Cystic Fibrosis Defective Response to Infection Involves Autophagy and Lipid Metabolism

 ,
,  and
and
Abstract
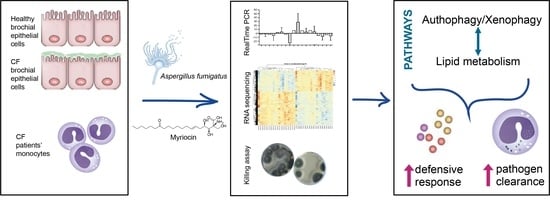
1. Introduction
2. Materials and Methods
2.1. Cells and Treatments
2.2. Aspergillus Fumigatus Culture
2.3. Cells Infection
2.4. PBMC Isolation and Infection
2.5. qRT-PCR
2.6. Statistical Analysis
2.7. RNA Extraction and Sequencing
2.8. Exploratory Data Analysis
2.9. RNA-Seq Data Analysis
3. Results
3.1. Myriocin Transcriptionally Regulates and Ameliorates the Response to A. fumigatus Infection in CF-Patients-Derived Monocytes
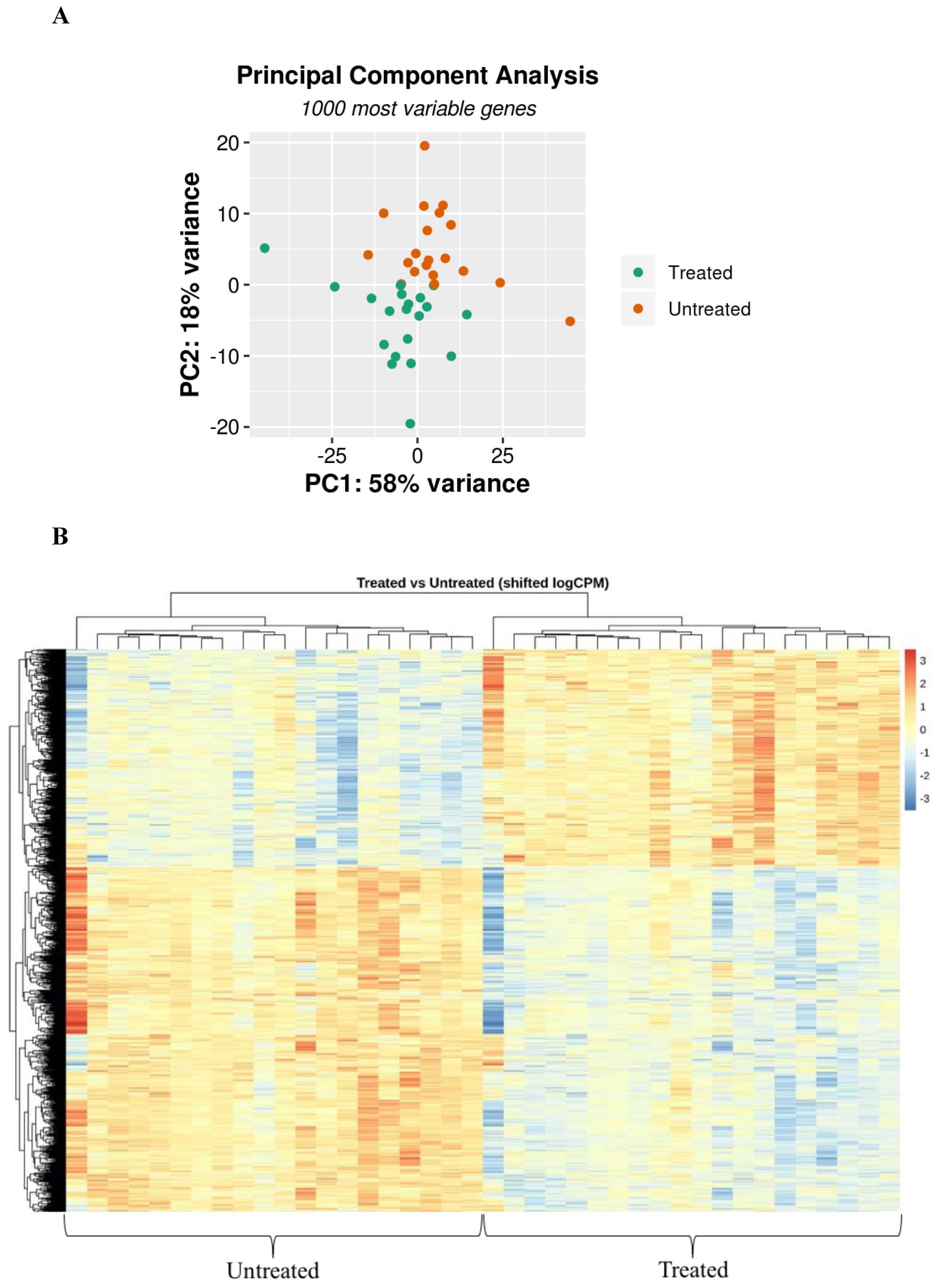
3.2. CF and Control Airways Epithelial Cell Lines Exhibit a Different Transcriptional Response to Infection
3.3. Myriocin Treatment Modulates CF Expression Profile under Infection
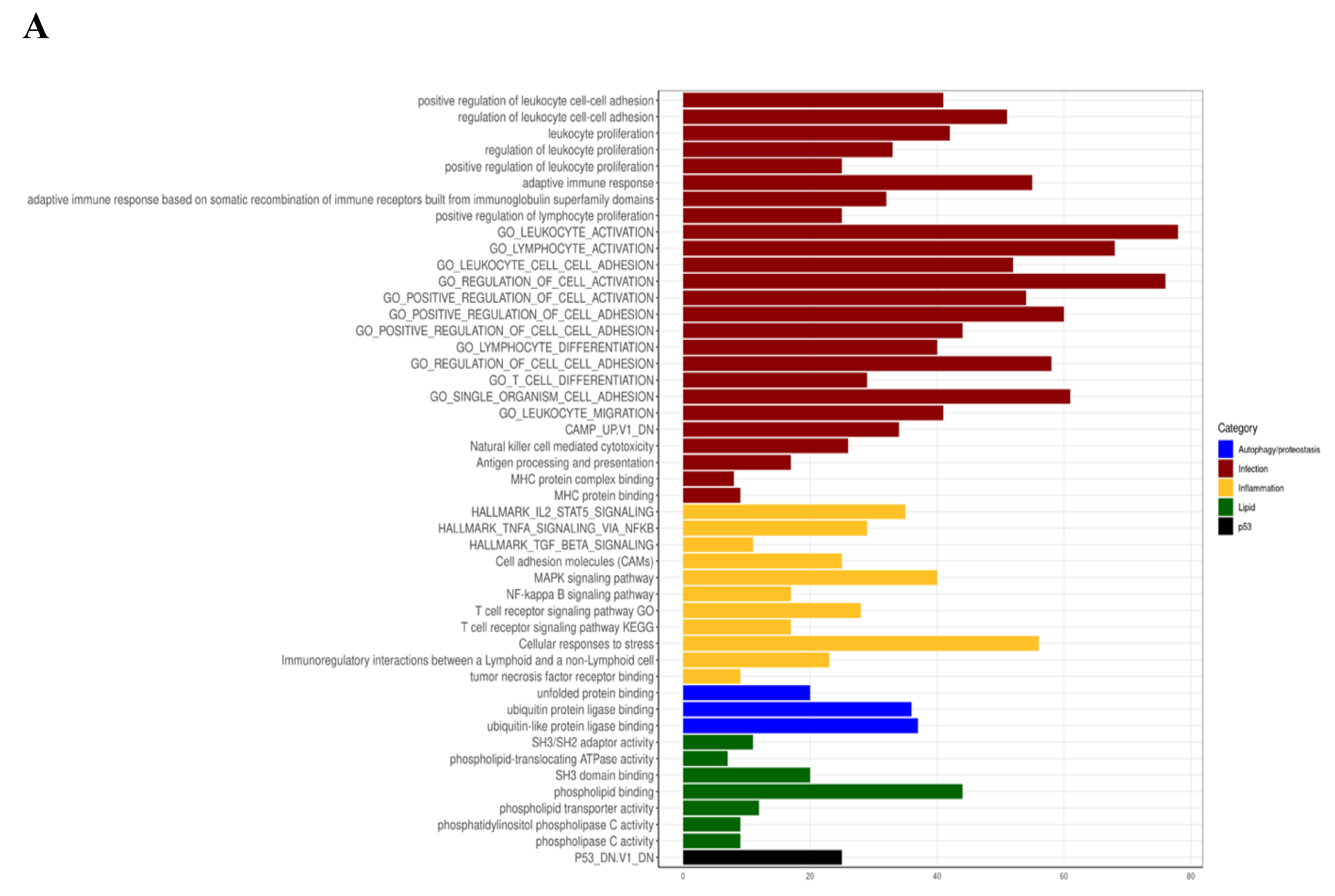
3.4. Myriocin Activates Gene Sets Involved in Inflammation, Infection, Autophagy/Proteostasis, and Lipid Metabolism in CF Bronchial Epithelial Infected Cells
4. Discussion
5. Strength and Limitations of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gelzo, M.; Sica, C.; Elce, A.; Dello Russo, A.; Iacotucci, P.; Carnovale, V.; Raia, V.; Salvatore, D.; Corso, G.; Castaldo, G. Reduced