The Chromatin Response to Double-Strand DNA Breaks and Their Repair


Abstract
1. Introduction
2. Tying up Loose Ends—Overview of DNA Double-Strand Break Repair Pathways
2.1. Sources of Damage
2.2. Pathways for Double-Strand DNA Breaks Repair
2.3. Classical Non-Homologous End Joining (c-NHEJ)
2.4. Homologous Recombination (HR)
2.5. Alternative End Joining (aEJ)
2.6. Single-Strand Annealing (SSA)
3. Chromatin Response to Double-Strand DNA Breaks
3.1. ATM and the Control of Early Events in DSB Repair
3.2. Setting up Chromatin for DSB Repair Pathway Choice
3.2.1. RNF8-RNF168-Mediated Recruitment of Repair Factors
3.2.2. BRCA1-BARD1-Dependent Repositioning of 53BP1 Promotes DNA End Resection
3.3. Recruitment of Effectors
3.3.1. Chromatin Changes that Promote c-NHEJ
3.3.2. Chromatin Events Promoting the Recruitment of HR Repair Factors
3.3.3. Chromatin Compaction in HR Repair
3.4. Dynamics of DSB Repair in Living Cells
4. Targeting the Chromatin Response to DSB DNA Damage
4.1. A New Hope: Synthetic Lethality and PARP1/2 Inhibitors
4.2. Targeting Chromatin Regulators
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Friedberg, E.C. A brief history of the DNA repair field. Cell Res. 2008, 18, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef]
- Peterson, C.L.; Almouzni, G. Nucleosome dynamics as modular systems that integrate DNA damage and repair. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e236. [Google Scholar] [CrossRef]
- Madhusudan, S.; Hickson, I.D. DNA repair inhibition: A selective tumour targeting strategy. Trends Mol. Med. 2005, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Raghavan, S.C. DNA double-strand break repair inhibitors as cancer therapeutics. Chem. Biol. 2015, 22, 17–29. [Google Scholar] [CrossRef]
- Cadet, J.; Douki, T.; Gasparutto, D.; Ravanat, J.L. Oxidative damage to DNA: Formation, measurement and biochemical features. Mutat. Res. 2003, 531, 5–23. [Google Scholar] [CrossRef]
- Stingele, J.; Jentsch, S. DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2015, 16, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Poulsen, H. Measurement of oxidatively generated base damage in cellular DNA and urine. Free Radic. Biol. Med. 2010, 48, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Sankar, T.S.; Wastuwidyaningtyas, B.D.; Dong, Y.; Lewis, S.A.; Wang, J.D. The nature of mutations induced by replication–transcription collisions. Nature 2016, 535, 178–181. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Lippert, M.J.; Kim, N.; Cho, J.E.; Larson, R.P.; Schoenly, N.E.; O’Shea, S.H.; Jinks-Robertson, S. Role for topoisomerase 1 in transcription-associated mutagenesis in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 698–703. [Google Scholar] [CrossRef]
- Soulas-Sprauel, P.; Rivera-Munoz, P.; Malivert, L.; Le Guyader, G.; Abramowski, V.; Revy, P.; de Villartay, J.P. V(D)J and immunoglobulin class switch recombinations: A paradigm to study the regulation of DNA end-joining. Oncogene 2007, 26, 7780–7791. [Google Scholar] [CrossRef] [PubMed]
- de Massy, B. Initiation of meiotic recombination: How and where? Conservation and specificities among eukaryotes. Annu. Rev. Genet. 2013, 47, 563–599. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Mahato, D.K.; Kamle, M.; Mohanta, T.K.; Kang, S.G. Aflatoxins: A Global Concern for Food Safety, Human Health and Their Management. Front. Microbiol. 2016, 7, 2170. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [PubMed]
- Hodskinson, M.R.; Bolner, A.; Sato, K.; Kamimae-Lanning, A.N.; Rooijers, K.; Witte, M.; Mahesh, M.; Silhan, J.; Petek, M.; Williams, D.M.; et al. Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature 2020, 579, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef]
- Maginn, E.N.; de Sousa, C.H.; Wasan, H.S.; Stronach, E.A. Opportunities for translation: Targeting DNA repair pathways in pancreatic cancer. Biochim. Biophys. Acta 2014, 1846, 45–54. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Lord, C.J. Dissecting PARP inhibitor resistance with functional genomics. Curr. Opin. Genet. Dev. 2019, 54, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Britton, S.; Coates, J.; Jackson, S.P. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013, 202, 579–595. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Lu, H.; Tippin, B.; Shimazaki, N.; Goodman, M.F.; Lieber, M.R. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. EMBO J. 2007, 26, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Grawunder, U.; Wilm, M.; Wu, X.; Kulesza, P.; Wilson, T.E.; Mann, M.; Lieber, M.R. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 1997, 388, 492–495. [Google Scholar] [CrossRef]
- Brouwer, I.; Sitters, G.; Candelli, A.; Heerema, S.J.; Heller, I.; de Melo, A.J.; Zhang, H.; Normanno, D.; Modesti, M.; Peterman, E.J.; et al. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature 2016, 535, 566–569. [Google Scholar] [CrossRef]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Yu, Y.; Riballo, E.; Douglas, P.; Walker, S.A.; Ye, R.; Härer, C.; Marchetti, C.; Morrice, N.; Jeggo, P.A.; et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 2006, 25, 3880–3889. [Google Scholar] [CrossRef]
- Grundy, G.J.; Rulten, S.L.; Zeng, Z.; Arribas-Bosacoma, R.; Iles, N.; Manley, K.; Oliver, A.; Caldecott, K.W. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J. 2013, 32, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Iles, N.; Rulten, S.; El-Khamisy, S.F.; Caldecott, K.W. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell. Biol. 2007, 27, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, N.K.; Williams, R.S.; Rakovszky, M.L.; Cui, D.; Green, R.; Karimi-Busheri, F.; Mani, R.S.; Galicia, S.; Koch, C.A.; Cass, C.E.; et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol. Cell 2005, 17, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Flett, F.J.; Ruksenaite, E.; Armstrong, L.A.; Bharati, S.; Carloni, R.; Morris, E.R.; Mackay, C.L.; Interthal, H.; Richardson, J.M. Structural basis for DNA 3’-end processing by human tyrosyl-DNA phosphodiesterase 1. Nat. Commun. 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Ahel, I.; Rass, U.; El-Khamisy, S.F.; Katyal, S.; Clements, P.M.; McKinnon, P.J.; Caldecott, K.W.; West, S.C. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 2006, 443, 713–716. [Google Scholar] [CrossRef]
- Nick McElhinny, S.A.; Havener, J.M.; Garcia-Diaz, M.; Juárez, R.; Bebenek, K.; Kee, B.L.; Blanco, L.; Kunkel, T.A.; Ramsden, D.A. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol. Cell 2005, 19, 357–366. [Google Scholar] [CrossRef]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Cannavo, E.; Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 2014, 514, 122–125. [Google Scholar] [CrossRef]
- Garcia, V.; Phelps, S.E.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef]
- Limbo, O.; Chahwan, C.; Yamada, Y.; de Bruin, R.A.; Wittenberg, C.; Russell, P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 2007, 28, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Melander, F.; Stucki, M.; Falck, J.; Bekker-Jensen, S.; Goldberg, M.; Lerenthal, Y.; Jackson, S.P.; Bartek, J.; Lukas, J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004, 23, 2674–2683. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Durocher, D. Push back to respond better: Regulatory inhibition of the DNA double-strand break response. Nat. Rev. Mol. Cell Biol. 2013, 14, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef]
- Kim, H.; Chen, J.; Yu, X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007, 316, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Ozsoy, A.Z.; Genschel, J.; Modrich, P.; Kowalczykowski, S.C. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. USA 2008, 105, 16906–16911. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef]
- Chen, H.; Lisby, M.; Symington, L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 2013, 50, 589–600. [Google Scholar] [CrossRef]
