Preterm Brain Injury, Antenatal Triggers, and Therapeutics: Timing Is Key

 , , and
, , and 
Abstract
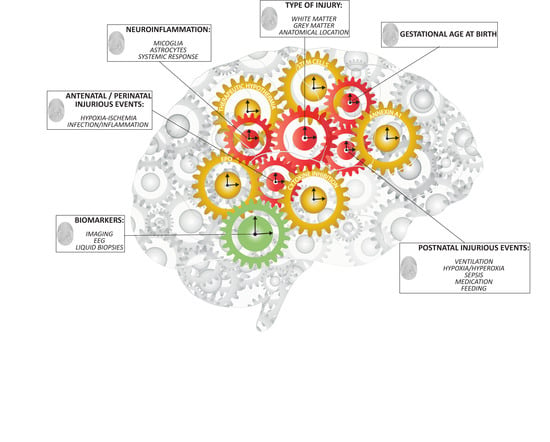
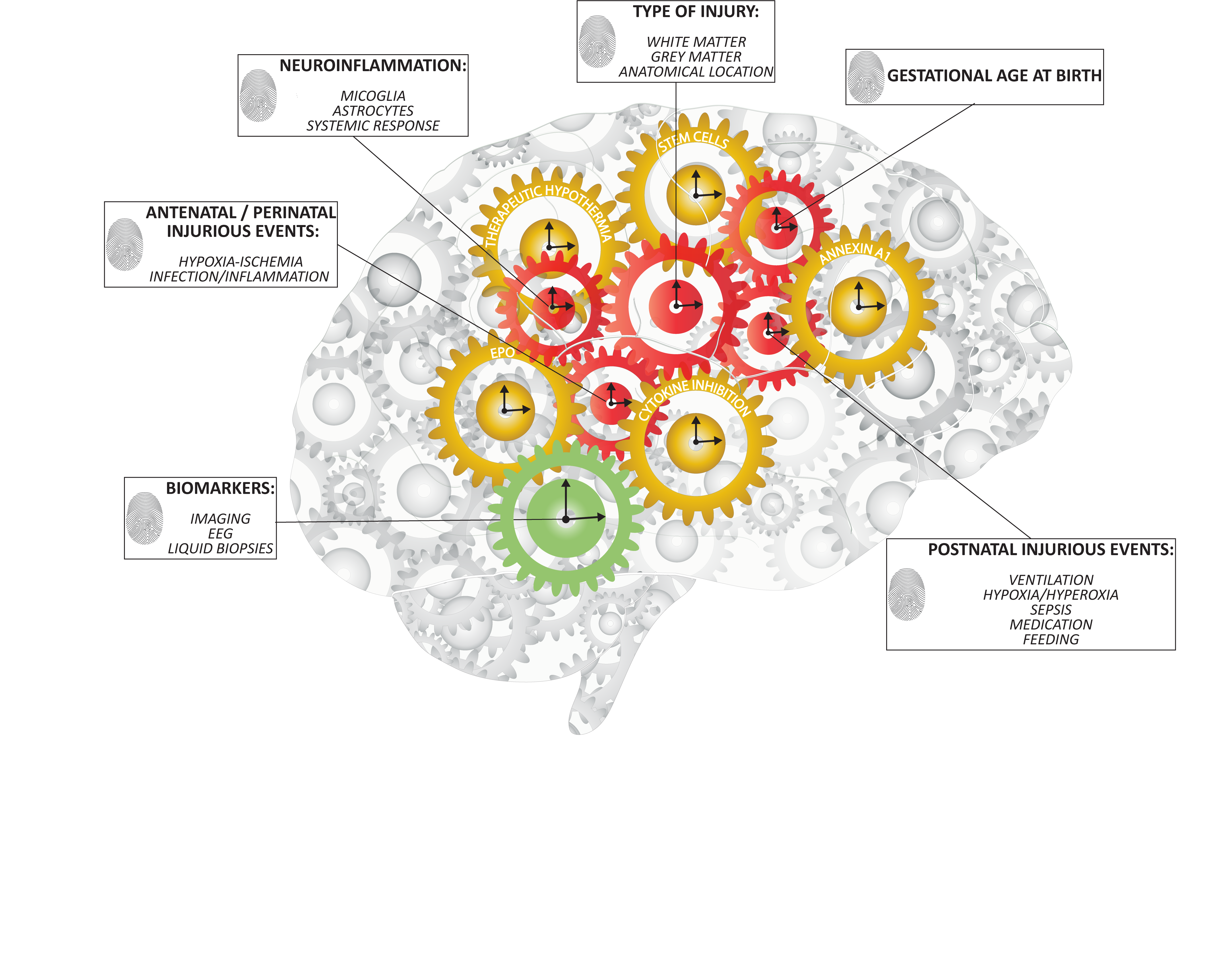
1. Patterns of Preterm Brain Injury
1.1. Diffuse White Matter Injury
1.2. Grey Matter Injury
1.3. Cerebellar Injury
2. Pathophysiology of Preterm Brain Injury
2.1. Intrauterine Infection and/or Inflammation
2.2. Fetal Immune Response Syndrome
2.3. Hypoxia-Ischemia
3. Preterm Brain Injury—Timing Is Key
3.1. Antenatal Inflammation
3.2. Hypoxia-Ischemia
3.3. Blood-Brain Barrier
3.4. Immune System Activation
3.4.1. Innate Immune System
3.4.2. Astrocytes
3.4.3. Adaptive Immunity
4. Treatment
4.1. Therapeutic Hypothermia for Preterm Hypoxia-Ischemia
4.2. Cell-Based Therapies
4.2.1. Stem Cells
4.2.2. Clinical Stem Cell Trials
4.2.3. Stem Cell Therapy—Timing Is Key
4.2.4. Stem Cell-Derived Extracellular Vesicles
4.3. Pharmacological Interventions
4.3.1. AnnexinA1
4.3.2. Cytokine Treatment
4.3.3. Recombinant Human Erythropoietin for Preterm Neonates
5. Future Perspectives—Closing the Translational Gap
5.1. Multi-Factorial Nature of Preterm Brain Injury
5.2. Biomarkers
5.2.1. Plasma Biomarkers
5.2.2. Imaging
5.2.3. EEG
5.3. Postnatal Factors
5.3.1. Ventilation-Induced Brain Injury
5.3.2. Feeding
5.3.3. Postnatal Medication
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Back, S.A.; Miller, S.P. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann. Neurol. 2014, 75, 469–486. [Google Scholar] [CrossRef] [PubMed]
- D’Apremont, I.; Marshall, G.; Musalem, C.; Mariani, G.; Musante, G.; Bancalari, A.; Fabres, J.; Mena, P.; Zegarra, J.; Tavosnanska, J. Trends in Perinatal Practices and Neonatal Outcomes of VLBW Infants during a 16-year Period at NEOCOSUR Centers. J. Pediatr. 2020. [Google Scholar] [CrossRef]
- Van Haastert, I.C.; Groenendaal, F.; Uiterwaal, C.S.; Termote, J.U.; van der Heide-Jalving, M.; Eijsermans, M.J.; Gorter, J.W.; Helders, P.J.; Jongmans, M.J.; de Vries, L.S. Decreasing incidence and severity of cerebral palsy in prematurely born children. J. Pediatr. 2011, 159, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Logitharajah, P.; Rutherford, M.A.; Cowan, F.M. Hypoxic-ischemic encephalopathy in preterm infants: Antecedent factors, brain imaging, and outcome. Pediatric Res. 2009, 66, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, M.A.; Supramaniam, V.; Ederies, A.; Chew, A.; Bassi, L.; Groppo, M.; Anjari, M.; Counsell, S.; Ramenghi, L.A. Magnetic resonance imaging of white matter diseases of prematurity. Neuroradiology 2010, 52, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Alexandrou, G.; Martensson, G.; Skiold, B.; Blennow, M.; Aden, U.; Vollmer, B. White matter microstructure is influenced by extremely preterm birth and neonatal respiratory factors. Acta Paediatr. 2014, 103, 48–56. [Google Scholar] [CrossRef]
- Jakovcevski, I.; Zecevic, N. Sequence of oligodendrocyte development in the human fetal telencephalon. Glia 2005, 49, 480–491. [Google Scholar] [CrossRef]
- Batalle, D.; O’Muircheartaigh, J.; Makropoulos, A.; Kelly, C.J.; Dimitrova, R.; Hughes, E.J.; Hajnal, J.V.; Zhang, H.; Alexander, D.C.; Edwards, A.D. Different patterns of cortical maturation before and after 38 weeks gestational age demonstrated by diffusion MRI in vivo. NeuroImage 2019, 185, 764–775. [Google Scholar] [CrossRef]
- Deoni, S.C.; Dean III, D.C.; Remer, J.; Dirks, H.; O’Muircheartaigh, J. Cortical maturation and myelination in healthy toddlers and young children. Neuroimage 2015, 115, 147–161. [Google Scholar] [CrossRef]
- Grydeland, H.; Walhovd, K.B.; Tamnes, C.K.; Westlye, L.T.; Fjell, A.M. Intracortical myelin links with performance variability across the human lifespan: Results from T1-and T2-weighted MRI myelin mapping and diffusion tensor imaging. J. Neurosci. 2013, 33, 18618–18630. [Google Scholar] [CrossRef]
- Ouyang, M.; Dubois, J.; Yu, Q.; Mukherjee, P.; Huang, H. Delineation of early brain development from fetuses to infants with diffusion MRI and beyond. Neuroimage 2019, 185, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Van Tilborg, E.; de Theije, C.G.M.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018, 66, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J.; Kinney, H.C.; Jensen, F.E.; Rosenberg, P.A. The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int. J. Dev. Neurosci. 2011, 29, 423–440. [Google Scholar] [CrossRef]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Gan, X.; Li, Y.; Rosenberg, P.A.; Volpe, J.J. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J. Neurosci. 1998, 18, 6241–6253. [Google Scholar] [CrossRef]
- Segovia, K.N.; McClure, M.; Moravec, M.; Luo, N.L.; Wan, Y.; Gong, X.; Riddle, A.; Craig, A.; Struve, J.; Sherman, L.S.; et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 2008, 63, 520–530. [Google Scholar] [CrossRef]
- Back, S.A.; Rosenberg, P.A. Pathophysiology of glia in perinatal white matter injury. Glia 2014, 62, 1790–1815. [Google Scholar] [CrossRef]
- Keunen, K.; Kersbergen, K.J.; Groenendaal, F.; Isgum, I.; de Vries, L.S.; Benders, M.J. Brain tissue volumes in preterm infants: Prematurity, perinatal risk factors and neurodevelopmental outcome: A systematic review. J. Matern. Fetal. Neonatal Med. 2012, 25, 89–100. [Google Scholar] [CrossRef]
- Volpe, J.J. Dysmaturation of Premature Brain: Importance, Cellular Mechanisms, and Potential Interventions. Pediatr. Neurol. 2019. [Google Scholar] [CrossRef]
- Galinsky, R.; Lear, C.A.; Dean, J.M.; Wassink, G.; Dhillon, S.K.; Fraser, M.; Davidson, J.O.; Bennet, L.; Gunn, A.J. Complex interactions between hypoxia-ischemia and inflammation in preterm brain injury. Dev. Med. Child. Neurol. 2018, 60, 126–133. [Google Scholar] [CrossRef]
- Riddle, A.; Maire, J.; Gong, X.; Chen, K.X.; Kroenke, C.D.; Hohimer, A.R.; Back, S.A. Differential susceptibility to axonopathy in necrotic and non-necrotic perinatal white matter injury. Stroke 2012, 43, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Van Tilborg, E.; Achterberg, E.J.M.; van Kammen, C.M.; van der Toorn, A.; Groenendaal, F.; Dijkhuizen, R.M.; Heijnen, C.J.; Vanderschuren, L.; Benders, M.; Nijboer, C.H.A. Combined fetal inflammation and postnatal hypoxia causes myelin deficits and autism-like behavior in a rat model of diffuse white matter injury. Glia 2018, 66, 78–93. [Google Scholar] [CrossRef] [PubMed]
- McClendon, E.; Chen, K.; Gong, X.; Sharifnia, E.; Hagen, M.; Cai, V.; Shaver, D.C.; Riddle, A.; Dean, J.M.; Gunn, A.J.; et al. Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann. Neurol. 2014, 75, 508–524. [Google Scholar] [CrossRef] [PubMed]
- McClendon, E.; Shaver, D.C.; Degener-O’Brien, K.; Gong, X.; Nguyen, T.; Hoerder-Suabedissen, A.; Molnar, Z.; Mohr, C.; Richardson, B.D.; Rossi, D.J.; et al. Transient Hypoxemia Chronically Disrupts Maturation of Preterm Fetal Ovine Subplate Neuron Arborization and Activity. J. Neurosci 2017, 37, 11912–11929. [Google Scholar] [CrossRef]
- Fowke, T.M.; Galinsky, R.; Davidson, J.O.; Wassink, G.; Karunasinghe, R.N.; Prasad, J.D.; Bennet, L.; Gunn, A.J.; Dean, J.M. Loss of interneurons and disruption of perineuronal nets in the cerebral cortex following hypoxia-ischaemia in near-term fetal sheep. Sci. Rep. 2018, 8, 1–13. [Google Scholar]
- Van Kooij, B.J.; Benders, M.J.; Anbeek, P.; Van Haastert, I.C.; De Vries, L.S.; Groenendaal, F. Cerebellar volume and proton magnetic resonance spectroscopy at term, and neurodevelopment at 2 years of age in preterm infants. Dev. Med. Child. Neurol. 2012, 54, 260–266. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Limperopoulos, C. Structure-function relationships in the developing cerebellum: Evidence from early-life cerebellar injury and neurodevelopmental disorders. Semin. Fetal Neonat. Med. 2016, 21, 356–364. [Google Scholar] [CrossRef]
- Pieterman, K.; White, T.J.; van den Bosch, G.E.; Niessen, W.J.; Reiss, I.K.M.; Tibboel, D.; Hoebeek, F.E.; Dudink, J. Cerebellar Growth Impairment Characterizes School-Aged Children Born Preterm without Perinatal Brain Lesions. Ajnr Am. J. Neuroradiol. 2018, 39, 956–962. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Bassan, H.; Gauvreau, K.; Robertson, R.L., Jr.; Sullivan, N.R.; Benson, C.B.; Avery, L.; Stewart, J.; Soul, J.S.; Ringer, S.A.; et al. Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics 2007, 120, 584–593. [Google Scholar] [CrossRef]
- Volpe, J.J. Cerebellum of the premature infant: Rapidly developing, vulnerable, clinically important. J. Child. Neurol. 2009, 24, 1085–1104. [Google Scholar] [CrossRef]