absorption and enhanced synthesis of cholesterol in patients with cystic fibrosis: A preliminary study of plasma sterols. Clin. Chem. Lab. Med. 2016, 54, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, V.; Milla, C.; Parks, E.J.; Schwarzenberg, S.J.; Moran, A. Abnormal lipid concentrations in cystic fibrosis. Am. J. Clin. Nutr. 2002, 75, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Villella, V.R.; Esposito, S.; Gavina, M.; Russo, I.; Silano, M.; Guido, S.; Pettoello-Mantovani, M.; Carnuccio, R.; Scholte, B.; et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on DeltaF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012, 8, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- Teichgraber, V.; Ulrich, M.; Endlich, N.; Riethmuller, J.; Wilker, B.; De Oliveira-Munding, C.C.; van Heeckeren, A.M.; Barr, M.L.; von Kurthy, G.; Schmid, K.W.; et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008, 14, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Mingione, A.; Dei Cas, M.; Bonezzi, F.; Caretti, A.; Piccoli, M.; Anastasia, L.; Ghidoni, R.; Paroni, R.; Signorelli, P. Inhibition of Sphingolipid Synthesis as a Phenotype-Modifying Therapy in Cystic Fibrosis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2020, 54, 110–125. [Google Scholar]
- Bruscia, E.M.; Bonfield, T.L. Innate and Adaptive Immunity in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 17–29. [Google Scholar] [CrossRef]
- Zhang, S.; Shrestha, C.L.; Kopp, B.T. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci. Rep. 2018, 8, 17066. [Google Scholar] [CrossRef]
- Caretti, A.; Torelli, R.; Perdoni, F.; Falleni, M.; Tosi, D.; Zulueta, A.; Casas, J.; Sanguinetti, M.; Ghidoni, R.; Borghi, E.; et al. Inhibition of ceramide de novo synthesis by myriocin produces the double effect of reducing pathological inflammation and exerting antifungal activity against A. fumigatus airways infection. Biochim. Biophys. Acta 2016, 1860, 1089–1097. [Google Scholar] [CrossRef]
- Leveque, M.; Le Trionnaire, S.; Del Porto, P.; Martin-Chouly, C. The impact of impaired macrophage functions in cystic fibrosis disease progression. J. Cyst. Fibros. 2017, 16, 443–453. [Google Scholar] [CrossRef]
- Tracy, M.C.; Moss, R.B. The myriad challenges of respiratory fungal infection in cystic fibrosis. Pediatr. Pulmonol. 2018, 53, S75–S85. [Google Scholar] [CrossRef]
- