.; Korolev, S.; Zatsepin, T.; Van Assche, J.; Daouad, F.; Wallet, C.; Rohr, O.; Gottikh, M. Analysis of RNA binding properties of human Ku protein reveals its interactions with 7SK snRNA and protein components of 7SK snRNP complex. Biochime 2020, 171–172, 110–123. [Google Scholar] [CrossRef]
- Chun, R.; Semmes, O.J.; Neuveut, C.; Jeang, K.T. Modulation of Sp1 Phosphorylation by Human Immunodeficiency Virus Type 1 Tat. J. Virol. 1998, 72, 2615–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.; Ochem, A.; Tyagi, M. DNA-dependent protein kinase interacts functionally with the RNA polymerase II complex recruited at the human immunodeficiency virus (HIV) long terminal repeat and plays an important role in HIV gene expression. J. Gen. Virol. 2011, 92, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.Y.; Khachigian, L.M. Sp1 Phosphorylation and Its Regulation of Gene Transcription. Mol. Cell. Biol. 2009, 29, 2483–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
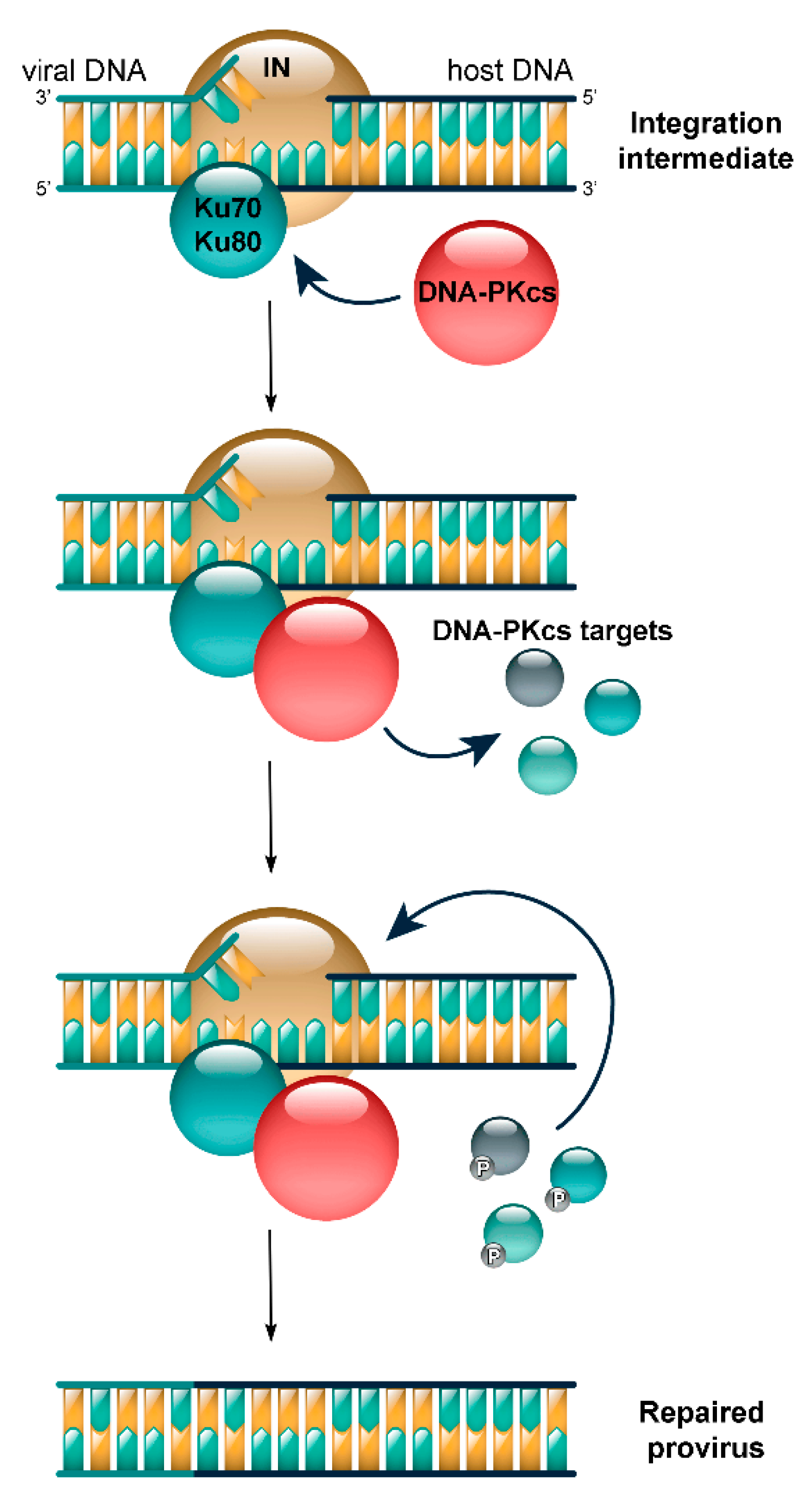
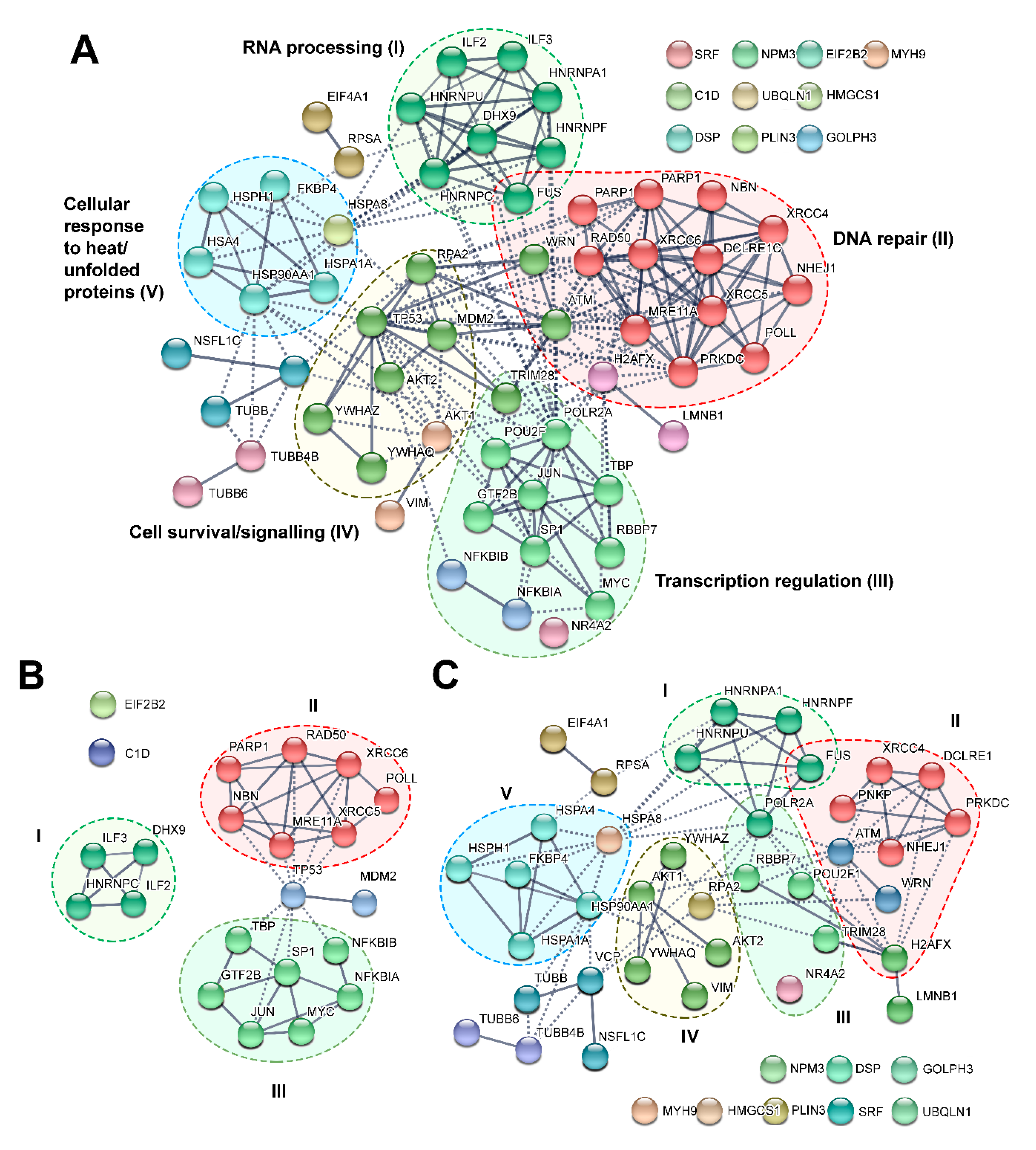

{kind=link}
{kind=link}
| Gene Name | Protein Name | Type of Experiment | DNA-PKcs Activation Method | Phosphorylation Event Validation | Identified Phosphorylation Sites | Effect of Phosphorylation (In Vivo) | Ref. |
|---|---|---|---|---|---|---|---|
| Akt1; Akt2; Akt3 | RAC-alpha serine/threonine-protein kinase | in vitro/in vivo | CpG ODN, UVB | Phosphorylated Akts activity; WB; 32P-incorporation | AKT1: T308, S473; AKT2: T309 | Cell survival after UVB treatment, Akt translocation to nucleus after CpG ODN treatment | [44,45] |
| ATM | Serine-protein kinase ATM | in vitro/in vivo | bleomycin | WB; 32P-incorporation; mutagenesis | S85/T86, T372/T373 and S1985/T1987/T1988 | Negative regulation of ATM | [19] |
| C1D | Nuclear nucleic acid-binding protein C1D | in vitro | C1D, dsDNA | 32P-incorporation | [46] | ||
| DCLRE1C | Protein Artemis | in vitro/in vivo | dsDNA, bleomycin | MS; changes in gel mobility of phosphorylated forms; 32P-incorporation; WB | S385, T410, S417, S503, S509, S516, S518, S572, S589, T601, S645, T676, S679, S688, T692 | Increase Artemis association with chromatin | [37,47,48] |
| DHX9 | ATP-dependent RNA helicase A | in vitro | poly(rG) | 32P-incorporation | [9] | ||
| DSP | Desmoplakin | in vivo | Dbait32H | ProQ-Diamond staining + MS | [16] | ||