- Carreira, A.; Kowalczykowski, S.C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 10448–10453. [Google Scholar] [CrossRef]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Renkawitz, J.; Lademann, C.A.; Jentsch, S. Mechanisms and principles of homology search during recombination. Nat. Rev. Mol. Cell Biol. 2014, 15, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, L.; Xu, Y.; Zhao, W.; Sung, P.; Wang, H.W. Cryo-EM structures of human RAD51 recombinase filaments during catalysis of DNA-strand exchange. Nat. Struct. Mol. Biol. 2017, 24, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Jian Ma, C.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365. [Google Scholar] [CrossRef]
- Kane, D.P.; Shusterman, M.; Rong, Y.; McVey, M. Competition between replicative and translesion polymerases during homologous recombination repair in Drosophila. PLoS Genet. 2012, 8, e1002659. [Google Scholar] [CrossRef] [PubMed]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e894. [Google Scholar] [CrossRef]
- Robert, I.; Dantzer, F.; Reina-San-Martin, B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med. 2009, 206, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Masani, S.; Han, L.; Meek, K.; Yu, K. Redundant function of DNA ligase 1 and 3 in alternative end-joining during immunoglobulin class switch recombination. Proc. Natl. Acad. Sci. USA 2016, 113, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. DNA end resection--unraveling the tail. DNA Repair 2011, 10, 344–348. [Google Scholar] [CrossRef]
- Symington, L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef]
- Mendez-Dorantes, C.; Bhargava, R.; Stark, J.M. Repeat-mediated deletions can be induced by a chromosomal break far from a repeat, but multiple pathways suppress such rearrangements. Genes Dev. 2018, 32, 524–536. [Google Scholar] [CrossRef]
- Setiaputra, D.; Durocher, D. Shieldin-the protector of DNA ends. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Andegeko, Y.; Moyal, L.; Mittelman, L.; Tsarfaty, I.; Shiloh, Y.; Rotman, G. Nuclear retention of ATM at sites of DNA double strand breaks. J. Biol. Chem. 2001, 276, 38224–38230. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Lukas, C.; Kitagawa, R.; Melander, F.; Kastan, M.B.; Bartek, J.; Lukas, J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006, 173, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.T.; Schwartz, R.A.; Stracker, T.H.; Lilley, C.E.; Lee, D.V.; Weitzman, M.D. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003, 22, 6610–6620. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef]
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174. [Google Scholar] [CrossRef]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef]
- Khoury-Haddad, H.; Guttmann-Raviv, N.; Ipenberg, I.; Huggins, D.; Jeyasekharan, A.D.; Ayoub, N. PARP1-dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E728–E737. [Google Scholar] [CrossRef]
- Gupta, A.; Sharma, G.G.; Young, C.S.; Agarwal, M.; Smith, E.R.; Paull, T.T.; Lucchesi, J.C.; Khanna, K.K.; Ludwig, T.; Pandita, T.K. Involvement of human MOF in ATM function. Mol. Cell. Biol. 2005, 25, 5292–5305. [Google Scholar] [CrossRef]
- Kim, Y.C.; Gerlitz, G.; Furusawa, T.; Catez, F.; Nussenzweig, A.; Oh, K.S.; Kraemer, K.H.; Shiloh, Y.; Bustin, M. Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Nat. Cell Biol. 2009, 11, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G(2)-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G(2)-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Shema, E.; Moyal, L.; Oren, M. RNF20-RNF40: A ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett. 2011, 585, 2795–2802. [Google Scholar] [CrossRef]
- Feng, J.; Shen, W.H. Dynamic regulation and function of histone monoubiquitination in plants. Front. Plant Sci. 2014, 5, 83. [Google Scholar] [CrossRef]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef]
- So, C.C.; Ramachandran, S.; Martin, A. E3 Ubiquitin Ligases RNF20 and RNF40 Are Required for Double-Stranded Break (DSB) Repair: Evidence for Monoubiquitination of Histone H2B Lysine 120 as a Novel Axis of DSB Signaling and Repair. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef]
- Ramachandran, S.; Haddad, D.; Li, C.; Le, M.X.; Ling, A.K.; So, C.C.; Nepal, R.M.; Gommerman, J.L.; Yu, K.; Ketela, T.; et al. The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated γH2AX Formation. Cell Rep. 2016, 15, 1554–1565. [Google Scholar] [CrossRef]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef]
- Kato, A.; Komatsu, K. RNF20-SNF2H Pathway of Chromatin Relaxation in DNA Double-Strand Break Repair. Genes 2015, 6, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Toiber, D.; Erdel, F.; Bouazoune, K.; Silberman, D.M.; Zhong, L.; Mulligan, P.; Sebastian, C.; Cosentino, C.; Martinez-Pastor, B.; Giacosa, S.; et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 2013, 51, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Künzel, J.; Löbrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef]
- Iannelli, F.; Galbiati, A.; Capozzo, I.; Nguyen, Q.; Magnuson, B.; Michelini, F.; D’Alessandro, G.; Cabrini, M.; Roncador, M.; Francia, S.; et al. A damaged genome’s transcriptional landscape through multilayered expression profiling around in situ-mapped DNA double-strand breaks. Nat. Commun. 2017, 8, 15656. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 2004, 14, 1703–1711. [Google Scholar] [CrossRef]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Polo, S.E.; Almouzni, G. Prime, repair, restore: The active role of chromatin in the DNA damage response. Mol. Cell 2012, 46, 722–734. [Google Scholar] [CrossRef]
- Cao, L.L.; Wei, F.; Du, Y.; Song, B.; Wang, D.; Shen, C.; Lu, X.; Cao, Z.; Yang, Q.; Gao, Y.; et al. ATM-mediated KDM2A phosphorylation is required for the DNA damage repair. Oncogene 2016, 35, 402. [Google Scholar] [CrossRef]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J.; et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 2012, 45, 505–516. [Google Scholar] [CrossRef]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef]
- Wang, J.; Gong, Z.; Chen, J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 2011, 193, 267–273. [Google Scholar] [CrossRef]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Diaz, C.; Orthwein, A.; Leung, C.C.; Huang, H.; Landry, M.C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Luijsterburg, M.S.; Acs, K.; Ackermann, L.; Wiegant, W.W.; Bekker-Jensen, S.; Larsen, D.H.; Khanna, K.K.; van Attikum, H.; Mailand, N.; Dantuma, N.P. A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure. EMBO J. 2012, 31, 2511–2527. [Google Scholar] [CrossRef] [PubMed]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A., Jr.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.L.; Portoso, M.; Mata, J.; Bahler, J.; Allshire, R.C.; Kouzarides, T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 2004, 119, 603–614. [Google Scholar] [CrossRef]
- Schotta, G.; Sengupta, R.; Kubicek, S.; Malin, S.; Kauer, M.; Callen, E.; Celeste, A.; Pagani, M.; Opravil, S.; De La Rosa-Velazquez, I.A.; et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008, 22, 2048–2061. [Google Scholar] [CrossRef]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Bergsagel, P.L.; Wang, L.; You, Z.; Lou, Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar] [CrossRef]
- Hartlerode, A.J.; Guan, Y.; Rajendran, A.; Ura, K.; Schotta, G.; Xie, A.; Shah, J.V.; Scully, R. Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks. PLoS ONE 2012, 7, e49211. [Google Scholar] [CrossRef]
- Nishioka, K.; Rice, J.C.; Sarma, K.; Erdjument-Bromage, H.; Werner, J.; Wang, Y.; Chuikov, S.; Valenzuela, P.; Tempst, P.; Steward, R.; et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 2002, 9, 1201–1213. [Google Scholar] [CrossRef]
- Beck, D.B.; Oda, H.; Shen, S.S.; Reinberg, D. PR-Set7 and H4K20me1: At the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012, 26, 325–337. [Google Scholar] [CrossRef]
- Yang, H.; Pesavento, J.J.; Starnes, T.W.; Cryderman, D.E.; Wallrath, L.L.; Kelleher, N.L.; Mizzen, C.A. Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J. Biol. Chem. 2008, 283, 12085–12092. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, S.; Schotta, G.; Sørensen, C.S. Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013, 41, 2797–2806. [Google Scholar] [CrossRef]
- Min, J.; Allali-Hassani, A.; Nady, N.; Qi, C.; Ouyang, H.; Liu, Y.; MacKenzie, F.; Vedadi, M.; Arrowsmith, C.H. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 2007, 14, 1229–1230. [Google Scholar] [CrossRef] [PubMed]
- Acs, K.; Luijsterburg, M.S.; Ackermann, L.; Salomons, F.A.; Hoppe, T.; Dantuma, N.P. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011, 18, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Thompson, J.R.; Botuyan, M.V.; Mer, G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 2008, 15, 109–111. [Google Scholar] [CrossRef]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012, 31, 1865–1878. [Google Scholar] [CrossRef]
- Jacquet, K.; Fradet-Turcotte, A.; Avvakumov, N.; Lambert, J.P.; Roques, C.; Pandita, R.K.; Paquet, E.; Herst, P.; Gingras, A.C.; Pandita, T.K.; et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol. Cell 2016, 62, 409–421. [Google Scholar] [CrossRef]
- Drane, P.; Brault, M.E.; Cui, G.; Meghani, K.; Chaubey, S.; Detappe, A.; Parnandi, N.; He, Y.; Zheng, X.F.; Botuyan, M.V.; et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 2017, 543, 211–216. [Google Scholar] [CrossRef]
- Wakeman, T.P.; Wang, Q.; Feng, J.; Wang, X.F. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J. 2012, 31, 2169–2181. [Google Scholar] [CrossRef]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Köhn, M.; Chatterjee, C.; Chiang, K.P.; Pratt, M.R.; Muir, T.W. Structure-activity analysis of semisynthetic nucleosomes: Mechanistic insights into the stimulation of Dot1L by ubiquitylated histone H2B. Acs Chem. Biol. 2009, 4, 958–968. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Kim, J.; Chatterjee, C.; Roeder, R.G.; Muir, T.W. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 2008, 453, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Holt, M.T.; Ohashi, N.; Zhao, A.; Müller, M.M.; Wang, B.; Muir, T.W. Evidence that ubiquitylated H2B corrals hDot1L on the nucleosomal surface to induce H3K79 methylation. Nat. Commun. 2016, 7, 10589. [Google Scholar] [CrossRef] [PubMed]
- Conde, F.; Refolio, E.; Cordón-Preciado, V.; Cortés-Ledesma, F.; Aragón, L.; Aguilera, A.; San-Segundo, P.A. The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics 2009, 182, 437–446. [Google Scholar] [CrossRef]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef]
- Wu, W.; Nishikawa, H.; Fukuda, T.; Vittal, V.; Asano, M.; Miyoshi, Y.; Klevit, R.E.; Ohta, T. Interaction of BARD1 and HP1 Is Required for BRCA1 Retention at Sites of DNA Damage. Cancer Res. 2015, 75, 1311–1321. [Google Scholar] [CrossRef]
- Hu, Y.; Scully, R.; Sobhian, B.; Xie, A.; Shestakova, E.; Livingston, D.M. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011, 25, 685–700. [Google Scholar] [CrossRef]
- Savage, K.I.; Harkin, D.P. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015, 282, 630–646. [Google Scholar] [CrossRef]
- Ng, H.M.; Wei, L.; Lan, L.; Huen, M.S. The Lys63-deubiquitylating Enzyme BRCC36 Limits DNA Break Processing and Repair. J. Biol. Chem. 2016, 291, 16197–16207. [Google Scholar] [CrossRef]
- Aleksandrov, R.; Dotchev, A.; Poser, I.; Krastev, D.; Georgiev, G.; Panova, G.; Babukov, Y.; Danovski, G.; Dyankova, T.; Hubatsch, L.; et al. Protein Dynamics in Complex DNA Lesions. Mol. Cell 2018, 69, 1046–1061.e1045. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, S.; Michelena, J.; Teloni, F.; Imhof, R.; Altmeyer, M. Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell Rep. 2017, 19, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, M.; de Krijger, I.; Serrat, J.; Moatti, N.; Fortunato, D.; Hoekman, L.; Bleijerveld, O.B.; Altelaar, A.F.M.; Jacobs, J.J.L. H4K20me2 distinguishes pre-replicative from post-replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1-RIF1-MAD2L2. Cell Cycle 2018, 17, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.H.; Hakim, O.; et al. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014, 8, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xu, Y.; Gursoy-Yuzugullu, O.; Price, B.D. The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. FEBS Lett. 2012, 586, 3920–3925. [Google Scholar] [CrossRef]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci 2012, 125, 3529–3534. [Google Scholar] [CrossRef]
- Lorick, K.L.; Jensen, J.P.; Fang, S.; Ong, A.M.; Hatakeyama, S.; Weissman, A.M. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. USA 1999, 96, 11364–11369. [Google Scholar] [CrossRef]
- Ruffner, H.; Joazeiro, C.A.; Hemmati, D.; Hunter, T.; Verma, I.M. Cancer-predisposing mutations within the RING domain of BRCA1: Loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc. Natl. Acad. Sci. USA 2001, 98, 5134–5139. [Google Scholar] [CrossRef]
- Xia, Y.; Pao, G.M.; Chen, H.W.; Verma, I.M.; Hunter, T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem. 2003, 278, 5255–5263. [Google Scholar] [CrossRef]
- Kalb, R.; Mallery, D.L.; Larkin, C.; Huang, J.T.; Hiom, K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 2014, 8, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Awad, S.; Ryan, D.; Prochasson, P.; Owen-Hughes, T.; Hassan, A.H. The Snf2 homolog Fun30 acts as a homodimeric ATP-dependent chromatin-remodeling enzyme. J. Biol. Chem. 2010, 285, 9477–9484. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cui, D.; Papusha, A.; Zhang, X.; Chu, C.D.; Tang, J.; Chen, K.; Pan, X.; Ira, G. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature 2012, 489, 576–580. [Google Scholar] [CrossRef]
- Costelloe, T.; Louge, R.; Tomimatsu, N.; Mukherjee, B.; Martini, E.; Khadaroo, B.; Dubois, K.; Wiegant, W.W.; Thierry, A.; Burma, S.; et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 2012, 489, 581–584. [Google Scholar] [CrossRef]
- Eapen, V.V.; Sugawara, N.; Tsabar, M.; Wu, W.H.; Haber, J.E. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol. Cell. Biol. 2012, 32, 4727–4740. [Google Scholar] [CrossRef]
- Bantele, S.C.; Ferreira, P.; Gritenaite, D.; Boos, D.; Pfander, B. Targeting of the Fun30 nucleosome remodeller by the Dpb11 scaffold facilitates cell cycle-regulated DNA end resection. eLife 2017, 6. [Google Scholar] [CrossRef]
- Chen, X.; Niu, H.; Yu, Y.; Wang, J.; Zhu, S.; Zhou, J.; Papusha, A.; Cui, D.; Pan, X.; Kwon, Y.; et al. Enrichment of Cdk1-cyclins at DNA double-strand breaks stimulates Fun30 phosphorylation and DNA end resection. Nucleic Acids Res. 2016, 44, 2742–2753. [Google Scholar] [CrossRef]
- Uckelmann, M.; Densham, R.M.; Baas, R.; Winterwerp, H.H.K.; Fish, A.; Sixma, T.K.; Morris, J.R. USP48 restrains resection by site-specific cleavage of the BRCA1 ubiquitin mark from H2A. Nat. Commun. 2018, 9, 229. [Google Scholar] [CrossRef]
- Alagoz, M.; Katsuki, Y.; Ogiwara, H.; Ogi, T.; Shibata, A.; Kakarougkas, A.; Jeggo, P. SETDB1, HP1 and SUV39 promote repositioning of 53BP1 to extend resection during homologous recombination in G2 cells. Nucleic Acids Res. 2015, 43, 7931–7944. [Google Scholar] [CrossRef]
- Keogh, M.C.; Kim, J.A.; Downey, M.; Fillingham, J.; Chowdhury, D.; Harrison, J.C.; Onishi, M.; Datta, N.; Galicia, S.; Emili, A.; et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature 2006, 439, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Allis, C.D.; Nussenzweig, A. Phosphorylation of histone H2B at DNA double-strand breaks. J. Exp. Med. 2004, 199, 1671–1677. [Google Scholar] [CrossRef]
- Utley, R.T.; Lacoste, N.; Jobin-Robitaille, O.; Allard, S.; Côté, J. Regulation of NuA4 histone acetyltransferase activity in transcription and DNA repair by phosphorylation of histone H4. Mol. Cell. Biol. 2005, 25, 8179–8190. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.L.; Turner, F.B.; Krishnamoorthy, T.; Wolner, B.; Ahn, S.H.; Foley, M.; Dorsey, J.A.; Peterson, C.L.; Berger, S.L.; Allis, C.D. Phosphorylation of histone H4 serine 1 during DNA damage requires casein kinase II in S. cerevisiae. Curr. Biol. 2005, 15, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Z.; Dong, L.; Tang, M.; Zhang, P.; Zhang, C.; Cao, Z.; Zhu, Q.; Chen, Y.; Wang, H.; et al. Histone H1 acetylation at lysine 85 regulates chromatin condensation and genome stability upon DNA damage. Nucleic Acids Res. 2018, 46, 7716–7730. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Tashiro, S.; Kakino, A.; Shima, H.; Jacob, N.; Amunugama, R.; Yoder, K.; Izumi, S.; Kuraoka, I.; Tanaka, K.; et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol. Cell. Biol. 2007, 27, 7028–7040. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e256. [Google Scholar] [CrossRef]