- Wu, Y.; Stoodley, C.; Brossard-Racine, M.; Kapse, K.; Vezina, G.; Murnick, J.; du Plessis, A.J.; Limperopoulos, C. Altered local cerebellar and brainstem development in preterm infants. NeuroImage 2020, 116702. [Google Scholar] [CrossRef] [PubMed]
- Hutton, L.C.; Yan, E.; Yawno, T.; Castillo-Melendez, M.; Hirst, J.J.; Walker, D.W. Injury of the developing cerebellum: A brief review of the effects of endotoxin and asphyxial challenges in the late gestation sheep fetus. Cerebellum 2014, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Polglase, G.R.; Hooper, S.B.; Black, M.J.; Moss, T.J. The consequences of chorioamnionitis: Preterm birth and effects on development. J. Pregnancy 2013, 2013, 412831. [Google Scholar] [CrossRef] [PubMed]
- Yap, V.; Perlman, J.M. Mechanisms of brain injury in newborn infants associated with the fetal inflammatory response syndrome. Semin. Fetal Neonat. Med. 2020. [Google Scholar] [CrossRef]
- Kim, C.J.; Romero, R.; Chaemsaithong, P.; Chaiyasit, N.; Yoon, B.H.; Kim, Y.M. Acute chorioamnionitis and funisitis: Definition, pathologic features, and clinical significance. Am. J. Obs. Gynecol 2015, 213, S29–S52. [Google Scholar] [CrossRef]
- Goldenberg, R.L.; Hauth, J.C.; Andrews, W.W. Intrauterine infection and preterm delivery. N. Engl. J. Med. 2000, 342, 1500–1507. [Google Scholar] [CrossRef]
- Romero, R.; Espinoza, J.; Goncalves, L.F.; Kusanovic, J.P.; Friel, L.A.; Nien, J.K. Inflammation in preterm and term labour and delivery. Semin Fetal Neonatal Med. 2006, 11, 317–326. [Google Scholar] [CrossRef]
- Elovitz, M.A.; Brown, A.G.; Breen, K.; Anton, L.; Maubert, M.; Burd, I. Intrauterine inflammation, insufficient to induce parturition, still evokes fetal and neonatal brain injury. Int. J. Dev. Neurosci. 2011, 29, 663–671. [Google Scholar] [CrossRef]
- Arayici, S.; Kadioglu Simsek, G.; Oncel, M.Y.; Eras, Z.; Canpolat, F.E.; Oguz, S.S.; Uras, N.; Zergeroglu, S.; Dilmen, U. The effect of histological chorioamnionitis on the short-term outcome of preterm infants ≤ 32 weeks: A single-center study . J. Matern. Fetal Neonatal. Med. 2014, 27, 1129–1133. [Google Scholar] [CrossRef]
- Lu, H.Y.; Zhang, Q.; Wang, Q.X.; Lu, J.Y. Contribution of Histologic Chorioamnionitis and Fetal Inflammatory Response Syndrome to Increased Risk of Brain Injury in Infants With Preterm Premature Rupture of Membranes. Pediatr. Neurol. 2016, 61, 94–98 e91. [Google Scholar] [CrossRef]
- Korzeniewski, S.J.; Romero, R.; Cortez, J.; Pappas, A.; Schwartz, A.G.; Kim, C.J.; Kim, J.S.; Kim, Y.M.; Yoon, B.H.; Chaiworapongsa, T.; et al. A “multi-hit” model of neonatal white matter injury: Cumulative contributions of chronic placental inflammation, acute fetal inflammation and postnatal inflammatory events. J. Perinat. Med. 2014, 42, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Soraisham, A.S.; Trevenen, C.; Wood, S.; Singhal, N.; Sauve, R. Histological chorioamnionitis and neurodevelopmental outcome in preterm infants. J. Perinatol 2013, 33, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Gomez, R.; Romero, R.; Ghezzi, F.; Yoon, B.H.; Mazor, M.; Berry, S.M. The fetal inflammatory response syndrome. Am. J. Obs. Gynecol 1998, 179, 194–202. [Google Scholar] [CrossRef]
- Yoon, B.H.; Jun, J.K.; Romero, R.; Park, K.H.; Gomez, R.; Choi, J.H.; Kim, I.O. Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha), neonatal brain white matter lesions, and cerebral palsy. Am. J. Obs. Gynecol 1997, 177, 19–26. [Google Scholar] [CrossRef]
- Pappas, A.; Kendrick, D.E.; Shankaran, S.; Stoll, B.J.; Bell, E.F.; Laptook, A.R.; Walsh, M.C.; Das, A.; Hale, E.C.; Newman, N.S.; et al. Chorioamnionitis and early childhood outcomes among extremely low-gestational-age neonates. Jama Pediatr 2014, 168, 137–147. [Google Scholar] [CrossRef]
- Malaeb, S.; Dammann, O. Fetal inflammatory response and brain injury in the preterm newborn. J. Child. Neurol 2009, 24, 1119–1126. [Google Scholar] [CrossRef]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef]
- Manuck, T.A.; Rice, M.M.; Bailit, J.L.; Grobman, W.A.; Reddy, U.M.; Wapner, R.J.; Thorp, J.M.; Caritis, S.N.; Prasad, M.; Tita, A.T. Preterm neonatal morbidity and mortality by gestational age: A contemporary cohort. Am. J. Obstet. Gynecol. 2016, 215, 103.e101–103.e114. [Google Scholar] [CrossRef]
- Graham, E.M.; Ruis, K.A.; Hartman, A.L.; Northington, F.J.; Fox, H.E. A systematic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. Am. J. Obstet. Gynecol. 2008, 199, 587–595. [Google Scholar] [CrossRef]
- Rees, S.; Inder, T. Fetal and neonatal origins of altered brain development. Early Hum. Dev. 2005, 81, 753–761. [Google Scholar] [CrossRef]
- Wang, Q.; Lv, H.; Lu, L.; Ren, P.; Li, L. Neonatal hypoxic-ischemic encephalopathy: Emerging therapeutic strategies based on pathophysiologic phases of the injury. J. Matern. Fetal Neonatal. Med. 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Roelfsema, V.; Bennet, L.; George, S.; Wu, D.; Guan, J.; Veerman, M.; Gunn, A.J. Window of opportunity of cerebral hypothermia for postischemic white matter injury in the near-term fetal sheep. J. Cereb. Blood Flow Metab. 2004, 24, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Fleiss, B.; Gressens, P. Tertiary mechanisms of brain damage: A new hope for treatment of cerebral palsy? Lancet Neurol. 2012, 11, 556–566. [Google Scholar] [CrossRef]
- Gussenhoven, R.; Ophelders, D.R.M.G.; Kemp, M.W.; Payne, M.S.; Spiller, O.B.; Beeton, M.L.; Stock, S.J.; Cillero-Pastor, B.; Barré, F.P.Y.; Heeren, R.M.A.; et al. The Paradoxical Effects of Chronic Intra-Amniotic Ureaplasma parvum Exposure on Ovine Fetal Brain Development. Dev. Neurosci. 2017, 39, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Ophelders, D.R.; Gussenhoven, R.; Lammens, M.; Küsters, B.; Kemp, M.W.; Newnham, J.P.; Payne, M.S.; Kallapur, S.G.; Jobe, A.H.; Zimmermann, L.J. Neuroinflammation and structural injury of the fetal ovine brain following intra-amniotic Candida albicans exposure. J. Neuroinflamm. 2016, 13, 1–12. [Google Scholar] [CrossRef]
- Gussenhoven, R.; Westerlaken, R.J.J.; Ophelders, D.; Jobe, A.H.; Kemp, M.W.; Kallapur, S.G.; Zimmermann, L.J.; Sangild, P.T.; Pankratova, S.; Gressens, P.; et al. Chorioamnionitis, neuroinflammation, and injury: Timing is key in the preterm ovine fetus. J. Neuroinflamm. 2018, 15, 113. [Google Scholar] [CrossRef]
- Carson, M.J.; Thrash, J.C.; Walter, B. The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 2006, 6, 237–245. [Google Scholar] [CrossRef]
- Volpe, J.J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res. 2001, 50, 553–562. [Google Scholar] [CrossRef]
- Prout, A.P.; Frasch, M.G.; Veldhuizen, R.A.; Hammond, R.; Ross, M.G.; Richardson, B.S. Systemic and cerebral inflammatory response to umbilical cord occlusions with worsening acidosis in the ovine fetus. Am. J. Obs. Gynecol. 2010, 202, 82.e81–89. [Google Scholar] [CrossRef]
- Brew, N.; Azhan, A.; den Heijer, I.; Boomgardt, M.; Davies, G.I.; Nitsos, I.; Miller, S.L.; Walker, A.M.; Walker, D.W.; Wong, F.Y. Dopamine treatment during acute hypoxia is neuroprotective in the developing sheep brain. Neuroscience 2016, 316, 82–93. [Google Scholar] [CrossRef]
- Brew, N.; Nakamura, S.; Hale, N.; Azhan, A.; Davies, G.I.; Nitsos, I.; Miller, S.L.; Walker, D.W.; Wong, F.Y. Dobutamine treatment reduces inflammation in the preterm fetal sheep brain exposed to acute hypoxia. Pediatr. Res. 2018, 84, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Wassink, G.; Davidson, J.O.; Dhillon, S.K.; Fraser, M.; Galinsky, R.; Bennet, L.; Gunn, A.J. Partial white and grey matter protection with prolonged infusion of recombinant human erythropoietin after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 2017, 37, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Jellema, R.K.; Passos, V.L.; Zwanenburg, A.; Ophelders, D.R.; De Munter, S.; Vanderlocht, J.; Germeraad, W.T.; Kuypers, E.; Collins, J.J.; Cleutjens, J.P. Cerebral inflammation and mobilization of the peripheral immune system following global hypoxia-ischemia in preterm sheep. J. Neuroinflamm. 2013, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jellema, R.K.; Lima Passos, V.; Ophelders, D.R.M.G.; Wolfs, T.G.A.M.; Zwanenburg, A.; De Munter, S.; Nikiforou, M.; Collins, J.J.P.; Kuypers, E.; Bos, G.M.J.; et al. Systemic G-CSF attenuates cerebral inflammation and hypomyelination but does not reduce seizure burden in preterm sheep exposed to global hypoxia–ischemia. Exp. Neurol. 2013, 250, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Jellema, R.K.; Wolfs, T.G.; Lima Passos, V.; Zwanenburg, A.; Ophelders, D.R.; Kuypers, E.; Hopman, A.H.; Dudink, J.; Steinbusch, H.W.; Andriessen, P.; et al. Mesenchymal stem cells induce T-cell tolerance and protect the preterm brain after global hypoxia-ischemia. PLoS ONE 2013, 8, e73031. [Google Scholar] [CrossRef]
- Ophelders, D.R.M.G.; Wolfs, T.G.A.M.; Jellema, R.K.; Zwanenburg, A.; Andriessen, P.; Delhaas, T.; Ludwig, A.-K.; Radtke, S.; Peters, V.; Janssen, L.; et al. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Protect the Fetal Brain After Hypoxia-Ischemia. Stem Cells Transl. Med. 2016, 5, 754–763. [Google Scholar] [CrossRef]
- Jellema, R.K.; Ophelders, D.R.G.; Zwanenburg, A.; Nikiforou, M.; Delhaas, T.; Andriessen, P.; Mays, R.W.; Deans, R.; Germeraad, W.T.; Wolfs, T.G. Multipotent adult progenitor cells for hypoxic-ischemic injury in the preterm brain. J. Neuroinflamm. 2015, 12, 1–14. [Google Scholar] [CrossRef]
- Davidson, J.O.; Drury, P.P.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS ONE 2014, 9, e96558. [Google Scholar] [CrossRef]
- Abbasi, H.; Drury, P.P.; Lear, C.A.; Gunn, A.J.; Davidson, J.O.; Bennet, L.; Unsworth, C.P. EEG sharp waves are a biomarker of striatal neuronal survival after hypoxia-ischemia in preterm fetal sheep. Sci. Rep. 2018, 8, 16312. [Google Scholar] [CrossRef]
- Yawno, T.; Mahen, M.; Li, J.; Fahey, M.C.; Jenkin, G.; Miller, S.L. The beneficial effects of melatonin administration following hypoxia-ischemia in preterm fetal sheep. Front. Cell. Neurosci. 2017, 11, 296. [Google Scholar] [CrossRef]
- Li, J.; Yawno, T.; Sutherland, A.E.; Gurung, S.; Paton, M.; McDonald, C.; Tiwari, A.; Pham, Y.; Castillo-Melendez, M.; Jenkin, G.; et al. Preterm umbilical cord blood derived mesenchymal stem/stromal cells protect preterm white matter brain development against hypoxia-ischemia. Exp. Neurol. 2018, 308, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yawno, T.; Sutherland, A.; Loose, J.; Nitsos, I.; Bischof, R.; Castillo-Melendez, M.; McDonald, C.A.; Wong, F.Y.; Jenkin, G. Preterm white matter brain injury is prevented by early administration of umbilical cord blood cells. Exp. Neurol. 2016, 283, 179–187. [Google Scholar] [CrossRef] [PubMed]
- van den Heuij, L.G.; Fraser, M.; Miller, S.L.; Jenkin, G.; Wallace, E.M.; Davidson, J.O.; Lear, C.A.; Lim, R.; Wassink, G.; Gunn, A.J. Delayed intranasal infusion of human amnion epithelial cells improves white matter maturation after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 2019, 39, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Bennet, L.; Roelfsema, V.; George, S.; Dean, J.M.; Emerald, B.S.; Gunn, A.J. The effect of cerebral hypothermia on white and grey matter injury induced by severe hypoxia in preterm fetal sheep. J. Physiol. 2007, 578, 491–506. [Google Scholar] [CrossRef]
- Riddle, A.; Dean, J.; Buser, J.R.; Gong, X.; Maire, J.; Chen, K.; Ahmad, T.; Cai, V.; Nguyen, T.; Kroenke, C.D. Histopathological correlates of magnetic resonance imaging–defined chronic perinatal white matter injury. Ann. Neurol. 2011, 70, 493–507. [Google Scholar] [CrossRef]
- Buser, J.R.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 2012, 71, 93–109. [Google Scholar] [CrossRef]
- Davies, A.; Wassink, G.; Bennet, L.; Gunn, A.J.; Davidson, J.O. Can we further optimize therapeutic hypothermia for hypoxic-ischemic encephalopathy? Neural Regen. Res. 2019, 14, 1678. [Google Scholar]
- Lee, W.L.A.; Michael-Titus, A.T.; Shah, D.K. Hypoxic-Ischaemic Encephalopathy and the Blood-Brain Barrier in Neonates. Dev. Neurosci. 2017, 39, 49–58. [Google Scholar] [CrossRef]
- Malaeb, S.N.; Sadowska, G.B.; Stonestreet, B.S. Effects of maternal treatment with corticosteroids on tight junction protein expression in the cerebral cortex of the ovine fetus with and without exposure to in utero brain ischemia. Brain Res. 2007, 1160, 11–19. [Google Scholar] [CrossRef][Green Version]
- Chen, X.; Threlkeld, S.W.; Cummings, E.E.; Juan, I.; Makeyev, O.; Besio, W.G.; Gaitanis, J.; Banks, W.A.; Sadowska, G.B.; Stonestreet, B.S. Ischemia–reperfusion impairs blood–brain barrier function and alters tight junction protein expression in the ovine fetus. Neuroscience 2012, 226, 89–100. [Google Scholar] [CrossRef]
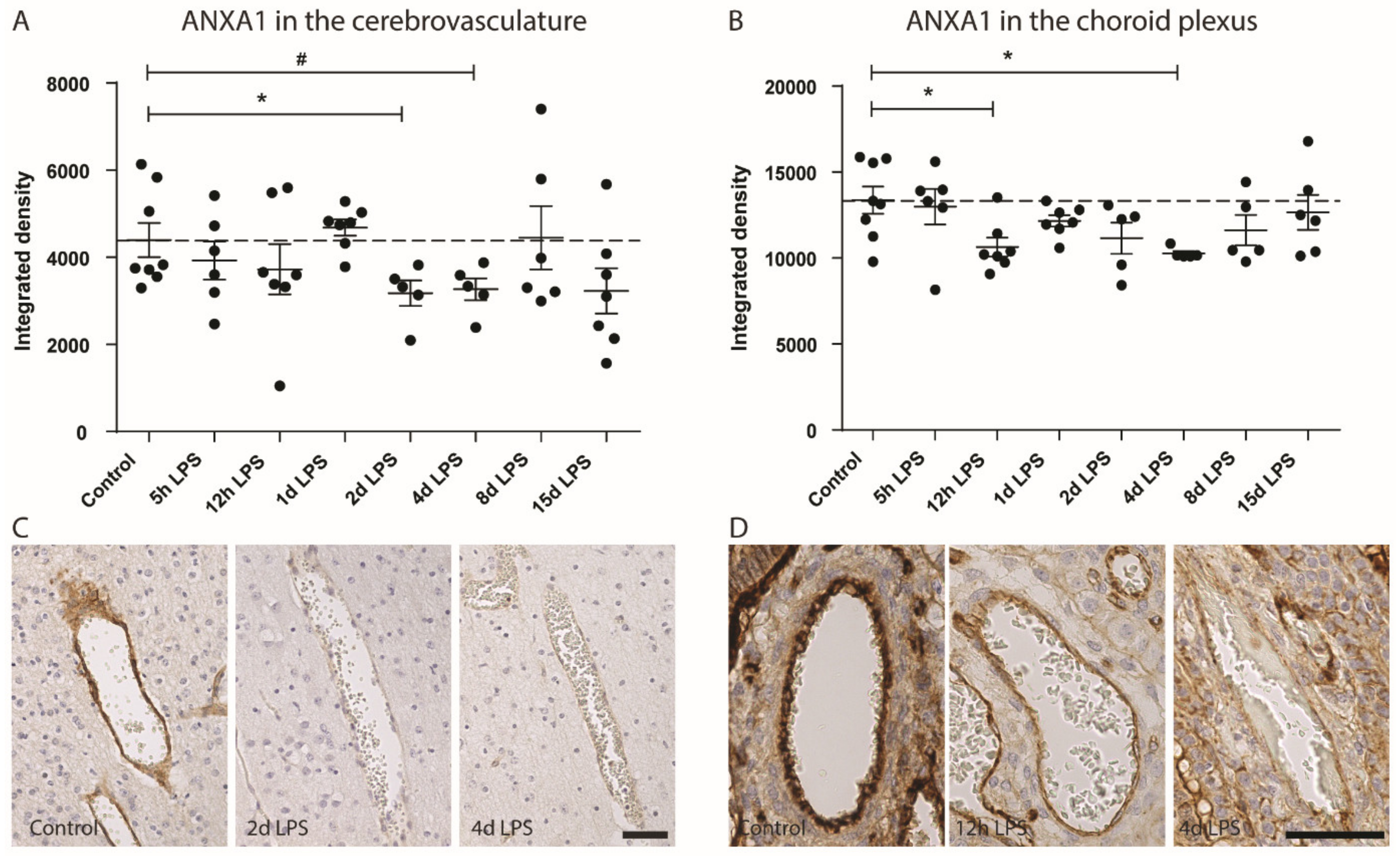
- Gussenhoven, R.; Klein, L.; Ophelders, D.; Habets, D.H.J.; Giebel, B.; Kramer, B.W.; Schurgers, L.J.; Reutelingsperger, C.P.M.; Wolfs, T. Annexin A1 as Neuroprotective Determinant for Blood-Brain Barrier Integrity in Neonatal Hypoxic-Ischemic Encephalopathy. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Loiola, R.A.; Wickstead, E.S.; Solito, E.; McArthur, S. Estrogen Promotes Pro-resolving Microglial Behavior and Phagocytic Cell Clearance Through the Actions of Annexin A1. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Loiola, R.; Shah, U.N.; Gentleman, S.M.; Solito, E.; Sastre, M. The anti-inflammatory Annexin A1 induces the clearance and degradation of the amyloid-beta peptide. J. Neuroinflamm. 2016, 13, 234. [Google Scholar] [CrossRef] [PubMed]
- McArthur, S.; Cristante, E.; Paterno, M.; Christian, H.; Roncaroli, F.; Gillies, G.; Solito, E. Annexin A1: A Central Player in the Anti-Inflammatory and Neuroprotective Role of Microglia. J. Immunol. 2010, 185, 6317–6328. [Google Scholar] [CrossRef]
- Mallard, C.; Ek, C.J.; Vexler, Z.S. The myth of the immature barrier systems in the developing brain: Role in perinatal brain injury. J. Physiol. 2018, 596, 5655–5664. [Google Scholar] [CrossRef]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.-U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef]
- Fathali, N.; Ostrowski, R.P.; Hasegawa, Y.; Lekic, T.; Tang, J.; Zhang, J.H. Splenic immune cells in experimental neonatal hypoxia–ischemia. Transl. Stroke Res. 2013, 4, 208–219. [Google Scholar] [CrossRef]
- Baburamani, A.A.; Supramaniam, V.G.; Hagberg, H.; Mallard, C. Microglia toxicity in preterm brain injury. Reprod. Toxicol. 2014, 48, 106–112. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef]
- Czeh, M.; Gressens, P.; Kaindl, A.M. The yin and yang of microglia. Dev. Neurosci. 2011, 33, 199–209. [Google Scholar] [CrossRef]
- Lai, J.C.Y.; Rocha-Ferreira, E.; Ek, C.J.; Wang, X.; Hagberg, H.; Mallard, C. Immune responses in perinatal brain injury. Brain Behav. Immun. 2017, 63, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Ziemka-Nalecz, M.; Jaworska, J.; Zalewska, T. Insights Into the Neuroinflammatory Responses After Neonatal Hypoxia-Ischemia. J. Neuropathol. Exp. Neurol. 2017, 76, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Di Martino, E.; Mukai, T.; Tsuji, S.; Murakami, T.; Harris, R.A.; Blomgren, K.; Åden, U. Aggravated brain injury after neonatal hypoxic ischemia in microglia-depleted mice. J. Neuroinflamm. 2020, 17, 111. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hébert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef]
- Faustino, J.V.; Wang, X.; Johnson, C.E.; Klibanov, A.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.-V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. BrainBehav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Hellström Erkenstam, N.; Smith, P.L.P.; Fleiss, B.; Nair, S.; Svedin, P.; Wang, W.; Boström, M.; Gressens, P.; Hagberg, H.; Brown, K.L.; et al. Temporal Characterization of Microglia/Macrophage Phenotypes in a Mouse Model of Neonatal Hypoxic-Ischemic Brain Injury. Front. Cell. Neurosci. 2016, 10. [Google Scholar] [CrossRef]
- Serdar, M.; Kempe, K.; Rizazad, M.; Herz, J.; Bendix, I.; Felderhoff-Müser, U.; Sabir, H. Early Pro-inflammatory Microglia Activation After Inflammation-Sensitized Hypoxic-Ischemic Brain Injury in Neonatal Rats. Front. Cell. Neurosci. 2019, 13, 237. [Google Scholar] [CrossRef]
- Jenkins, D.D.; Rollins, L.G.; Perkel, J.K.; Wagner, C.L.; Katikaneni, L.P.; Bass, W.T.; Kaufman, D.A.; Horgan, M.J.; Languani, S.; Givelichian, L.; et al. Serum cytokines in a clinical trial of hypothermia for neonatal hypoxic-ischemic encephalopathy. J. Cereb Blood Flow Metab 2012, 32, 1888–1896. [Google Scholar] [CrossRef]
- Johnston, M.V.; Fatemi, A.; Wilson, M.A.; Northington, F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol 2011, 10, 372–382. [Google Scholar] [CrossRef]
- Smith, P.L.P.; Mottahedin, A.; Svedin, P.; Mohn, C.-J.; Hagberg, H.; Ek, J.; Mallard, C. Peripheral myeloid cells contribute to brain injury in male neonatal mice. J. Neuroinflamm. 2018, 15, 301. [Google Scholar] [CrossRef] [PubMed]
- Zachariae, C.O.C.; Larsen, C.G.; Matsushima, K. Monocyte Chemoattractant Protein 1 (MCP-1). In Encyclopedia of Immunology, 2nd ed.; Delves, P.J., Ed.; Elsevier: Oxford, UK, 1998; pp. 1748–1750. [Google Scholar]