| EIF2B2 | Translation initiation factor eIF-2B subunit beta | in vitro | dsDNA | 32P-incorporation | [49] | ||
| EIF4A1 | Eukaryotic initiation factor 4A-I | in vivo | Dbait32H | ProQ-Diamond staining + MS | [16] | ||
| FKBP4 | Peptidyl-prolyl cis-trans isomerase FKBP4 | in vivo | Dbait32H | ProQ-Diamond staining + MS | [16] | ||
| FUS | RNA-binding protein FUS | in vitro/in vivo | Calicheamicin γ1, Dbait32H | WB; changes in gel mobility of phosphorylated forms; mutagenesis | S/T-Q located in N-terminal region of FUS (1–165 aa) | Translocation to cytoplasm | [50] |
| GOLPH3 | Golgi phosphoprotein 3 | in vitro/in vivo | Camptothecin, doxorubicin, IR | MS; 32P-incorporation | T143 | Cell survival following DNA damage | [51] |
| GTF2B | Transcription initiation factor IIB | in vitro | dsDNA | 32P-incorporation | [52] | ||
| H2AFX | Histone H2AX | in vitro/in vivo | dsDNA, IR, Dbait32H | WB; IF; changes in gel mobility of phosphorylated form | S139 | Assembly of DNA repair proteins at the DNA-damage sites | [16,53,54,55,56,57,58] |
| HMGCS1 | Hydroxymethylglutaryl-CoA synthase, cytoplasmic | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| HNRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 | in vitro/in vivo | dsDNA, hTR, hnRNP | 32P-incorporation; WB; mutagenesis | S95, S192 | Essential for capping of the newly replicated telomeres and prevention of telomeric aberrations | [5,9,59] |
| HNRNPC | Heterogeneous nuclear ribo-nucleoproteins C1/C2 | in vitro | hnRNP | 32P-incorporation | [9] | ||
| HNRNPF | Heterogeneous nuclear ribo-nucleoprotein F | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| HNRNPU | Heterogeneous nuclear ribo-nucleoprotein U | in vitro/in vivo | dsDNA, etoposide, Calicheamicin γ1 | MS; WB; changes in gel mobility of phosphorylated form | S59 | [60,61] | |
| HSP90AA1 | Heat shock protein HSP 90-alpha | in vitro/in vivo | dsDNA; Dbait32H; IR | ProQ-Diamond + MS; 32P-incorporation; WB | T5, T7 | pThr7-HSP90α accumulates at repair foci, that is necessary for maintenance of γ-H2AX Foci and efficient DNA repair | [16,62,63,64] |
| HSPA1A | Heat shock 70 kDa protein 1A | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| HSPA4 | Heat shock 70 kDa protein 4 | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| HSPA8 | Heat shock cognate 71 kDa protein | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| HSPH1 | Heat shock protein 105 kDa | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| ILF2 | Interleukin enhancer-binding factor 2 | in vitro | dsDNA | 32P-incorporation | [49] | ||