- Tamburini, B.A.; Tyler, J.K. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell. Biol. 2005, 25, 4903–4913. [Google Scholar] [CrossRef]
- Ogiwara, H.; Ui, A.; Otsuka, A.; Satoh, H.; Yokomi, I.; Nakajima, S.; Yasui, A.; Yokota, J.; Kohno, T. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene 2011, 30, 2135–2146. [Google Scholar] [CrossRef]
- Das, C.; Lucia, M.S.; Hansen, K.C.; Tyler, J.K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009, 459, 113–117. [Google Scholar] [CrossRef]
- Chen, C.C.; Carson, J.J.; Feser, J.; Tamburini, B.; Zabaronick, S.; Linger, J.; Tyler, J.K. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 2008, 134, 231–243. [Google Scholar] [CrossRef]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, M.M.; Madabhushi, R.; Pan, L.; Chen, Y.; Kim, D.; Gao, J.; Ahanonu, B.; Pao, P.C.; Qiu, Y.; Zhao, Y.; et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci. 2013, 16, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hébert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef]
- Yan, Q.; Dutt, S.; Xu, R.; Graves, K.; Juszczynski, P.; Manis, J.P.; Shipp, M.A. BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol. Cell 2009, 36, 110–120. [Google Scholar] [CrossRef]
- Gong, F.; Clouaire, T.; Aguirrebengoa, M.; Legube, G.; Miller, K.M. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell Biol. 2017, 216, 1959–1974. [Google Scholar] [CrossRef]
- Mosammaparast, N.; Kim, H.; Laurent, B.; Zhao, Y.; Lim, H.J.; Majid, M.C.; Dango, S.; Luo, Y.; Hempel, K.; Sowa, M.E.; et al. The histone demethylase LSD1/KDM1A promotes the DNA damage response. J. Cell Biol. 2013, 203, 457–470. [Google Scholar] [CrossRef]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008, 453, 682–686. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Ui, A.; Nagaura, Y.; Yasui, A. Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol. Cell 2015, 58, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Abu-Zhayia, E.R.; Awwad, S.W.; Ben-Oz, B.M.; Khoury-Haddad, H.; Ayoub, N. CDYL1 fosters double-strand break-induced transcription silencing and promotes homology-directed repair. J. Mol. Cell Biol. 2018, 10, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Fnu, S.; Williamson, E.A.; De Haro, L.P.; Brenneman, M.; Wray, J.; Shaheen, M.; Radhakrishnan, K.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. USA 2011, 108, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef]
- Carvalho, S.; Vítor, A.C.; Sridhara, S.C.; Martins, F.B.; Raposo, A.C.; Desterro, J.M.; Ferreira, J.; de Almeida, S.F. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. eLife 2014, 3, e02482. [Google Scholar] [CrossRef]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sørensen, C.S.; Petersen, N.H.; Sorensen, P.H.; Lukas, C.; Bartek, J.; et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, J.H.; Kim, S.J.; Kwon, S.J.; Kwon, J. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J. 2010, 29, 1434–1445. [Google Scholar] [CrossRef]
- Watanabe, R.; Ui, A.; Kanno, S.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef]
- Kwon, J.; Morshead, K.B.; Guyon, J.R.; Kingston, R.E.; Oettinger, M.A. Histone acetylation and hSWI/SNF remodeling act in concert to stimulate V(D)J cleavage of nucleosomal DNA. Mol. Cell 2000, 6, 1037–1048. [Google Scholar] [CrossRef]
- Morshead, K.B.; Ciccone, D.N.; Taverna, S.D.; Allis, C.D.; Oettinger, M.A. Antigen receptor loci poised for V(D)J rearrangement are broadly associated with BRG1 and flanked by peaks of histone H3 dimethylated at lysine 4. Proc. Natl. Acad. Sci. USA 2003, 100, 11577–11582. [Google Scholar] [CrossRef] [PubMed]
- Patenge, N.; Elkin, S.K.; Oettinger, M.A. ATP-dependent remodeling by SWI/SNF and ISWI proteins stimulates V(D)J cleavage of 5 S arrays. J. Biol. Chem. 2004, 279, 35360–35367. [Google Scholar] [CrossRef]
- Chai, B.; Huang, J.; Cairns, B.R.; Laurent, B.C. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev. 2005, 19, 1656–1661. [Google Scholar] [CrossRef]
- Sinha, M.; Watanabe, S.; Johnson, A.; Moazed, D.; Peterson, C.L. Recombinational repair within heterochromatin requires ATP-dependent chromatin remodeling. Cell 2009, 138, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Wiest, N.E.; Houghtaling, S.; Sanchez, J.C.; Tomkinson, A.E.; Osley, M.A. The SWI/SNF ATP-dependent nucleosome remodeler promotes resection initiation at a DNA double-strand break in yeast. Nucleic Acids Res. 2017, 45, 5887–5900. [Google Scholar] [CrossRef]
- Adkins, N.L.; Swygert, S.G.; Kaur, P.; Niu, H.; Grigoryev, S.A.; Sung, P.; Wang, H.; Peterson, C.L. Nucleosome-like, Single-stranded DNA (ssDNA)-Histone Octamer Complexes and the Implication for DNA Double Strand Break Repair. J. Biol. Chem. 2017, 292, 5271–5281. [Google Scholar] [CrossRef]
- Strålfors, A.; Walfridsson, J.; Bhuiyan, H.; Ekwall, K. The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet. 2011, 7, e1001334. [Google Scholar] [CrossRef]
- Durand-Dubief, M.; Will, W.R.; Petrini, E.; Theodorou, D.; Harris, R.R.; Crawford, M.R.; Paszkiewicz, K.; Krueger, F.; Correra, R.M.; Vetter, A.T.; et al. SWI/SNF-like chromatin remodeling factor Fun30 supports point centromere function in S. cerevisiae. PLoS Genet. 2012, 8, e1002974. [Google Scholar] [CrossRef]
- Shen, X.; Mizuguchi, G.; Hamiche, A.; Wu, C. A chromatin remodelling complex involved in transcription and DNA processing. Nature 2000, 406, 541–544. [Google Scholar] [CrossRef]
- Shen, X.; Ranallo, R.; Choi, E.; Wu, C. Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol. Cell 2003, 12, 147–155. [Google Scholar] [CrossRef]
- Gospodinov, A.; Vaissiere, T.; Krastev, D.B.; Legube, G.; Anachkova, B.; Herceg, Z. Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol. Cell. Biol. 2011, 31, 4735–4745. [Google Scholar] [CrossRef] [PubMed]
- van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Tsukuda, T.; Fleming, A.B.; Nickoloff, J.A.; Osley, M.A. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 2005, 438, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Hur, S.K.; Kwon, J. Human INO80 chromatin-remodelling complex contributes to DNA double-strand break repair via the expression of Rad54B and XRCC3 genes. Biochem. J. 2010, 431, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Krebs, J.E.; Peterson, C.L. Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev. 2006, 20, 2437–2449. [Google Scholar] [CrossRef] [PubMed]
- Lademann, C.A.; Renkawitz, J.; Pfander, B.; Jentsch, S. The INO80 Complex Removes H2A.Z to Promote Presynaptic Filament Formation during Homologous Recombination. Cell Rep. 2017, 19, 1294–1303. [Google Scholar] [CrossRef]
- Li, X.; Tyler, J.K. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. eLife 2016, 5. [Google Scholar] [CrossRef]
- Poli, J.; Gerhold, C.B.; Tosi, A.; Hustedt, N.; Seeber, A.; Sack, R.; Herzog, F.; Pasero, P.; Shimada, K.; Hopfner, K.P.; et al. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev. 2016, 30, 337–354. [Google Scholar] [CrossRef]
- Dong, S.; Han, J.; Chen, H.; Liu, T.; Huen, M.S.Y.; Yang, Y.; Guo, C.; Huang, J. The human SRCAP chromatin remodeling complex promotes DNA-end resection. Curr. Biol. 2014, 24, 2097–2110. [Google Scholar] [CrossRef]
- van Attikum, H.; Fritsch, O.; Gasser, S.M. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 2007, 26, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Kari, V.; Mansour, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016, 17, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Ahel, D.; Horejsí, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiácovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010, 29, 3130–3139. [Google Scholar] [CrossRef]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; van Attikum, H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 2010, 190, 741–749. [Google Scholar] [CrossRef]
- Lan, L.; Ui, A.; Nakajima, S.; Hatakeyama, K.; Hoshi, M.; Watanabe, R.; Janicki, S.M.; Ogiwara, H.; Kohno, T.; Kanno, S.; et al. The ACF1 complex is required for DNA double-strand break repair in human cells. Mol. Cell 2010, 40, 976–987. [Google Scholar] [CrossRef]
- Mehrotra, P.V.; Ahel, D.; Ryan, D.P.; Weston, R.; Wiechens, N.; Kraehenbuehl, R.; Owen-Hughes, T.; Ahel, I. DNA repair factor APLF is a histone chaperone. Mol. Cell 2011, 41, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, E.C.; Hargreaves, D.C.; Miller, E.L.; Cui, K.; Korshunov, A.; Kool, M.; Pfister, S.; Cho, Y.J.; Zhao, K.; Crabtree, G.R. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 2013, 497, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.; Tischfield, J.A.; Serrano, L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef]
- McCord, R.A.; Michishita, E.; Hong, T.; Berber, E.; Boxer, L.D.; Kusumoto, R.; Guan, S.; Shi, X.; Gozani, O.; Burlingame, A.L.; et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging 2009, 1, 109–121. [Google Scholar] [CrossRef]
- Pai, C.C.; Deegan, R.S.; Subramanian, L.; Gal, C.; Sarkar, S.; Blaikley, E.J.; Walker, C.; Hulme, L.; Bernhard, E.; Codlin, S.; et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat. Commun. 2014, 5, 4091. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J. Biol. Chem. 2017, 292, 11951–11959. [Google Scholar] [CrossRef]
- Clouaire, T.; Legube, G. A Snapshot on the Cis Chromatin Response to DNA Double-Strand Breaks. Trends Genet. 2019, 35, 330–345. [Google Scholar] [CrossRef]
- Caron, M.C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.P.; Langelier, M.F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [PubMed]
- Taty-Taty, G.C.; Chailleux, C.; Quaranta, M.; So, A.; Guirouilh-Barbat, J.; Lopez, B.S.; Bertrand, P.; Trouche, D.; Canitrot, Y. Control of alternative end joining by the chromatin remodeler p400 ATPase. Nucleic Acids Res. 2016, 44, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Adkins, N.L.; Niu, H.; Sung, P.; Peterson, C.L. Nucleosome dynamics regulates DNA processing. Nat. Struct. Mol. Biol. 2013, 20, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Shim, E.Y.; Hong, S.J.; Oum, J.H.; Yanez, Y.; Zhang, Y.; Lee, S.E. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol. Cell. Biol. 2007, 27, 1602–1613. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.; Papamichos-Chronakis, M.; Peterson, C.L. DNA repair choice defines a common pathway for recruitment of chromatin regulators. Nat. Commun. 2013, 4, 2084. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Caron, P.; Legube, G.; Paull, T.T. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014, 42, e19. [Google Scholar] [CrossRef]
- Gursoy-Yuzugullu, O.; Ayrapetov, M.K.; Price, B.D. Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc. Natl. Acad. Sci. USA 2015, 112, 7507–7512. [Google Scholar] [CrossRef]
- Mizuguchi, G.; Shen, X.; Landry, J.; Wu, W.H.; Sen, S.; Wu, C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 2004, 303, 343–348. [Google Scholar] [CrossRef]
- Papamichos-Chronakis, M.; Watanabe, S.; Rando, O.J.; Peterson, C.L. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 2011, 144, 200–213. [Google Scholar] [CrossRef]
- Kollárovič, G.; Topping, C.E.; Shaw, E.P.; Chambers, A.L. The human HELLS chromatin remodelling protein promotes end resection to facilitate homologous recombination and contributes to DSB repair within heterochromatin. Nucleic Acids Res. 2020, 48, 1872–1885. [Google Scholar] [CrossRef]
- Huang, T.H.; Fowler, F.; Chen, C.C.; Shen, Z.J.; Sleckman, B.; Tyler, J.K. The Histone Chaperones ASF1 and CAF-1 Promote MMS22L-TONSL-Mediated Rad51 Loading onto ssDNA during Homologous Recombination in Human Cells. Mol. Cell 2018, 69, 879–892.e875. [Google Scholar] [CrossRef] [PubMed]
- Courilleau, C.; Chailleux, C.; Jauneau, A.; Grimal, F.; Briois, S.; Boutet-Robinet, E.; Boudsocq, F.; Trouche, D.; Canitrot, Y. The chromatin remodeler p400 ATPase facilitates Rad51-mediated repair of DNA double-strand breaks. J. Cell Biol. 2012, 199, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Wang, R.; Chen, H.; Wang, X.; Xiao, T.; Boldogh, I.; Ba, X.; Han, L.; Zeng, X. BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. J. Cell Sci 2015, 128, 317–330. [Google Scholar] [CrossRef]
- Alonso-de Vega, I.; Paz-Cabrera, M.C.; Rother, M.B.; Wiegant, W.W.; Checa-Rodríguez, C.; Hernández-Fernaud, J.R.; Huertas, P.; Freire, R.; van Attikum, H.; Smits, V.A.J. PHF2 regulates homology-directed DNA repair by controlling the resection of DNA double strand breaks. Nucleic Acids Res. 2020, 48, 4915–4927. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, L.D.; LeClair, J.W.; Miller, K.N.; Strong, A.D.; Chan, H.L.; Oates, E.L.; Ligon, K.L.; Brennan, C.W.; Chheda, M.G. CHD4 regulates the DNA damage response and RAD51 expression in glioblastoma. Sci. Rep. 2019, 9, 4444. [Google Scholar] [CrossRef]
- Pan, M.R.; Hsieh, H.J.; Dai, H.; Hung, W.C.; Li, K.; Peng, G.; Lin, S.Y. Chromodomain helicase DNA-binding protein 4 (CHD4) regulates homologous recombination DNA repair, and its deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitor treatment. J. Biol. Chem. 2012, 287, 6764–6772. [Google Scholar] [CrossRef]
- Larsen, D.H.; Poinsignon, C.; Gudjonsson, T.; Dinant, C.; Payne, M.R.; Hari, F.J.; Rendtlew Danielsen, J.M.; Menard, P.; Sand, J.C.; Stucki, M.; et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell Biol. 2010, 190, 731–740. [Google Scholar] [CrossRef]
- Gong, F.; Chiu, L.Y.; Cox, B.; Aymard, F.; Clouaire, T.; Leung, J.W.; Cammarata, M.; Perez, M.; Agarwal, P.; Brodbelt, J.S.; et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015, 29, 197–211. [Google Scholar] [CrossRef]
- Gong, F.; Miller, K.M. Double duty: ZMYND8 in the DNA damage response and cancer. Cell Cycle 2018, 17, 414–420. [Google Scholar] [CrossRef]
- Savitsky, P.; Krojer, T.; Fujisawa, T.; Lambert, J.P.; Picaud, S.; Wang, C.Y.; Shanle, E.K.; Krajewski, K.; Friedrichsen, H.; Kanapin, A.; et al. Multivalent Histone and DNA Engagement by a PHD/BRD/PWWP Triple Reader Cassette Recruits ZMYND8 to K14ac-Rich Chromatin. Cell Rep. 2016, 17, 2724–2737. [Google Scholar] [CrossRef]
- Gursoy-Yuzugullu, O.; Carman, C.; Price, B.D. Spatially restricted loading of BRD2 at DNA double-strand breaks protects H4 acetylation domains and promotes DNA repair. Sci. Rep. 2017, 7, 12921. [Google Scholar] [CrossRef] [PubMed]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; van Lohuizen, M.; Ganesan, S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montaño, C.; Gorski, S.A.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007, 447, 730–734. [Google Scholar] [CrossRef]
- Ting, X.; Xia, L.; Yang, J.; He, L.; Si, W.; Shang, Y.; Sun, L. USP11 acts as a histone deubiquitinase functioning in chromatin reorganization during DNA repair. Nucleic Acids Res. 2019, 47, 9721–9740. [Google Scholar] [CrossRef]
- Caron, P.; van der Linden, J.; van Attikum, H. Bon voyage: A transcriptional journey around DNA breaks. DNA Repair 2019, 82, 102686. [Google Scholar] [CrossRef] [PubMed]
- Lashgari, A.; Millau, J.F.; Jacques, P.; Gaudreau, L. Global inhibition of transcription causes an increase in histone H2A.Z incorporation within gene bodies. Nucleic Acids Res. 2017, 45, 12715–12722. [Google Scholar] [CrossRef]
- Vermeulen, W. Dynamics of mammalian NER proteins. DNA Repair 2011, 10, 760–771. [Google Scholar] [CrossRef]
- Feuerhahn, S.; Egly, J.M. Tools to study DNA repair: What’s in the box? Trends Genet. 2008, 24, 467–474. [Google Scholar] [CrossRef]
- Kong, X.; Mohanty, S.K.; Stephens, J.; Heale, J.T.; Gomez-Godinez, V.; Shi, L.Z.; Kim, J.S.; Yokomori, K.; Berns, M.W. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res. 2009, 37, e68. [Google Scholar] [CrossRef]
- Zou, L.; Liu, D.; Elledge, S.J. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 13827–13832. [Google Scholar] [CrossRef]
- Ngo, G.H.; Lydall, D. The 9-1-1 checkpoint clamp coordinates resection at DNA double strand breaks. Nucleic Acids Res. 2015, 43, 5017–5032. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Gassman, N.R.; Wilson, S.H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair 2015, 31, 52–63. [Google Scholar] [CrossRef]
- Beneke, S. Regulation of chromatin structure by poly(ADP-ribosyl)ation. Front. Genet. 2012, 3, 169. [Google Scholar] [CrossRef]
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef]
- Messner, S.; Altmeyer, M.; Zhao, H.; Pozivil, A.; Roschitzki, B.; Gehrig, P.; Rutishauser, D.; Huang, D.; Caflisch, A.; Hottiger, M.O. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010, 38, 6350–6362. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.N.; Lord, C.J.; McCabe, N.; Farmer, H.; Turner, N.; Martin, N.M.; Jackson, S.P.; Smith, G.C.; Ashworth, A. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 139–148. [Google Scholar] [CrossRef]
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Bouberhan, S.; Philp, L.; Hill, S.; Al-Alem, L.F.; Rueda, B. Exploiting the Prevalence of Homologous Recombination Deficiencies in High-Grade Serous Ovarian Cancer. Cancers 2020, 12, 1206. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef]
- Zandarashvili, L.; Langelier, M.F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Nowsheen, S.; Maraboyina, S.; Xia, F. The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: A review. Cell Biosci. 2020, 10, 35. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Daley, J.M.; Sung, P. RIF1 in DNA break repair pathway choice. Mol. Cell 2013, 49, 840–841. [Google Scholar] [CrossRef]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e923. [Google Scholar] [CrossRef]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Arana, M.E.; Seki, M.; Wood, R.D.; Rogozin, I.B.; Kunkel, T.A. Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res. 2008, 36, 3847–3856. [Google Scholar] [CrossRef]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat. Commun. 2019, 10, 4423. [Google Scholar] [CrossRef]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef]
- Blasina, A.; Price, B.D.; Turenne, G.A.; McGowan, C.H. Caffeine inhibits the checkpoint kinase ATM. Curr. Biol. 1999, 9, 1135–1138. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar] [PubMed]
- Powell, S.N.; DeFrank, J.S.; Connell, P.; Eogan, M.; Preffer, F.; Dombkowski, D.; Tang, W.; Friend, S. Differential sensitivity of p53(-) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Res. 1995, 55, 1643–1648. [Google Scholar] [CrossRef]
- Yao, S.L.; Akhtar, A.J.; McKenna, K.A.; Bedi, G.C.; Sidransky, D.; Mabry, M.; Ravi, R.; Collector, M.I.; Jones, R.J.; Sharkis, S.J.; et al. Selective radiosensitization of p53-deficient cells by caffeine-mediated activation of p34cdc2 kinase. Nat. Med. 1996, 2, 1140–1143. [Google Scholar] [CrossRef]
- Bracey, T.S.; Williams, A.C.; Paraskeva, C. Inhibition of radiation-induced G2 delay potentiates cell death by apoptosis and/or the induction of giant cells in colorectal tumor cells with disrupted p53 function. Clin. Cancer Res. 1997, 3, 1371–1381. [Google Scholar]
- Newton, R.; Broughton, L.J.; Lind, M.J.; Morrison, P.J.; Rogers, H.J.; Bradbrook, I.D. Plasma and salivary pharmacokinetics of caffeine in man. Eur. J. Clin. Pharmacol. 1981, 21, 45–52. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Tibbetts, R.S.; Busby, E.C.; Kennedy, A.P.; Hill, D.E.; Abraham, R.T. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998, 58, 4375–4382. [Google Scholar]
- Price, B.D.; Youmell, M.B. The phosphatidylinositol 3-kinase inhibitor wortmannin sensitizes murine fibroblasts and human tumor cells to radiation and blocks induction of p53 following DNA damage. Cancer Res. 1996, 56, 246–250. [Google Scholar]
- Karve, S.; Werner, M.E.; Sukumar, R.; Cummings, N.D.; Copp, J.A.; Wang, E.C.; Li, C.; Sethi, M.; Chen, R.C.; Pacold, M.E.; et al. Revival of the abandoned therapeutic wortmannin by nanoparticle drug delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 8230–8235. [Google Scholar] [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef]
- Golding, S.E.; Rosenberg, E.; Adams, B.R.; Wignarajah, S.; Beckta, J.M.; O’Connor, M.J.; Valerie, K. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle 2012, 11, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Raso, A.; Mascelli, S.; Nozza, P.; Garrè, M.L.; Pitto, F.; Ravetti, J.L.; et al. Pharmacokinetics, pharmacodynamics and efficacy on pediatric tumors of the glioma radiosensitizer KU60019. Int. J. Cancer 2015, 136, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef]
- Mak, J.P.Y.; Ma, H.T.; Poon, R.Y.C. Synergism between ATM and PARP1 Inhibition Involves DNA Damage and Abrogating the G(2) DNA Damage Checkpoint. Mol. Cancer Ther. 2020, 19, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Ruzankina, Y.; Schoppy, D.W.; Asare, A.; Clark, C.E.; Vonderheide, R.H.; Brown, E.J. Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat. Genet. 2009, 41, 1144–1149. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar] [CrossRef]
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.P.; George, J.; Nowak, M.; Leeser, U.; Büttner, R.; Perner, S.; Peifer, M.; et al. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017, 77, 3040–3056. [Google Scholar] [CrossRef] [PubMed]
- Combès, E.; Andrade, A.F.; Tosi, D.; Michaud, H.A.; Coquel, F.; Garambois, V.; Desigaud, D.; Jarlier, M.; Coquelle, A.; Pasero, P.; et al. Inhibition of Ataxia-Telangiectasia Mutated and RAD3-Related (ATR) Overcomes Oxaliplatin Resistance and Promotes Antitumor Immunity in Colorectal Cancer. Cancer Res. 2019, 79, 2933–2946. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef]
- Willoughby, C.E.; Jiang, Y.; Thomas, H.D.; Willmore, E.; Kyle, S.; Wittner, A.; Phillips, N.; Zhao, Y.; Tudhope, S.J.; Prendergast, L.; et al. Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J. Clin. Investig. 2020, 130, 258–271. [Google Scholar] [CrossRef]
- Liu, Y.; Wan, W.Z.; Li, Y.; Zhou, G.L.; Liu, X.G. Recent development of ATP-competitive small molecule phosphatidylinostitol-3-kinase inhibitors as anticancer agents. Oncotarget 2017, 8, 7181–7200. [Google Scholar] [CrossRef] [PubMed]
- Leder, A.; Leder, P. Butyric acid, a potent inducer of erythroid differentiation in cultured erythroleukemic cells. Cell 1975, 5, 319–322. [Google Scholar] [CrossRef]
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, T.; Teng, Z.H.; Zhang, R.; Wang, J.B.; Mei, Q.B. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol. Ther. 2009, 8, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Blattmann, C.; Oertel, S.; Ehemann, V.; Thiemann, M.; Huber, P.E.; Bischof, M.; Witt, O.; Deubzer, H.E.; Kulozik, A.E.; Debus, J.; et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 237–245. [Google Scholar] [CrossRef]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef]
- Kachhap, S.K.; Rosmus, N.; Collis, S.J.; Kortenhorst, M.S.; Wissing, M.D.; Hedayati, M.; Shabbeer, S.; Mendonca, J.; Deangelis, J.; Marchionni, L.; et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS ONE 2010, 5, e11208. [Google Scholar] [CrossRef]
- Groselj, B.; Kerr, M.; Kiltie, A.E. Radiosensitisation of bladder cancer cells by panobinostat is modulated by Ku80 expression. Radiother. Oncol. 2013, 108, 429–433. [Google Scholar] [CrossRef]
- Krumm, A.; Barckhausen, C.; Kücük, P.; Tomaszowski, K.H.; Loquai, C.; Fahrer, J.; Krämer, O.H.; Kaina, B.; Roos, W.P. Enhanced Histone Deacetylase Activity in Malignant Melanoma Provokes RAD51 and FANCD2-Triggered Drug Resistance. Cancer Res. 2016, 76, 3067–3077. [Google Scholar] [CrossRef]
- Maertens, O.; Kuzmickas, R.; Manchester, H.E.; Emerson, C.E.; Gavin, A.G.; Guild, C.J.; Wong, T.C.; De Raedt, T.; Bowman-Colin, C.; Hatchi, E.; et al. MAPK Pathway Suppression Unmasks Latent DNA Repair Defects and Confers a Chemical Synthetic Vulnerability in BRAF-, NRAS-, and NF1-Mutant Melanomas. Cancer Discov. 2019, 9, 526–545. [Google Scholar] [CrossRef]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Csernok, A.; Aydin, S.; Kreienberg, R.; Wiesmüller, L.; Gatz, S.A. Role of SIRT1 in homologous recombination. DNA Repair 2010, 9, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.E.; Bhaskara, S.; Stengel, K.R.; Zhao, Y.; Sirbu, B.; Chagot, B.; Cortez, D.; Khabele, D.; Chazin, W.J.; Cooper, A.; et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS ONE 2013, 8, e68915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, X.; Qi, Y.; He, Y.; Du, W.; Pang, J.J. Histone Deacetylases Inhibitors in the Treatment of Retinal Degenerative Diseases: Overview and Perspectives. J. Ophthalmol. 2015, 2015, 250812. [Google Scholar] [CrossRef]
- Das Gupta, K.; Shakespear, M.R.; Iyer, A.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases in monocyte/macrophage development, activation and metabolism: Refining HDAC targets for inflammatory and infectious diseases. Clin. Transl. Immunol. 2016, 5, e62. [Google Scholar] [CrossRef]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef]