- Garcia-Bonilla, L.; Brea, D.; Benakis, C.; Lane, D.A.; Murphy, M.; Moore, J.; Racchumi, G.; Jiang, X.; Iadecola, C.; Anrather, J. Endogenous Protection from Ischemic Brain Injury by Preconditioned Monocytes. J. Neurosci. 2018, 38, 6722. [Google Scholar] [CrossRef] [PubMed]
- Hudome, S.; Palmer, C.; Roberts, R.L.; Mauger, D.; Housman, C.; Towfighi, J. The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr. Res. 1997, 41, 607–616. [Google Scholar] [CrossRef]
- Palmer, C.; Roberts, R.L.; Young, P.I. Timing of neutrophil depletion influences long-term neuroprotection in neonatal rat hypoxic-ischemic brain injury. Pediatr. Res. 2004, 55, 549–556. [Google Scholar] [CrossRef]
- Revuelta, M.; Elicegui, A.; Moreno-Cugnon, L.; Bührer, C.; Matheu, A.; Schmitz, T. Ischemic stroke in neonatal and adult astrocytes. Mech. Ageing Dev. 2019, 183, 111147. [Google Scholar] [CrossRef]
- Disdier, C.; Stonestreet, B.S. Hypoxic-ischemic-related cerebrovascular changes and potential therapeutic strategies in the neonatal brain. J. Neurosci. Res. 2020, 98, 1468–1484. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- El-Khoury, N.; Braun, A.; Hu, F.; Pandey, M.; Nedergaard, M.; Lagamma, E.F.; Ballabh, P. Astrocyte End-Feet in Germinal Matrix, Cerebral Cortex, and White Matter in Developing Infants. Pediatr. Res. 2006, 59, 673–679. [Google Scholar] [CrossRef]
- Bhalala, U.S.; Koehler, R.C.; Kannan, S. Neuroinflammation and neuroimmune dysregulation after acute hypoxic-ischemic injury of developing brain. Front. Pediatr 2015, 2, 144. [Google Scholar] [CrossRef]
- Leonardo, C.C.; Eakin, A.K.; Ajmo, J.M.; Collier, L.A.; Pennypacker, K.R.; Strongin, A.Y.; Gottschall, P.E. Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after hypoxia-ischemia in the neonatal rat. J. Neuroinflamm. 2008, 5, 34. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Shiow, L.R.; Favrais, G.; Schirmer, L.; Schang, A.-L.; Cipriani, S.; Andres, C.; Wright, J.N.; Nobuta, H.; Fleiss, B.; Gressens, P.; et al. Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia 2017, 65, 2024–2037. [Google Scholar] [CrossRef] [PubMed]
- Disdier, C.; Awa, F.; Chen, X.; Dhillon, S.K.; Galinsky, R.; Davidson, J.O.; Lear, C.A.; Bennet, L.; Gunn, A.J.; Stonestreet, B.S. Lipopolysaccharide-induced changes in the neurovascular unit in the preterm fetal sheep brain. J. Neuroinflamm. 2020, 17, 167. [Google Scholar] [CrossRef] [PubMed]
- Robel, S.; Sontheimer, H. Glia as drivers of abnormal neuronal activity. Nat. Neurosci. 2016, 19, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Burd, I.; Brown, A.; Gonzalez, J.M.; Chai, J.; Elovitz, M.A. A mouse model of term chorioamnionitis: Unraveling causes of adverse neurological outcomes. Reprod. Sci. 2011, 18, 900–907. [Google Scholar] [CrossRef]
- Yang, Y.; Higashimori, H.; Morel, L. Developmental maturation of astrocytes and pathogenesis of neurodevelopmental disorders. J. Neurodev. Disord. 2013, 5, 22. [Google Scholar] [CrossRef]
- Anastacio, J.B.R.; Sanches, E.F.; Nicola, F.; Odorcyk, F.; Fabres, R.B.; Netto, C.A. Phytoestrogen coumestrol attenuates brain mitochondrial dysfunction and long-term cognitive deficits following neonatal hypoxia–ischemia. Int. J. Dev. Neurosci. 2019, 79, 86–95. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, C.; Chen, J.; Luo, M.; Qu, Y.; Mu, D.; Chen, Q. Umbilical cord mesenchymal stem cells and umbilical cord blood mononuclear cells improve neonatal rat memory after hypoxia-ischemia. Behav. Brain Res. 2019, 362, 56–63. [Google Scholar] [CrossRef]
- Mari, C.; Odorcyk, F.; Sanches, E.; Wartchow, K.; Martini, A.; Nicola, F.; Zanotto, C.; Wyse, A.; Gonçalves, C.; Netto, C. Arundic acid administration protects astrocytes, recovers histological damage and memory deficits induced by neonatal hypoxia ischemia in rats. Int. J. Dev. Neurosci. 2019, 76, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Hagen, M.W.; Riddle, A.; McClendon, E.; Gong, X.; Shaver, D.; Srivastava, T.; Dean, J.M.; Bai, J.-Z.; Fowke, T.M.; Gunn, A.J.; et al. Role of recurrent hypoxia-ischemia in preterm white matter injury severity. PLoS ONE 2014, 9, e112800. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.T.; Wassink, G.; Galinsky, R.; Xu, B.; Mathai, S.; Dhillon, S.K.; van den Heuij, L.G.; Davidson, J.O.; Weaver-Mikaere, L.; Bennet, L.; et al. Protective effects of delayed intraventricular TLR7 agonist administration on cerebral white and gray matter following asphyxia in the preterm fetal sheep. Sci. Rep. 2019, 9, 9562. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Davidson, J.O.; Dean, J.M.; Green, C.R.; Bennet, L.; Gunn, A.J. Glia and hemichannels: Key mediators of perinatal encephalopathy. Neural Regen. Res. 2018, 13, 181. [Google Scholar]
- Nazmi, A.; Albertsson, A.M.; Rocha-Ferreira, E.; Zhang, X.; Vontell, R.; Zelco, A.; Rutherford, M.; Zhu, C.; Nilsson, G.; Mallard, C.; et al. Lymphocytes Contribute to the Pathophysiology of Neonatal Brain Injury. Front. Neurol. 2018, 9, 159. [Google Scholar] [CrossRef]
- Ortega, S.B.; Kong, X.; Venkataraman, R.; Savedra, A.M.; Kernie, S.G.; Stowe, A.M.; Raman, L. Perinatal chronic hypoxia induces cortical inflammation, hypomyelination, and peripheral myelin-specific T cell autoreactivity. J. Leukoc. Biol. 2016, 99, 21–29. [Google Scholar] [CrossRef]
- Herz, J.; Köster, C.; Crasmöller, M.; Abberger, H.; Hansen, W.; Felderhoff-Müser, U.; Bendix, I. Peripheral T cell depletion by FTY720 exacerbates hypoxic-ischemic brain injury in neonatal mice. Front. Immunol. 2018, 9, 1696. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2013. [Google Scholar] [CrossRef]
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N. Engl. J. Med. 2012, 366, 2085–2092. [Google Scholar] [CrossRef]
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N. Engl. J. Med. 2014, 371, 140–149. [Google Scholar] [CrossRef]
- Rao, R.; Trivedi, S.; Vesoulis, Z.; Liao, S.M.; Smyser, C.D.; Mathur, A.M. Safety and short-term outcomes of therapeutic hypothermia in preterm neonates 34–35 weeks gestational age with hypoxic-ischemic encephalopathy. J. Pediatr. 2017, 183, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.D.; Bennet, L.; Naylor, A.; George, S.A.; Dean, J.M.; Gunn, A.J. Effect of cerebral hypothermia and asphyxia on the subventricular zone and white matter tracts in preterm fetal sheep. Brain Res. 2012, 1469, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Osredkar, D.; Thoresen, M.; Maes, E.; Flatebo, T.; Elstad, M.; Sabir, H. Hypothermia is not neuroprotective after infection-sensitized neonatal hypoxic-ischemic brain injury. Resuscitation 2014, 85, 567–572. [Google Scholar] [CrossRef]
- Falck, M.; Osredkar, D.; Maes, E.; Flatebø, T.; Wood, T.R.; Sabir, H.; Thoresen, M. Hypothermic neuronal rescue from infection-sensitised hypoxic-ischaemic brain injury is pathogen dependent. Dev. Neurosci. 2017, 39, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Dalous, J.; Pansiot, J.; Pham, H.; Chatel, P.; Nadaradja, C.; D’Agostino, I.; Vottier, G.; Schwendimann, L.; Vanneaux, V.; Charriaut-Marlangue, C. Use of human umbilical cord blood mononuclear cells to prevent perinatal brain injury: A preclinical study. Stem Cells Dev. 2012, 22, 169–179. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Chang, Y.S.; Sung, D.K.; Sung, S.I.; Yoo, H.S.; Lee, J.H.; Oh, W.I.; Park, W.S. Mesenchymal stem cells prevent hydrocephalus after severe intraventricular hemorrhage. Stroke 2013, 44, 497–504. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Chang, Y.S.; Sung, S.I.; Park, W.S. Mesenchymal Stem Cells for Severe Intraventricular Hemorrhage in Preterm Infants: Phase I Dose-Escalation Clinical Trial. Stem Cells Transl Med. 2018, 7, 847–856. [Google Scholar] [CrossRef]
- Yasuhara, T.; Hara, K.; Maki, M.; Mays, R.W.; Deans, R.J.; Hess, D.C.; Carroll, J.E.; Borlongan, C.V. Intravenous grafts recapitulate the neurorestoration afforded by intracerebrally delivered multipotent adult progenitor cells in neonatal hypoxic-ischemic rats. J. Cereb Blood Flow Metab 2008, 28, 1804–1810. [Google Scholar] [CrossRef]
- Drommelschmidt, K.; Serdar, M.; Bendix, I.; Herz, J.; Bertling, F.; Prager, S.; Keller, M.; Ludwig, A.-K.; Duhan, V.; Radtke, S. Mesenchymal stem cell-derived extracellular vesicles ameliorate inflammation-induced preterm brain injury. Brain Behav. Immun. 2017, 60, 220–232. [Google Scholar] [CrossRef]
- Thomi, G.; Joerger-Messerli, M.; Haesler, V.; Muri, L.; Surbek, D.; Schoeberlein, A. Intranasally administered exosomes from umbilical cord stem cells have preventive neuroprotective effects and contribute to functional recovery after perinatal brain injury. Cells 2019, 8, 855. [Google Scholar] [CrossRef]
- Zhang, J.; Sadowska, G.B.; Chen, X.; Park, S.Y.; Kim, J.-E.; Bodge, C.A.; Cummings, E.; Lim, Y.-P.; Makeyev, O.; Besio, W.G. Anti–IL-6 neutralizing antibody modulates blood-brain barrier function in the ovine fetus. FASEB J. 2015, 29, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Dhillon, S.K.; Dean, J.M.; Davidson, J.O.; Lear, C.A.; Wassink, G.; Nott, F.; Kelly, S.B.; Fraser, M.; Yuill, C. Tumor necrosis factor inhibition attenuates white matter gliosis after systemic inflammation in preterm fetal sheep. J. Neuroinflamm. 2020, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sadowska, G.B.; Zhang, J.; Kim, J.-E.; Cummings, E.E.; Bodge, C.A.; Lim, Y.-P.; Makeyev, O.; Besio, W.G.; Gaitanis, J. Neutralizing anti-interleukin-1β antibodies modulate fetal blood–brain barrier function after ischemia. Neurobiol. Dis. 2015, 73, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hovanesian, V.; Naqvi, S.; Lim, Y.-P.; Tucker, R.; Donahue, J.E.; Stopa, E.G.; Stonestreet, B.S. Systemic infusions of anti-interleukin-1β neutralizing antibodies reduce short-term brain injury after cerebral ischemia in the ovine fetus. BrainBehav. Immun. 2018, 67, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Patra, A.; Chen, X.; Sadowska, G.B.; Zhang, J.; Lim, Y.-P.; Padbury, J.F.; Banks, W.A.; Stonestreet, B.S. Neutralizing anti-interleukin-1β antibodies reduce ischemia-related interleukin-1β transport across the blood–brain barrier in fetal sheep. Neuroscience 2017, 346, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Iwai, M.; Cao, G.; Yin, W.; Stetler, R.A.; Liu, J.; Chen, J. Erythropoietin promotes neuronal replacement through revascularization and neurogenesis after neonatal hypoxia/ischemia in rats. Stroke 2007, 38, 2795–2803. [Google Scholar] [CrossRef]
- Iwai, M.; Stetler, R.A.; Xing, J.; Hu, X.; Gao, Y.; Zhang, W.; Chen, J.; Cao, G. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke 2010, 41, 1032–1037. [Google Scholar] [CrossRef]
- Kellert, B.A.; McPherson, R.J.; Juul, S.E. A comparison of high-dose recombinant erythropoietin treatment regimens in brain-injured neonatal rats. Pediatric Res. 2007, 61, 451–455. [Google Scholar] [CrossRef]
- Alexander, M.; Hill, C.; Rosenkrantz, T.; Fitch, R. Evaluation of the therapeutic benefit of delayed administration of erythropoietin following early hypoxic-ischemic injury in rodents. Dev. Neurosci. 2012, 34, 515–524. [Google Scholar] [CrossRef]
- Keller, M.; Yang, J.; Griesmaier, E.; Gorna, A.; Sarkozy, G.; Urbanek, M.; Gressens, P.; Simbruner, G. Erythropoietin is neuroprotective against NMDA-receptor-mediated excitotoxic brain injury in newborn mice. Neurobiol. Dis. 2006, 24, 357–366. [Google Scholar] [CrossRef]
- Rees, S.; Hale, N.; De Matteo, R.; Cardamone, L.; Tolcos, M.; Loeliger, M.; Mackintosh, A.; Shields, A.; Probyn, M.; Greenwood, D. Erythropoietin is neuroprotective in a preterm ovine model of endotoxin-induced brain injury. J. Neuropathol. Exp. Neurol. 2010, 69, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Sun, H.; Xu, F.; Kang, W.; Gao, L.; Guo, J.; Zhang, Y.; Xia, L.; Wang, X.; Zhu, C. Recombinant human erythropoietin improves neurological outcomes in very preterm infants. Ann. Neurol. 2016, 80, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; Comstock, B.A.; Wadhawan, R.; Mayock, D.E.; Courtney, S.E.; Robinson, T.; Ahmad, K.A.; Bendel-Stenzel, E.; Baserga, M.; LaGamma, E.F. A Randomized Trial of Erythropoietin for Neuroprotection in Preterm Infants. N. Engl. J. Med. 2020, 382, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Davidson, J.O.; Dean, J.M.; Bennet, L.; Gunn, A.J. Cooling and immunomodulation for treating hypoxic-ischemic brain injury. Pediatrics Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wassink, G.; Gunn, E.R.; Drury, P.P.; Bennet, L.; Gunn, A.J. The mechanisms and treatment of asphyxial encephalopathy. Front. Neurosci. 2014, 8, 40. [Google Scholar] [CrossRef]
- Han, H.S.; Karabiyikoglu, M.; Kelly, S.; Sobel, R.A.; Yenari, M.A. Mild hypothermia inhibits nuclear factor-κB translocation in experimental stroke. J. Cereb. Blood Flow Metab. 2003, 23, 589–598. [Google Scholar] [CrossRef]
- Ceulemans, A.-G.; Zgavc, T.; Kooijman, R.; Hachimi-Idrissi, S.; Sarre, S.; Michotte, Y. Mild hypothermia causes differential, time-dependent changes in cytokine expression and gliosis following endothelin-1-induced transient focal cerebral ischemia. J. Neuroinflamm. 2011, 8, 60. [Google Scholar] [CrossRef]
- Gunn, A.J.; Gunn, T.R.; de Haan, H.H.; Williams, C.E.; Gluckman, P.D. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Investig. 1997, 99, 248–256. [Google Scholar] [CrossRef]
- Shankaran, S.; Barnes, P.D.; Hintz, S.R.; Laptook, A.R.; Zaterka-Baxter, K.M.; McDonald, S.A.; Ehrenkranz, R.A.; Walsh, M.C.; Tyson, J.E.; Donovan, E.F. Brain injury following trial of hypothermia for neonatal hypoxic–ischaemic encephalopathy. Arch. Dis. Child. -Fetal Neonatal Ed. 2012, 97, F398–F404. [Google Scholar]
- Azzopardi, D.; Group, T.S. Predictive value of the amplitude integrated EEG in infants with hypoxic ischaemic encephalopathy: Data from a randomised trial of therapeutic hypothermia. Arch. Dis. Child. -Fetal Neonatal Ed. 2014, 99, F80–F82. [Google Scholar] [CrossRef]
- Silverman, W.A.; Fertig, J.W.; Berger, A.P. The influence of the thermal environment upon the survival of newly born premature infants. Pediatrics 1958, 22, 876–886. [Google Scholar] [PubMed]
- Gunn, A.J.; Bennet, L. Brain cooling for preterm infants. Clin. Perinatol. 2008, 35, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Smit, E.; Liu, X.; Jary, S.; Cowan, F.; Thoresen, M. Cooling neonates who do not fulfil the standard cooling criteria–short-and long-term outcomes. Acta Paediatr. 2015, 104, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Kuban, K.C.; Joseph, R.M.; O’Shea, T.M.; Heeren, T.; Fichorova, R.N.; Douglass, L.; Jara, H.; Frazier, J.A.; Hirtz, D.; Rollins, J.V. Circulating inflammatory-associated proteins in the first month of life and cognitive impairment at age 10 years in children born extremely preterm. J. Pediatr. 2017, 180, 116–123.e111. [Google Scholar] [CrossRef]
- Mourvillier, B.; Tubach, F.; Van de Beek, D.; Garot, D.; Pichon, N.; Georges, H.; Lefevre, L.M.; Bollaert, P.-E.; Boulain, T.; Luis, D. Induced hypothermia in severe bacterial meningitis: A randomized clinical trial. Jama 2013, 310, 2174–2183. [Google Scholar] [CrossRef] [PubMed]
- Bennet, L.; Tan, S.; Van den Heuij, L.; Derrick, M.; Groenendaal, F.; van Bel, F.; Juul, S.; Back, S.A.; Northington, F.; Robertson, N.J. Cell therapy for neonatal hypoxia–ischemia and cerebral palsy. Ann. Neurol. 2012, 71, 589–600. [Google Scholar] [CrossRef]
- Titomanlio, L.; Kavelaars, A.; Dalous, J.; Mani, S.; El Ghouzzi, V.; Heijnen, C.; Baud, O.; Gressens, P. Stem cell therapy for neonatal brain injury: Perspectives and challenges. Ann. Neurol. 2011, 70, 698–712. [Google Scholar] [CrossRef]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Regeneration of the ischemic brain by engineered stem cells: Fuelling endogenous repair processes. Brain Res. Rev. 2009, 61, 1–13. [Google Scholar] [CrossRef]
- Gortner, L.; Felderhoff-Müser, U.; Monz, D.; Bieback, K.; Klüter, H.; Jellema, R.; Kramer, B.; Keller, M.; Reiss, I.; Horn, P. Regenerative therapies in neonatology: Clinical perspectives. Klin. Pädiatrie 2012, 224, 233. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Weiss, M.D. Baby STEPS: A giant leap for cell therapy in neonatal brain injury. Pediatric Res. 2011, 70, 3–9. [Google Scholar] [CrossRef]
- Donega, V.; van Velthoven, C.T.; Nijboer, C.H.; Kavelaars, A.; Heijnen, C.J. The endogenous regenerative capacity of the damaged newborn brain: Boosting neurogenesis with mesenchymal stem cell treatment. J. Cereb. Blood Flow Metab. 2013, 33, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.; Kavelaars, A.; Heijnen, C.J. Mesenchymal stem cells as a treatment for neonatal ischemic brain damage. Pediatric Res. 2012, 71, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Fleiss, B.; Guillot, P.V.; Titomanlio, L.; Baud, O.; Hagberg, H.; Gressens, P. Stem cell therapy for neonatal brain injury. Clin. Perinatol. 2014, 41, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jahagirdar, B.N.; Reinhardt, R.L.; Schwartz, R.E.; Keene, C.D.; Ortiz-Gonzalez, X.R.; Reyes, M.; Lenvik, T.; Lund, T.; Blackstad, M. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Le Blanc, K.; Mougiakakos, D. Multipotent mesenchymal stromal cells and the innate immune system. Nat. Rev. Immunol. 2012, 12, 383–396. [Google Scholar] [CrossRef]
- Bieback, K.; Schallmoser, K.; Klüter, H.; Strunk, D. Clinical protocols for the isolation and expansion of mesenchymal stromal cells. Transfus. Med. Hemotherapy 2008, 35, 286–294. [Google Scholar] [CrossRef]
- Portmann-Lanz, C.B.; Schoeberlein, A.; Huber, A.; Sager, R.; Malek, A.; Holzgreve, W.; Surbek, D.V. Placental mesenchymal stem cells as potential autologous graft for pre-and perinatal neuroregeneration. Am. J. Obstet. Gynecol. 2006, 194, 664–673. [Google Scholar] [CrossRef]
- Uccelli, A.; Benvenuto, F.; Laroni, A.; Giunti, D. Neuroprotective features of mesenchymal stem cells. Best Pract. Res. Clin. Haematol. 2011, 24, 59–64. [Google Scholar] [CrossRef]
- Siegel, G.; Schäfer, R.; Dazzi, F. The immunosuppressive properties of mesenchymal stem cells. Transplantation 2009, 87, S45–S49. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Repeated mesenchymal stem cell treatment after neonatal hypoxia–ischemia has distinct effects on formation and maturation of new neurons and oligodendrocytes leading to restoration of damage, corticospinal motor tract activity, and sensorimotor function. J. Neurosci. 2010, 30, 9603–9611. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.J.; Couillard-Despres, S.; Pedre, X.; Ploetz, S.; Caioni, M.; Lois, C.; Bogdahn, U.; Aigner, L. Mesenchymal stem cells instruct oligodendrogenic fate decision on adult neural stem cells. Stem Cells 2006, 24, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Steffenhagen, C.; Dechant, F.-X.; Oberbauer, E.; Furtner, T.; Weidner, N.; Küry, P.; Aigner, L.; Rivera, F.J. Mesenchymal stem cells prime proliferating adult neural progenitors toward an oligodendrocyte fate. Stem Cells Dev. 2011, 21, 1838–1851. [Google Scholar] [CrossRef]
- Walker, P.A.; Shah, S.K.; Jimenez, F.; Gerber, M.H.; Xue, H.; Cutrone, R.; Hamilton, J.A.; Mays, R.W.; Deans, R.; Pati, S. Intravenous multipotent adult progenitor cell therapy for traumatic brain injury: Preserving the blood brain barrier via an interaction with splenocytes. Exp. Neurol. 2010, 225, 341–352. [Google Scholar] [CrossRef]
- Walker, P.A.; Bedi, S.S.; Shah, S.K.; Jimenez, F.; Xue, H.; Hamilton, J.A.; Smith, P.; Thomas, C.P.; Mays, R.W.; Pati, S. Intravenous multipotent adult progenitor cell therapy after traumatic brain injury: Modulation of the resident microglia population. J. Neuroinflamm. 2012, 9, 228. [Google Scholar] [CrossRef]
- DePaul, M.A.; Palmer, M.; Lang, B.T.; Cutrone, R.; Tran, A.P.; Madalena, K.M.; Bogaerts, A.; Hamilton, J.A.; Deans, R.J.; Mays, R.W. Intravenous multipotent adult progenitor cell treatment decreases inflammation leading to functional recovery following spinal cord injury. Sci. Rep. 2015, 5, 16795. [Google Scholar] [CrossRef]
- Donega, V.; Nijboer, C.H.; van Tilborg, G.; Dijkhuizen, R.M.; Kavelaars, A.; Heijnen, C.J. Intranasally administered mesenchymal stem cells promote a regenerative niche for repair of neonatal ischemic brain injury. Exp. Neurol. 2014, 261, 53–64. [Google Scholar] [CrossRef]
- Donega, V.; van Velthoven, C.T.; Nijboer, C.H.; van Bel, F.; Kas, M.J.; Kavelaars, A.; Heijnen, C.J. Intranasal mesenchymal stem cell treatment for neonatal brain damage: Long-term cognitive and sensorimotor improvement. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Wagenaar, N.; Nijboer, C.H.; van Bel, F. Repair of neonatal brain injury: Bringing stem cell-based therapy into clinical practice. Dev Med. Child. Neurol 2017, 59, 997–1003. [Google Scholar] [CrossRef]
- Van Velthoven, C.T.; Kavelaars, A.; Van Bel, F.; Heijnen, C.J. Nasal administration of stem cells: A promising novel route to treat neonatal ischemic brain damage. Pediatric Res. 2010, 68, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Oppliger, B.; Joerger-Messerli, M.; Mueller, M.; Reinhart, U.; Schneider, P.; Surbek, D.V.; Schoeberlein, A. Intranasal delivery of umbilical cord-derived mesenchymal stem cells preserves myelination in perinatal brain damage. Stem Cells Dev. 2016, 25, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Finder, M.; Boylan, G.B.; Twomey, D.; Ahearne, C.; Murray, D.M.; Hallberg, B. Two-Year Neurodevelopmental Outcomes After Mild Hypoxic Ischemic Encephalopathy in the Era of Therapeutic Hypothermia. Jama Pediatr. 2019. [Google Scholar] [CrossRef]
- Kabat, M.; Bobkov, I.; Kumar, S.; Grumet, M. Trends in mesenchymal stem cell clinical trials 2004–2018: Is efficacy optimal in a narrow dose range? Stem Cells Transl. Med. 2020, 9, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.C.; Wechsler, L.R.; Clark, W.M.; Savitz, S.I.; Ford, G.A.; Chiu, D.; Yavagal, D.R.; Uchino, K.; Liebeskind, D.S.; Auchus, A.P.; et al. Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol 2017, 16, 360–368. [Google Scholar] [CrossRef]
- Cotten, C.M.; Murtha, A.P.; Goldberg, R.N.; Grotegut, C.A.; Smith, P.B.; Goldstein, R.F.; Fisher, K.A.; Gustafson, K.E.; Waters-Pick, B.; Swamy, G.K.; et al. Feasibility of autologous cord blood cells for infants with hypoxic-ischemic encephalopathy. J. Pediatr 2014, 164, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Chang, Y.S.; Kim, J.H.; Sung, S.I.; Park, W.S. Two-Year Follow-Up Outcomes of Premature Infants Enrolled in the Phase I Trial of Mesenchymal Stem Cells Transplantation for Bronchopulmonary Dysplasia. J. Pediatr 2017, 185, 49–54. [Google Scholar] [CrossRef]
- Chang, Y.S.; Ahn, S.Y.; Yoo, H.S.; Sung, S.I.; Choi, S.J.; Oh, W.I.; Park, W.S. Mesenchymal stem cells for bronchopulmonary dysplasia: Phase 1 dose-escalation clinical trial. J. Pediatr 2014, 164, 966–972. [Google Scholar] [CrossRef]
- Powell, S.B.; Silvestri, J.M. Safety of Intratracheal Administration of Human Umbilical Cord Blood Derived Mesenchymal Stromal Cells in Extremely Low Birth Weight Preterm Infants. J. Pediatr. 2019. [Google Scholar] [CrossRef]
- Sun, J.M.; Song, A.W.; Case, L.E.; Mikati, M.A.; Gustafson, K.E.; Simmons, R.; Goldstein, R.; Petry, J.; McLaughlin, C.; Waters-Pick, B.; et al. Effect of Autologous Cord Blood Infusion on Motor Function and Brain Connectivity in Young Children with Cerebral Palsy: A Randomized, Placebo-Controlled Trial. Stem Cells Transl Med. 2017, 6, 2071–2078. [Google Scholar] [CrossRef]
- Bi, M.; Wang, J.; Zhang, Y.; Li, L.; Wang, L.; Yao, R.; Duan, S.; Tong, S.; Li, J. Bone mesenchymal stem cells transplantation combined with mild hypothermia improves the prognosis of cerebral ischemia in rats. PLoS ONE 2018, 13, e0197405. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Koster, C.; Reinboth, B.S.; Dzietko, M.; Hansen, W.; Sabir, H.; van Velthoven, C.; Bendix, I.; Felderhoff-Muser, U. Interaction between hypothermia and delayed mesenchymal stem cell therapy in neonatal hypoxic-ischemic brain injury. Brain Behav Immun 2018, 70, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Chen, T.S.; Lim, S.K. Mesenchymal stem cell exosome: A novel stem cell-based therapy for cardiovascular disease. Regen Med. 2011, 6, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, S.; Andrzejewska, A.; Strzemecki, D.; Muraca, M.; Janowski, M.; Lukomska, B. Human bone marrow mesenchymal stem cell-derived extracellular vesicles attenuate neuroinflammation evoked by focal brain injury in rats. J. Neuroinflamm. 2019, 16, 216. [Google Scholar] [CrossRef]
- Saunders, N.R.; Daneman, R.; Dziegielewska, K.M.; Liddelow, S.A. Transporters of the blood-brain and blood-CSF interfaces in development and in the adult. Mol Asp. Med. 2013, 34, 742–752. [Google Scholar] [CrossRef]
- Moretti, R.; Pansiot, J.; Bettati, D.; Strazielle, N.; Ghersi-Egea, J.F.; Damante, G.; Fleiss, B.; Titomanlio, L.; Gressens, P. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci 2015, 9, 40. [Google Scholar] [CrossRef]
- Smith, S.F. Lipocortin 1: Glucocorticoids caught in the act? Thorax 1996, 51, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic Potential in Microvascular Disease. Front. Immunol 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed]
- Cristante, E.; McArthur, S.; Mauro, C.; Maggioli, E.; Romero, I.A.; Wylezinska-Arridge, M.; Couraud, P.O.; Lopez-Tremoleda, J.; Christian, H.C.; Weksler, B.B.; et al. Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. Proc. Natl. Acad. Sci. USA 2013, 110, 832–841. [Google Scholar] [CrossRef]