| ILF3 | Interleukin enhancer-binding factor 3 | in vitro | dsDNA | 32P-incorporation | [49] | ||
| JUN | Transcription factor AP-1 | in vitro | dsDNA | 32P-incorporation; mutagenesis | S249 | [65] | |
| LMNB1 | Lamin-B1 | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| MDM2 | E3 ubiquitin-protein ligase Mdm2 | in vitro | dsDNA | 32P-incorporation; mutagenesis | S17 | Mdm-2 Phosphorylation by DNA-PK Prevents Interaction with p53 | [66] |
| MRE11 | Double-strand break repair protein MRE11 | in vitro | dsDNA | 32P-incorporation | [19] | ||
| MYC | Myc proto-oncogene protein | in vitro | dsDNA | 32P-incorporation | [67] | ||
| MYH9 | Myosin-9 | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| NBN | Nibrin | in vitro | dsDNA | 32P-incorporation | [19] | ||
| NFKBIA | NF-kappa-B inhibitor alpha | in vitro | 32P-incorporation; MS | S36, T273 | [68] | ||
| NFKBIB | NF-kappa-B inhibitor beta | in vitro | 32P-incorporation | [68] | |||
| NHEJ1 | Non-homologous end-joining factor 1 (XLF) | in vitro/in vivo | dsDNA, IR | MS; WB; 32P-incorporation | S245 | Dispensable for DSB repair | [69] |
| NPM3 | Nucleoplasmin-3 | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| NR4A2 | Nuclear receptor subfamily 4 group A member 3 | in vitro/in vivo | dsDNA, IR | WB; MS; IF; mutagenesis | S337 | Promotes DSB repair | [70] |
| NSFL1C | NSFL1 cofactor p47 | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| PARP1 | Poly-(ADP-ribose) polymerase 1 | in vitro | dsDNA | 32P-incorporation | [71] | ||
| PLIN3 | Perilipin-3 | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| PNKP | Bifunctional polynucleotide phosphatase/kinase | in vitro/in vivo | IR | WB; MS; 32P-incorporation; mutagenesis | S114, S126 | Regulates DSB repair | [72] |
| POLL | DNA polymerase lambda, involved in BER, NHEJ and HR | in vitro | dsDNA | WB; 32P-incorporation; mutagenesis | T204 | [73] | |
| POLR2A | DNA-directed RNA polymerase II subunit RPB1 | in vitro/in vivo | dsDNA; unknown transcriptiona signal | WB; 32P-incorporation | Heptapeptide repeats of CTD; S2; S5 | Increase transcription efficiency | [14,74] |
| POU2F1 | POU domain, class 2, transcription factor 1 (octamer transcription factor 1, Oct-1) | in vitro/in vivo | dsDNA, IR, zeocin | 32P-incorporation | Stabilizes Oct-1, decreases Oct-1 dependent transcription | [75] | |
| PRKDC | DNA-dependent protein kinase catalytic subunit | in vivo | Dbait32H | WB; IF; MS; 32P-incorporation; mutagenesis | S2056, T2609, S2612, T2620, S2624, T2638, T2647; S3205; S3821; S4046; T4102 | [16,33,36,37] | |
| RAD50 | DNA repair protein RAD50 | in vitro | dsDNA | 32P-incorporation | [19] | ||
| RBBP7 | Histone-binding protein RBBP7 | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| RPA2 | Replication protein A 32 kDa subunit | in vitro/in vivo | Camptothecin, UV, 4NQO, Etoposide | WB; mutagenesis; 32P-incorporation; changes in gel mobility of phosphorylated forms | S4, S8, T21 | Regulates fork restart, new origin firing, HR, mitotic catastrophe, and cell survival in response to replication stress. RPA2 hyperphosphorylation by DNA-PK in response to DSBs blocks unscheduled homologous recombination and delays mitotic entry. | [76,77,78,79] |
| RPSA | 40S ribosomal protein SA | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| SP1 | Transcription factor Sp1 | in vitro | dsDNA | 32P-incorporation | [80] | ||
| SRF | Serum response factor | in vitro/in vivo | IR | 32P-incorporation; two-dimensional separation of phosphopeptides on thin-layer cellulose plates | S435, S446 | [81] | |
| TBP | TATA-box-binding protein | in vitro | dsDNA | 32P-incorporation | [52] | ||
| TP53 | Cellular tumor antigen p53 | in vitro | dsDNA | WB; 32P-incorporation; SPR | S6, S15, S37, S46, S166 | [82,83,84,85] | |
| TRIM28 | Transcription intermediary factor 1-beta | in vivo | IR; Heat-shock induced gene transcription | WB | S824 | TRIM28 phosphorylation induces chromatin changes in response to DNA breaks. | [13,86,87] |
| TRIM28 stabilizes Pol II pausing, and its release depends on the S824 phosphorylation. | |||||||
| TUBB | Tubulin beta chain | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| TUBB2C | Tubulin beta-4B chain | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| TUBB6 | Tubulin beta-6 chain | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| UBQLN1 | Ubiquilin-1 | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| VCP | Transitional endoplasmic reticulum ATPase | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| VIM | Vimentin | in vitro/in vivo | Dbait32H | ProQ-Diamond + MS; WB; 32P-incorporation | S459 | Regulates cell migration and adhesion | [16] |
| WRN | Werner syndrome ATP-dependent helicase | in vitro/in vivo | dsDNA; bleomycin; 4NQO | WB; MS; 32P-incorporation; mutagenesis | S440, S467 | Inhibits both the helicase and exonuclease activities of WRN. Phosphorylation of S440 and S467 are important for relocalization of WRN to nucleoli, and that it is required for efficient DSB repair. | [38,39,88] |
| XRCC4 | DNA repair protein XRCC4 | in vitro/in vivo | dsDNA, IR | MS; 32P-incorporation; mutagenesis | S260, S318, S320 | Not essential for DSB repair | [34,40,41,42,43] |
| XRCC5 | XRCC5 X-ray repair cross-complementing protein 5 (Ku80) | in vitro | dsDNA | WB; MS; 32P-incorporation; amino acid sequencing | S577, S580, T715 | [35,89] | |
| XRCC6 | XRCC6 X-ray repair cross-complementing protein 6 (Ku70) | in vitro | dsDNA | WB; MS; 32P-incorporation; amino acid sequencing | S6 | [35,89] | |
| YWHAQ | 14-3-3 protein theta | in vivo | Dbait32H | ProQ-Diamond + MS | [16] | ||
| YWHAZ | 14-3-3 protein zeta/delta | in vivo | Dbait32H | ProQ-Diamond + MS | [16] |
| Gene Name (Common Protein Name) | Role in HIV Life Cycle | Comments/Life Cycle Step | Publications |
|---|---|---|---|
| AKT | Positive | Cell survival during HIV infection | [111,112] |
| ATM | Positive | Post-integrational repair (indirect evidence) | [101,106,108,113] |
| DCLRE1C (Artemis) | Positive | No data | [114] |
| DHX9 | Positive | Reverse transcription | [115,116] |
| EIF4A1 | Positive | Reverse transcription | [117] |
| FUS | Negative | LTR-dependent transcription | [118] |
| H2AFX (H2AX) | Dispensable | H2AFX is phosphorylated during integration, but not essential for HIV replication | [119] |
| HNRNPA1 | Complex | HIV transcription, viral mRNA splicing, mRNA transport | [120,121] |
| HSP90AA1 | Positive | Transcription, capsid core stability | [122] |
| JUN | Complex | Transcription. c-Jun enhances Tat-mediated LTR transcription but suppresses basal LTR transcription without Tat | [123] |
| MDM2 | Complex | 1. Positive regulator of early replicative stages in macrophages by inhibition of p53 activity; | [124,125,126] |
| 2. Negative regulation of Vif stability, removes its counteracting effect on the APOBEC3G restriction factor; | |||
| 3. Positive regulation of Tat activity | |||
| MRE11 | Controversial | Integration, pre-integration steps, post-integrational DNA repair (indirect and controversial evidences) | [106,110] |
| MYC | Complex | 1. Positive regulation of cDNA nuclear transport; | [127,128] |
| 2. c-Myc and Sp1 contribute to proviral latency. Negative regulation of transcription from LTR promoter | |||
| NBN (Nibrin) | Controversial | Integration, pre-integration steps, post-integrational DNA repair (indirect and controversial evidences) | [106,110] |
| NFKBIA (IκBα) | Negative | IκBα but not IκBβ suppress latent-active transcription transition | [129] |
| PARP1 | Controversial | 1. Early replicative steps (integration and/or post-integrational DNA-repair (indirect evidence) | [130,131,132,133,134,135,136,137,138] |
| 2. LTR-dependent transcription | |||
| POU2F1 (Oct-1) | Negative | Repress LTR-mediated transcription | [139] |
| RBBP7 | Negative | LTR-mediated transcription | [140] |
| SP1 | Positive | c-Myc and Sp1 contribute to proviral latency | [141] |
| TBP | Positive | LTR-mediated transcription | [142,143] |
| TP53 (p53) | Negative | 1. Reverse transcription | [144,145,146,147,148] |
| 2. LTR-mediated transcription | |||
| 3. Cell survival during HIV-infection | |||
| TRIM28 | Negative | Promotes HIV-1 Latency. DNA-PKcs dependent phosphorylation reactivates LTR mediated transcription | [14,149] |
| VIM | Positive | No data | [150] |
| WRN | Positive | LTR-mediated transcription | [151,152] |
| XRCC4 | Positive | early replicative stages | [103] |
| XRCC5 (Ku80) | Complex | 1. LTR-mediated transcription | [10,101,102,103,104,105,106,107] |
| 2. Integration, post-integrational DNA repair (direct evidence) | |||
| XRCC6 (Ku70) | Positive | Post-integrational DNA repair (direct evidence) | [10] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anisenko, A.; Kan, M.; Shadrina, O.; Brattseva, A.; Gottikh, M. Phosphorylation Targets of DNA-PK and Their Role in HIV-1 Replication. Cells 2020, 9, 1907. https://doi.org/10.3390/cells9081907
Anisenko A, Kan M, Shadrina O, Brattseva A, Gottikh M. Phosphorylation Targets of DNA-PK and Their Role in HIV-1 Replication. Cells. 2020; 9(8):1907. https://doi.org/10.3390/cells9081907
Chicago/Turabian StyleAnisenko, Andrey, Marina Kan, Olga Shadrina, Anna Brattseva, and Marina Gottikh. 2020. "Phosphorylation Targets of DNA-PK and Their Role in HIV-1 Replication" Cells 9, no. 8: 1907. https://doi.org/10.3390/cells9081907
APA StyleAnisenko, A., Kan, M., Shadrina, O., Brattseva, A., & Gottikh, M. (2020). Phosphorylation Targets of DNA-PK and Their Role in HIV-1 Replication. Cells, 9(8), 1907. https://doi.org/10.3390/cells9081907