- Heideman, M.R.; Wilting, R.H.; Yanover, E.; Velds, A.; de Jong, J.; Kerkhoven, R.M.; Jacobs, H.; Wessels, L.F.; Dannenberg, J.H. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 2013, 121, 2038–2050. [Google Scholar] [CrossRef]
- Santoro, F.; Botrugno, O.A.; Dal Zuffo, R.; Pallavicini, I.; Matthews, G.M.; Cluse, L.; Barozzi, I.; Senese, S.; Fornasari, L.; Moretti, S.; et al. A dual role for Hdac1: Oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 2013, 121, 3459–3468. [Google Scholar] [CrossRef]
- Hemshekhar, M.; Sebastin Santhosh, M.; Kemparaju, K.; Girish, K.S. Emerging roles of anacardic acid and its derivatives: A pharmacological overview. Basic Clin. Pharmacol. Toxicol. 2012, 110, 122–132. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Swaminathan, V.; Ranganathan, A.; Kundu, T.K. Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 2003, 278, 19134–19140. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Kohno, T. CBP and p300 histone acetyltransferases contribute to homologous recombination by transcriptionally activating the BRCA1 and RAD51 genes. PLoS ONE 2012, 7, e52810. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Price, B.D. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 2006, 580, 4353–4356. [Google Scholar] [CrossRef] [PubMed]
- Eliseeva, E.D.; Valkov, V.; Jung, M.; Jung, M.O. Characterization of novel inhibitors of histone acetyltransferases. Mol. Cancer Ther. 2007, 6, 2391–2398. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Altaf, M.; Varier, R.A.; Swaminathan, V.; Ravindran, A.; Sadhale, P.P.; Kundu, T.K. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J. Biol. Chem. 2004, 279, 33716–33726. [Google Scholar] [CrossRef]
- Oike, T.; Ogiwara, H.; Torikai, K.; Nakano, T.; Yokota, J.; Kohno, T. Garcinol, a histone acetyltransferase inhibitor, radiosensitizes cancer cells by inhibiting non-homologous end joining. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 815–821. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef]
- Ogiwara, H.; Ui, A.; Shiotani, B.; Zou, L.; Yasui, A.; Kohno, T. Curcumin suppresses multiple DNA damage response pathways and has potency as a sensitizer to PARP inhibitor. Carcinogenesis 2013, 34, 2486–2497. [Google Scholar] [CrossRef]
- Kumar, B.; Yadav, A.; Hideg, K.; Kuppusamy, P.; Teknos, T.N.; Kumar, P. A novel curcumin analog (H-4073) enhances the therapeutic efficacy of cisplatin treatment in head and neck cancer. PLoS ONE 2014, 9, e93208. [Google Scholar] [CrossRef]
- Santer, F.R.; Höschele, P.P.; Oh, S.J.; Erb, H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef]
- Wang, Y.M.; Gu, M.L.; Meng, F.S.; Jiao, W.R.; Zhou, X.X.; Yao, H.P.; Ji, F. Histone acetyltransferase p300/CBP inhibitor C646 blocks the survival and invasion pathways of gastric cancer cell lines. Int. J. Oncol. 2017, 51, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- de Boer, J.; Walf-Vorderwülbecke, V.; Williams, O. In focus: MLL-rearranged leukemia. Leukemia 2013, 27, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Klaus, C.R.; Iwanowicz, D.; Johnston, D.; Campbell, C.A.; Smith, J.J.; Moyer, M.P.; Copeland, R.A.; Olhava, E.J.; Scott, M.P.; Pollock, R.M.; et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther. 2014, 350, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.S.; Chakraborty, A.; Grigsby, S.M.; Umeano, A.C.; Liao, C.; Moukha-Chafiq, O.; Pathak, V.; Mathew, B.; Lee, Y.T.; Dou, Y.; et al. Identification of DOT1L inhibitors by structure-based virtual screening adapted from a nucleoside-focused library. Eur. J. Med. Chem. 2020, 189, 112023. [Google Scholar] [CrossRef]
- Byun, W.S.; Kim, W.K.; Yoon, J.S.; Jarhad, D.B.; Jeong, L.S.; Lee, S.K. Antiproliferative and Antimigration Activities of Fluoro-Neplanocin A via Inhibition of Histone H3 Methylation in Triple-Negative Breast Cancer. Biomolecules 2020, 10, 530. [Google Scholar] [CrossRef]
- Yang, L.; Lei, Q.; Li, L.; Yang, J.; Dong, Z.; Cui, H. Silencing or inhibition of H3K79 methyltransferase DOT1L induces cell cycle arrest by epigenetically modulating c-Myc expression in colorectal cancer. Clin. Epigenetics 2019, 11, 199. [Google Scholar] [CrossRef]
- Yang, C.; Chen, Z.; Yu, H.; Liu, X. Inhibition of Disruptor of Telomeric Silencing 1-Like Alleviated Renal Ischemia and Reperfusion Injury-Induced Fibrosis by Blocking PI3K/AKT-Mediated Oxidative Stress. Drug Des. Dev. Ther. 2019, 13, 4375–4387. [Google Scholar] [CrossRef]
- Williams, D.E.; Dalisay, D.S.; Li, F.; Amphlett, J.; Maneerat, W.; Chavez, M.A.; Wang, Y.A.; Matainaho, T.; Yu, W.; Brown, P.J.; et al. Nahuoic acid A produced by a Streptomyces sp. isolated from a marine sediment is a selective SAM-competitive inhibitor of the histone methyltransferase SETD8. Org. Lett. 2013, 15, 414–417. [Google Scholar] [CrossRef]
- Ma, A.; Yu, W.; Li, F.; Bleich, R.M.; Herold, J.M.; Butler, K.V.; Norris, J.L.; Korboukh, V.; Tripathy, A.; Janzen, W.P.; et al. Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J. Med. Chem. 2014, 57, 6822–6833. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, K.D.; Mitchell, T.R.; Upadhyay, A.K.; Jakob, C.G.; Jhala, M.A.; Comess, K.M.; Lasko, L.M.; Li, C.; Tuzon, C.T.; Dai, Y.; et al. The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat. Chem. Biol. 2017, 13, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Liang, D.; Jiang, N.; Zhou, J.; Long, X.; Wang, X.; Wu, M.; Wu, C.; Bao, J. Computer-aided screening for suppressor of variegation 4-20 homolog 1 inhibitors and their preliminary activity validation in human osteosarcoma. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gieni, R.S.; Ismail, I.H.; Campbell, S.; Hendzel, M.J. Polycomb group proteins in the DNA damage response: A link between radiation resistance and “stemness”. Cell Cycle 2011, 10, 883–894. [Google Scholar] [CrossRef]
- Simó-Riudalbas, L.; Esteller, M. Targeting the histone orthography of cancer: Drugs for writers, erasers and readers. Br. J. Pharmacol. 2015, 172, 2716–2732. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Ismail, I.H.; Young, L.C.; Poirier, G.G.; Hendzel, M.J. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle 2013, 12, 2675–2683. [Google Scholar] [CrossRef]
- Jeusset, L.M.; McManus, K.J. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells 2019, 8, 165. [Google Scholar] [CrossRef]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef] [PubMed]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Möbitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA Damage in Stem Cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef]
- Mandal, P.K.; Blanpain, C.; Rossi, D.J. DNA damage response in adult stem cells: Pathways and consequences. Nat. Rev. Mol. Cell Biol. 2011, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zou, R.S.; He, S.; Nihongaki, Y.; Li, X.; Razavi, S.; Wu, B.; Ha, T. Very fast CRISPR on demand. Science 2020, 368, 1265–1269. [Google Scholar] [CrossRef] [PubMed]





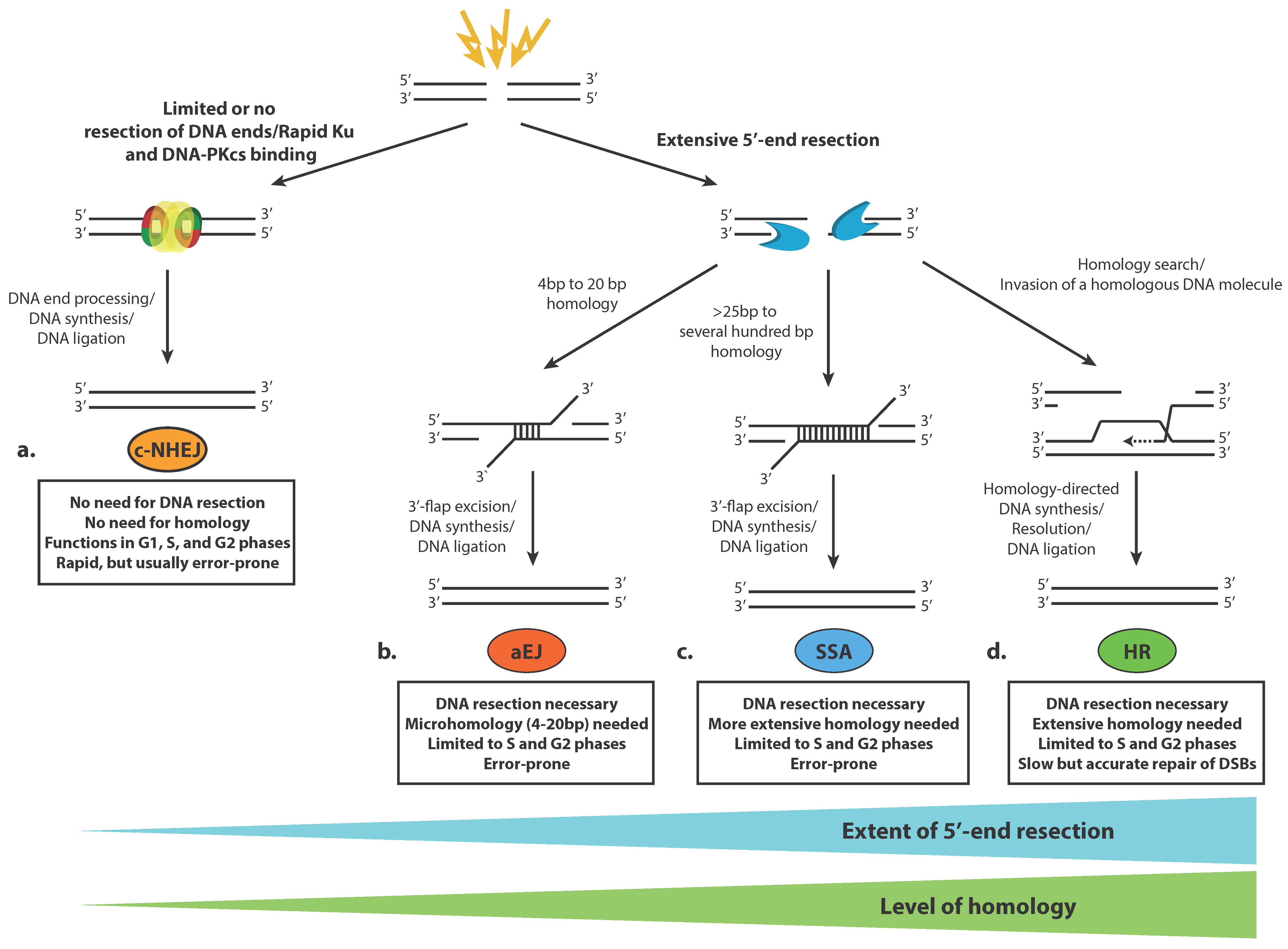{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Function | Model Organism | References |
|---|---|---|---|
| Phosphorylation | |||
| H2A.X S139 (H2A S129) | Primary beacon of damaged chromatin, necessary for the recruitment of chromatin regulators and repair factors | H. sapiens, M. muntjak, X. laevis, D. melanogaster; S. cerevisiae | [119,181] |
| H2A.X Y142 | Inhibitory to MDC1 binding, a switch between DNA repair and apoptosis | H. sapiens, M. musculus | [127,128] |
| H2BS14 | Phosphorylated in response to DNA damage, associated with chromatin condensation and apoptosis | M. musculus | [182] |
| H4 S1 | Inhibitory to NuA4-mediated acetylation of H4, participates in NHEJ in yeast | S. cerevisiae | [183,184] |
| Acetylation | |||
| H1K85 | Recruits HP1 and impedes HR and NHEJ | H. sapiens | [185] |
| H2A.X K5 | Mediates release of phosphorylated H2A.X | H. sapiens | [186] |
| H2BK120 | Promotes histone H1 eviction, and 53BP1 accumulation over γH2A.X | M. musculus, H. sapiens | [109,187] |
| H3K9,14,18,23,27 | GCN5-mediated acetylation in yeast triggered by HR repair. Changes dynamically during DNA repair | S. cerevisiae | [188] |
| H3K14 | Globally increased in HMGN-dependent manner in response to IR. Regulates ATM activation | M. musculus | [101] |