- McArthur, S.; Loiola, R.A.; Maggioli, E.; Errede, M.; Virgintino, D.; Solito, E. The restorative role of annexin A1 at the blood-brain barrier. Fluids Barriers Cns 2016, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Aly, H.; Khashaba, M.T.; El-Ayouty, M.; El-Sayed, O.; Hasanein, B.M. IL-1beta, IL-6 and TNF-alpha and outcomes of neonatal hypoxic ischemic encephalopathy. Brain Dev. 2006, 28, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Disdier, C.; Chen, X.; Kim, J.-E.; Threlkeld, S.W.; Stonestreet, B.S. Anti-Cytokine Therapy to Attenuate Ischemic-Reperfusion Associated Brain Injury in the Perinatal Period. Brain Sci 2018, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Oorschot, D.E.; Sizemore, R.J.; Amer, A.R. Treatment of Neonatal Hypoxic-Ischemic Encephalopathy with Erythropoietin Alone, and Erythropoietin Combined with Hypothermia: History, Current Status, and Future Research. Int. J. Mol. Sci. 2020, 21, 1487. [Google Scholar] [CrossRef] [PubMed]
- Rees, S.; Harding, R.; Walker, D. The biological basis of injury and neuroprotection in the fetal and neonatal brain. Int. J. Dev. Neurosci. 2011, 29, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Kumral, A.; Baskin, H.; Yesilirmak, D.C.; Ergur, B.U.; Aykan, S.; Genc, S.; Genc, K.; Yilmaz, O.; Tugyan, K.; Giray, O. Erythropoietin attenuates lipopolysaccharide-induced white matter injury in the neonatal rat brain. Neonatology 2007, 92, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, A.; Aher, S.M. Early erythropoiesis-stimulating agents in preterm or low birth weight infants. Cochrane Database Syst. Rev. 2017, 2017. [Google Scholar] [CrossRef]
- Back, S.A. White matter injury in the preterm infant: Pathology and mechanisms. Acta Neuropathol. 2017, 134, 331–349. [Google Scholar] [CrossRef]
- Wong, H.S.; Santhakumaran, S.; Cowan, F.M.; Modi, N.; Group, M.f.N.I. Developmental assessments in preterm children: A meta-analysis. Pediatrics 2016, 138, e20160251. [Google Scholar] [CrossRef]
- Fleiss, B.; Tann, C.J.; Degos, V.; Sigaut, S.; Van Steenwinckel, J.; Schang, A.L.; Kichev, A.; Robertson, N.J.; Mallard, C.; Hagberg, H.; et al. Inflammation-induced sensitization of the brain in term infants. Dev Med. Child. Neurol 2015, 57 Suppl. 3, 17–28. [Google Scholar] [CrossRef]
- Hagberg, H.; Dammann, O.; Mallard, C.; Leviton, A. Preconditioning and the developing brain. Semin Perinatol 2004, 28, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.; Svedin, P.; Ardalan, M.; Levy, O.; Ek, C.J.; Mallard, C.; Lai, J.C. Staphylococcus epidermidis sensitizes perinatal hypoxic-ischemic brain injury in male but not female mice. Front. Immunol. 2020, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Leviton, A.; Fichorova, R.N.; O’Shea, T.M.; Kuban, K.; Paneth, N.; Dammann, O.; Allred, E.N.; Investigators, E.S. Two-hit model of brain damage in the very preterm newborn: Small for gestational age and postnatal systemic inflammation. Pediatr Res. 2013, 73, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch. Dis Child. Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef]
- Miller, S.L.; Huppi, P.S.; Mallard, C. The consequences of fetal growth restriction on brain structure and neurodevelopmental outcome. J. Physiol 2016, 594, 807–823. [Google Scholar] [CrossRef]
- Barton, S.K.; Tolcos, M.; Miller, S.L.; Christoph-Roehr, C.; Schmolzer, G.M.; Moss, T.J.; Hooper, S.B.; Wallace, E.M.; Polglase, G.R. Ventilation-Induced Brain Injury in Preterm Neonates: A Review of Potential Therapies. Neonatology 2016, 110, 155–162. [Google Scholar] [CrossRef]
- Barnett, M.L.; Tusor, N.; Ball, G.; Chew, A.; Falconer, S.; Aljabar, P.; Kimpton, J.A.; Kennea, N.; Rutherford, M.; David Edwards, A.; et al. Exploring the multiple-hit hypothesis of preterm white matter damage using diffusion MRI. Neuroimage Clin 2018, 17, 596–606. [Google Scholar] [CrossRef]
- Yoon, B.H.; Romero, R.; Park, J.S.; Kim, M.; Oh, S.Y.; Kim, C.J.; Jun, J.K. The relationship among inflammatory lesions of the umbilical cord (funisitis), umbilical cord plasma interleukin 6 concentration, amniotic fluid infection, and neonatal sepsis. Am. J. Obs. Gynecol 2000, 183, 1124–1129. [Google Scholar] [CrossRef]
- Ophelders, D.R.M.G.; Boots, A.W.; Hütten, M.C.; Al-Nasiry, S.; Jellema, R.K.; Spiller, O.B.; van Schooten, F.J.; Smolinska, A.; Wolfs, T.G.A.M. Screening of chorioamnionitis using volatile organic compound (VOC) detection in exhaled breath: A pre-clinical proof of concept study. In preparation.
- Saha, S.; Pagnozzi, A.; Bourgeat, P.; George, J.M.; Bradford, D.; Colditz, P.B.; Boyd, R.N.; Rose, S.E.; Fripp, J.; Pannek, K. Predicting motor outcome in preterm infants from very early brain diffusion MRI using a deep learning convolutional neural network (CNN) model. NeuroImage 2020, 116807. [Google Scholar] [CrossRef]
- Inder, T.E.; Buckland, L.; Williams, C.E.; Spencer, C.; Gunning, M.I.; Darlow, B.A.; Volpe, J.J.; Gluckman, P.D. Lowered electroencephalographic spectral edge frequency predicts the presence of cerebral white matter injury in premature infants. Pediatrics 2003, 111, 27–33. [Google Scholar] [CrossRef]
- Wikström, S.; Pupp, I.H.; Rosén, I.; Norman, E.; Fellman, V.; Ley, D.; Hellström-Westas, L. Early single-channel aEEG/EEG predicts outcome in very preterm infants. Acta Paediatr. 2012, 101, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, H.; Kubota, T.; Hayashi, N.; Hayakawa, M.; Takemoto, K.; Kato, Y.; Okumura, A. Absent cyclicity on aEEG within the first 24 h is associated with brain damage in preterm infants. Neuropediatrics 2010, 41, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Pillay, K.; Dereymaeker, A.; Jansen, K.; Naulaers, G.; De Vos, M. Applying a data-driven approach to quantify eeG maturational deviations in preterms with normal and abnormal neurodevelopmental outcomes. Sci. Rep. 2020, 10, 1–14. [Google Scholar]
- Chiesa, C.; Pellegrini, G.; Panero, A.; De Luca, T.; Assumma, M.; Signore, F.; Pacifico, L. Umbilical cord interleukin-6 levels are elevated in term neonates with perinatal asphyxia. Eur. J. Clin Invest. 2003, 33, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Waje-Andreassen, U.; Krakenes, J.; Ulvestad, E.; Thomassen, L.; Myhr, K.M.; Aarseth, J.; Vedeler, C.A. IL-6: An early marker for outcome in acute ischemic stroke. Acta Neurol Scand 2005, 111, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Vaisbuch, E.; Romero, R.; Gomez, R.; Kusanovic, J.P.; Mazaki-Tovi, S.; Chaiworapongsa, T.; Hassan, S.S. An elevated fetal interleukin-6 concentration can be observed in fetuses with anemia due to Rh alloimmunization: Implications for the understanding of the fetal inflammatory response syndrome. J. Matern Fetal Neonatal Med. 2011, 24, 391–396. [Google Scholar] [CrossRef]
- Rodney, T.; Osier, N.; Gill, J. Pro- and anti-inflammatory biomarkers and traumatic brain injury outcomes: A review. Cytokine 2018, 110, 248–256. [Google Scholar] [CrossRef]
- Jin, C.; Londono, I.; Mallard, C.; Lodygensky, G.A. New means to assess neonatal inflammatory brain injury. J. Neuroinflamm. 2015, 12, 180. [Google Scholar] [CrossRef]
- Kersbergen, K.J.; Benders, M.J.; Groenendaal, F.; Koopman-Esseboom, C.; Nievelstein, R.A.; van Haastert, I.C.; de Vries, L.S. Different patterns of punctate white matter lesions in serially scanned preterm infants. PLoS ONE 2014, 9, e108904. [Google Scholar] [CrossRef]
- Ment, L.R.; Vohr, B.R. Preterm birth and the developing brain. Lancet Neurol 2008, 7, 378–379. [Google Scholar] [CrossRef]
- Annink, K.V.; de Vries, L.S.; Groenendaal, F.; Vijlbrief, D.C.; Weeke, L.C.; Roehr, C.C.; Lequin, M.; Reiss, I.; Govaert, P.; Benders, M.J. The development and validation of a cerebral ultrasound scoring system for infants with hypoxic-ischaemic encephalopathy. Pediatric Res. 2020, 87, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Govaert, P.; Roehr, C.C.; Gressens, P. Cranial ultrasound by neonatologists. Pediatr. Res. 2020, 87, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Dudink, J.; Steggerda, S.J.; Horsch, S. State-of-the-art neonatal cerebral ultrasound: Technique and reporting. Pediatric Res. 2020, 87, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Demené, C.; Mairesse, J.; Baranger, J.; Tanter, M.; Baud, O. Ultrafast Doppler for neonatal brain imaging. Neuroimage 2019, 185, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Deffieux, T.; Demene, C.; Pernot, M.; Tanter, M. Functional ultrasound neuroimaging: A review of the preclinical and clinical state of the art. Curr. Opin. Neurobiol. 2018, 50, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.J.; Thayyil, S.; Cady, E.B.; Raivich, G. Magnetic resonance spectroscopy biomarkers in term perinatal asphyxial encephalopathy: From neuropathological correlates to future clinical applications. Curr. Pediatr. Rev. 2014, 10, 37–47. [Google Scholar] [CrossRef]
- Thayyil, S.; Chandrasekaran, M.; Taylor, A.; Bainbridge, A.; Cady, E.B.; Chong, W.K.; Murad, S.; Omar, R.Z.; Robertson, N.J. Cerebral magnetic resonance biomarkers in neonatal encephalopathy: A meta-analysis. Pediatrics 2010, 125, e382–e395. [Google Scholar] [CrossRef]
- Lally, P.J.; Montaldo, P.; Oliveira, V.; Soe, A.; Swamy, R.; Bassett, P.; Mendoza, J.; Atreja, G.; Kariholu, U.; Pattnayak, S.; et al. Magnetic resonance spectroscopy assessment of brain injury after moderate hypothermia in neonatal encephalopathy: A prospective multicentre cohort study. Lancet Neurol. 2019, 18, 35–45. [Google Scholar] [CrossRef]
- Glass, H.C.; Kan, J.; Bonifacio, S.L.; Ferriero, D.M. Neonatal seizures: Treatment practices among term and preterm infants. Pediatric Neurol. 2012, 46, 111–115. [Google Scholar] [CrossRef]
- Azzopardi, D. Clinical applications of cerebral function monitoring in neonates. Semin. Fetal Neonatal Med. 2015, 20, 154–163. [Google Scholar] [CrossRef]
- Uria-Avellanal, C.; Marlow, N.; Rennie, J.M. Outcome following neonatal seizures. Semin. Fetal Neonatal Med. 2013, 18, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.J.; Volpe, J.J. Encephalopathy of prematurity: Clinical-neurological features, diagnosis, imaging, prognosis, therapy. In Volpe’s Neurology of the Newborn; Elsevier: Amsterdam, The Netherlands, 2018; pp. 425–457. [Google Scholar]
- Roberts, D.; Brown, J.; Medley, N.; Dalziel, S.R. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sweet, D.G.; Carnielli, V.; Greisen, G.; Hallman, M.; Ozek, E.; Te Pas, A.; Plavka, R.; Roehr, C.C.; Saugstad, O.D.; Simeoni, U. European consensus guidelines on the management of respiratory distress syndrome–2019 update. Neonatology 2019, 115, 432–450. [Google Scholar] [CrossRef] [PubMed]
- Polglase, G.R.; Nitsos, I.; Baburamani, A.A.; Crossley, K.J.; Slater, M.K.; Gill, A.W.; Allison, B.J.; Moss, T.J.; Pillow, J.J.; Hooper, S.B. Inflammation in utero exacerbates ventilation-induced brain injury in preterm lambs. J. Appl. Physiol. 2012, 112, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Barton, S.K.; Moss, T.J.; Hooper, S.B.; Crossley, K.J.; Gill, A.W.; Kluckow, M.; Zahra, V.; Wong, F.Y.; Pichler, G.; Galinsky, R. Protective ventilation of preterm lambs exposed to acute chorioamnionitis does not reduce ventilation-induced lung or brain injury. PLoS ONE 2014, 9, e112402. [Google Scholar] [CrossRef]
- Hortensius, L.M.; Van Elburg, R.M.; Nijboer, C.H.; Benders, M.J.; De Theije, C.G.M. Postnatal nutrition to improve brain development in the preterm infant: A systematic review from bench to bedside. Front. Physiol. 2019, 10, 961. [Google Scholar] [CrossRef]
- Keunen, K.; Van Elburg, R.M.; Van Bel, F.; Benders, M.J. Impact of nutrition on brain development and its neuroprotective implications following preterm birth. Pediatr. Res. 2015, 77, 148–155. [Google Scholar] [CrossRef]
- Doyle, L.W.; Ehrenkranz, R.A.; Halliday, H.L. Dexamethasone treatment in the first week of life for preventing bronchopulmonary dysplasia in preterm infants: A systematic review. Neonatology 2010, 98, 217–224. [Google Scholar] [CrossRef]
- Kersbergen, K.J.; de Vries, L.S.; van Kooij, B.J.; Išgum, I.; Rademaker, K.J.; van Bel, F.; Hüppi, P.S.; Dubois, J.; Groenendaal, F.; Benders, M.J. Hydrocortisone treatment for bronchopulmonary dysplasia and brain volumes in preterm infants. J. Pediatr. 2013, 163, 666–671. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Setup | Model/Clinical Setting | Main Findings | Reference | |
|---|---|---|---|---|
| Hypothermia | Meta-analysis 11 randomized controlled trials (>35 weeks GA) | HI + HT |
| [129] |
| Clinical 208 infants >35 weeks GA | HI + HT |
| [130] | |
| Clinical 314 infants >35 weeks GA | HI + HT |
| [131] | |
| Clinical 31 infants 34–35 weeks GA | HI + HT |
| [132] | |
| Preclinical Preterm (0.7 GA) ovine fetuses | HI + HT |
| [133] | |
| Preclinical Preterm (0.7 GA) ovine fetuses | HI + HT |
| [74] | |
| Experimental P7 rat | LPS i.p. + left carotid artery occlusion + HT |
| [134] | |
| Experimental P7 rat | PAM3CSK4 i.p. + left carotid artery occlusion + HT |
| [135] | |
| Stem Cells | Experimental P5 rat | Intracranial ibotenate + hUCB-MNCs i.p./i.v. |
| [136] |
| Preclinical Preterm (0.7 GA) ovine fetuses | HI + MSC i.v. |
| [65] | |
| Preclinical Preterm (0.7 GA) ovine fetuses | HI + MAPC i.v. |
| [67] | |
| Preclinical Preterm (0.7 GA) ovine fetuses | HI + hAECs i.n. |
| [73] | |
| Experimental P4 Rat | IVH +UCBC i.c.v. |
| [137] | |
| Clinical trial (NCT02274428, phase I), IVH; mean GA 26.1 ± 0.7 weeks | IVH + UCBC i.c.v. |
| [138] | |
| Experimental P7 rat | Unilateral HI + MAPC i.v./i.c.v. |
| [139] | |
| Extracellular Vesicles | Experimental, P3 rat | LPS i.p. + MSC-EV i.p. |
| [140] |
| Preclinical Preterm (0.7 GA) ovine fetuses | HI + MSC-EV i.v. |
| [66] | |
| Experimental P2 rat | LPS i.p. + HI + MSC i.n. |
| [141] | |
| Cytokine Treatment | Preclinical Preterm (0.86 GA) ovine fetuses | Carotid occlusion + anti-IL-6 antibody i.v. |
| [142] |
| Preclinical Preterm (0.7 GA) ovine fetuses | LPS i.v. + TNF-α antibody i.v. |
| [143] | |
| Preclinical Preterm (0.86 GA) ovine fetuses | Carotid occlusion + anti-IL-1β antibody i.v. |
| [144] | |
| Preclinical Preterm (0.86 GA) ovine fetuses | Carotid occlusion + anti-IL-1β antibody i.v. |
| [145] | |
| Preclinical Preterm (0.86 GA) ovine fetuses | Carotid occlusion + anti-IL-1β antibody i.v. |
| [146] | |
| hrEPO | Experimental P7 rat | Unilateral HI + hrEPO i.p. |
| [147] |
| Experimental P7 rat | Unilateral HI + hrEPO i.p. |
| [148] | |
| Experimental P7 rat | Unilateral HI + hrEPO s.c. |
| [149] | |
| Experimental P7 rat | Unilateral HI + hrEPO i.p. |
| [150] | |
| Experimental P5 mouse | PVL (ibotenic acid intra-cranial) + EPO |
| [151] | |
| Preclinical Preterm (0.7 GA) ovine fetuses | HI (UCO) + hrEPO i.v. |
| [62] | |
| Preclinical Preterm (0.7 GA) ovine fetuses | LPS i.v. + hrEPO i.v. |
| [152] | |
| Clinical trial NCT02036073; 800 infants ≤32-weeks | Extreme/very preterm + hrEPO |
| [153] | |
| Clinical trial NCT0137827; extremely preterm infants, (n = 741) | Extreme preterm + hrEPO |
| [154] |
| Setup | Model/Clinical Setting | Main Findings | Reference | |
|---|---|---|---|---|
| Plasma Biomarkers | Clinical n = 155 <37 weeks GA Cord blood IL-6 | Preterm birth |
| [43] |
| Clinical 94 patients with preterm labor Amniotic fluid IL-6, TNF-α, IL-1β | Preterm birth |
| [44] | |
| Clinical 315 patients with preterm labor (20–35 weeks’ GA) Cord blood IL-6 | Preterm birth |
| [229] | |
| Preclinical Intra-amniotic LPS exposure of fetal sheep IL-6, IL-8 (0.7–0.8 GA) | Inflammation (LPS exposure) |
| [56] | |
| VOC | Preclinical Ovine chorioamnionitis model (0.8 GA) | Infection (Ureaplasma parvum) |
| [230] |
| Imaging | Clinical Infants < 31 weeks GA (n = 77) MRI | Very preterm |
| [231] |
| EEG | Clinical Very low birth weight infants (n = 95) | Extreme preterm/very preterm |
| [232] |
| Clinical Median GA 25 weeks (22–30 weeks; n = 94) | Extreme preterm/very preterm |
| [233] | |
| Clinical GA between 27 and 32 weeks (n = 12) | Very preterm/moderate preterm |
| [234] | |
| Clinical GA < 30 weeks GA (n = 65) | Very preterm |
| [235] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ophelders, D.R.M.G.; Gussenhoven, R.; Klein, L.; Jellema, R.K.; Westerlaken, R.J.J.; Hütten, M.C.; Vermeulen, J.; Wassink, G.; Gunn, A.J.; Wolfs, T.G.A.M. Preterm Brain Injury, Antenatal Triggers, and Therapeutics: Timing Is Key. Cells 2020, 9, 1871. https://doi.org/10.3390/cells9081871
Ophelders DRMG, Gussenhoven R, Klein L, Jellema RK, Westerlaken RJJ, Hütten MC, Vermeulen J, Wassink G, Gunn AJ, Wolfs TGAM. Preterm Brain Injury, Antenatal Triggers, and Therapeutics: Timing Is Key. Cells. 2020; 9(8):1871. https://doi.org/10.3390/cells9081871
Chicago/Turabian StyleOphelders, Daan R.M.G., Ruth Gussenhoven, Luise Klein, Reint K. Jellema, Rob J.J. Westerlaken, Matthias C. Hütten, Jeroen Vermeulen, Guido Wassink, Alistair J. Gunn, and Tim G.A.M. Wolfs. 2020. "Preterm Brain Injury, Antenatal Triggers, and Therapeutics: Timing Is Key" Cells 9, no. 8: 1871. https://doi.org/10.3390/cells9081871
APA StyleOphelders, D. R. M. G., Gussenhoven, R., Klein, L., Jellema, R. K., Westerlaken, R. J. J., Hütten, M. C., Vermeulen, J., Wassink, G., Gunn, A. J., & Wolfs, T. G. A. M. (2020). Preterm Brain Injury, Antenatal Triggers, and Therapeutics: Timing Is Key. Cells, 9(8), 1871. https://doi.org/10.3390/cells9081871