| H3K18 | Controls the recruitment of Ku proteins | H. sapiens | [189] |
| H3K56 | Recruits the chromatin remodeler SNF2H and promotes chromatin relaxation early in the repair process. Forms foci that colocalize with sites of DNA repair Deacetylated rapidly after break induction by HDAC1 and 2, necessary for NHEJ | M. musculus, Drosophila, H. sapiens, S. cerevisiae | [112,190,191] |
| H3, H4–N terminus | Acetylated by TRRAP-TIP60 control the recruitment of 53BP1, RAD51, and BRCA1 to DNA damage sites | H. sapiens | [192] |
| H4K5,8,12,16 | Controls the recruitment of Ku proteins. GCN5-mediated acetylation in yeast triggered by HR repair. Changes dynamically during DNA repair | S. cerevisiae, H. sapiens | [188,189] |
| H4K16 | Deacetylated rapidly after break induction by HDAC1 and -2 promoting NHEJ. Involved in the activation of ATM in response to ionizing radiation | M. musculus, H. sapiens | [101,193] |
| Ubiquitylation | |||
| H1 | Serves as a recruitment signal for RNF168, needed for interaction with downstream effectors | H. sapiens, M. musculus, Gallus gallus | [58,59,60,132] |
| H2AK13-15 | Critically important for 53BP1 and BRCA1 recruitment | H. sapiens, M. musculus, Gallus gallus | [59,60,129,132,194] |
| H2AK119 | Necessary to silence transcription in the vicinity of DSBs and promote HR repair | H. sapiens, G. gallus | [116,195,196] |
| H2BK120 | Essential for the timely accumulation of NHEJ and HR proteins at DSB sites (53BP1 and BRCA1 foci formation; XRCC4 and Ku80 recruitment); mediates chromatin relaxation | H. sapiens, M. musculus | [107,109,111] |
| H2AK125, 127, 129 | BRCA1/BARD1-mediated ubiquitination. Required for SMARCAD1 binding; promotes DSB end resection. | S. cerevisae, H. sapiens | [168,169,170,172] |
| H4K91 | Necessary for H4K20 methylation and 53BP1 foci formation at sites of DNA damage | H. sapiens | [197] |
| H4K119 | Induced by H2A.XK5Ac, necessary for the release of phosphorylated H2A.X. | H. sapiens | [186] |
| Methylation | |||
| H3K4me0 | Required for ZMYND8-NuRD binding to lesions. NuRD promotes HR repair | H. sapiens | [198] |
| H3K4me2 | Reduced at DNA damage sites by LSD1 to promote 53BP1 and BRCA1 foci formation | H. sapiens, M. musculus | [199] |
| H3K9me3 | Necessary for binding of HP1β, which is released early during DDR. Directs interaction with TIP60 and controls the recruitment and activity of ATM. Targeted by KDM4D to promote DSB repair | M. musculus, H. sapiens | [98,99,200,201] |
| H3K27me3 | Carried out by the EZH2 methyltransferase subunit of PRC2. Required for transcriptional repression around DSBs | H. sapiens | [117,202,203] |
| H3K36me | Promotes chromatin binding of NHEJ factors | H. sapiens | [204] |
| H3K36me2 | Increases MRN binding around DSB. Promotes BARD1 binding. Enhances the recruitment of Ku70. | H. sapiens | [125,157,204] |
| H3 K36me3 | Stimulates HR repair. Promotes resection via LEDGF-mediated CtIP recruitment | H. sapiens | [187,205,206,207,208] |
| H3 K79me | Promotes binding of 53BP1 | S. cerevisiae, H. sapiens | [134,149,150,151] |
| H4 K20me | Necessary for the accumulation of 53BP1 at DSBs. | S. pombe, M. musculus, H. sapiens | [135,136,137] |
| H4 K20me2 | Required for the binding of L3MBTL1 and TIP60 complex, which compete with 53BP1 for binding sites at DSBs. Necessary for 53BP1 binding | H. sapiens | [143,144,147,163,164] |
| Chromatin Remodeler | Function | Model Organism | References |
|---|---|---|---|
| SWI/SNF-family | BRG1 stimulates H2A.X phosphorylation. Binds H2AX-containing nucleosomes via acetylated H3. | H. sapiens | [209] |
| BRM1 subunit is required for the recruitment of Ku70 and Ku80. BRM1 needs acetylation of H3 by CBP and p300 for its recruitment. | H. sapiens | [189] | |
| BAF complex controls the accumulation of Ku70 | H. sapiens | [210] | |
| Human SWI/SNF complex participates in V(D)J recombination. | H. sapiens | [211,212,213] | |
| Yeast SWI/SNF complex participates in HR repair by remodeling nucleosomes at the donor locus. Disrupts heterochromatin by evicting Sir3 from a heterochromatic donor in silent mating-type loci in yeast to facilitate HR repair. Absence of functional SWI/SNF impaired recruitment of MRX and significantly delayed the initiation of DNA end resection. | H. sapiens, S. cerevisiae | [214,215,216] | |
| RSC complex is required following synapsis in recombination repair | H. sapiens | [214] | |
| SMARCAD1 is required for the repositioning of 53BP1 away from BRCA1 stimulating HR repair. SMARCAD1 and Fun30 are targeted by CDK1 and together with TopBP1 and yeast Dpb11 facilitate cell cycle-dependent DNA end resection. | H. sapiens, S. cerevisiae | [172,174,176,177,178] | |
| Fun 30 is associated with resection in yeast; influences the distribution of H2A.Z genome-wide and particularly in centromeric, pericentromeric, and subtelomeric chromatin. | S. cerevisiae, H. sapiens | [174,175,176,217,218,219] | |
| INO80 family | Deficiency of the INO80 complex leads to hypersensitivity to DSB-inducing agents in yeast and HR defects in mammalian cells. | S. cerevisiae, H. sapiens | [220,221,222] |
| INO80 is necessary for the initial 5′- resection at DSB ends prior to strand invasion in both yeast and mammals. | H. sapiens, S. pombe, S. cerevisiae | [222,223] | |
| In yeast INO80 mutants loading of Rad51 and Rad52 repair proteins was defective; In higher eukaryotes, INO80 depletion reduces Rad54B and XRCC3 transcription | S. cerevisiae, H. sapiens | [224,225] | |
| INO80 complex participates in the maintenance of H2AX phosphorylation levels by antagonizing the SWR1 remodeler. | S. cerevisiae | [226] | |
| Stimulate Rad51 binding to resected DNA during HR repair. | S. cerevisiae | [227] | |
| INO80 promotes nucleosome disassembly (manifested by removal of histone H3 from the genome) during NHEJ | H. sapiens | [228] | |
| Participate in chromatin-bound RNAPIIs degradation in yeast | S. cerevisiae | [229] | |
| Low levels of SRCAP impair resection due to defective CtIP recruitment | H. sapiens | [230] | |
| SWR1 complex maintains H2AX levels by replacing it with H2AZ. Involved in NHEJ, facilitates the recruitment of Ku proteins. | S. cerevisiae | [226,231] | |
| p400 complex deposits H2AZ required for the loading of Ku70/Ku80 | H. sapiens | [232] | |
| CHD family | CHD1 is required for the recruitment of CtIP and HR repair | H. sapiens | [233] |
| CHD1B (ALC1) localizes to DSBs in PAR-dependent manner and interacts with Ku70, XRCC1, and DNA-PKcs. | H. sapiens | [234] | |
| CHD2 is involved in the deposition of histone variant H3.3 at sites of DNA damage and efficient assembly of NHEJ complexes | H. sapiens | [235] | |
| CHD4, together with RNF8, creates a chromatin environment that is permissive to the assembly of checkpoint and repair machinery at DSBs | H. sapiens | [133,236] | |
| NuRD complex is implicated in promoting HR repair by repressing transcription at DSBs Recruited to DSBs in a poly(ADP-ribose)-dependent manner, stimulates recruitment of RNF168 and BRCA1 to DSBs. Interacts with Ku70 and is required for the recruitment of Ku proteins at DSBs | H. sapiens, C. elegans H. sapiens H. sapiens, S. pombe | [237,238,239,240,241] | |
| ISWI family | SNF2H promoteschromatin relaxation early in the repair process; SNF2H is recruited to RNF20-ubiquitylated H2B and its depletion impairs DNA end processing and recruitment of RAD51 and BRCA1. | H. sapiens | [110,112] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleksandrov, R.; Hristova, R.; Stoynov, S.; Gospodinov, A. The Chromatin Response to Double-Strand DNA Breaks and Their Repair. Cells 2020, 9, 1853. https://doi.org/10.3390/cells9081853
Aleksandrov R, Hristova R, Stoynov S, Gospodinov A. The Chromatin Response to Double-Strand DNA Breaks and Their Repair. Cells. 2020; 9(8):1853. https://doi.org/10.3390/cells9081853
Chicago/Turabian StyleAleksandrov, Radoslav, Rossitsa Hristova, Stoyno Stoynov, and Anastas Gospodinov. 2020. "The Chromatin Response to Double-Strand DNA Breaks and Their Repair" Cells 9, no. 8: 1853. https://doi.org/10.3390/cells9081853
APA StyleAleksandrov, R., Hristova, R., Stoynov, S., & Gospodinov, A. (2020). The Chromatin Response to Double-Strand DNA Breaks and Their Repair. Cells, 9(8), 1853. https://doi.org/10.3390/cells9081853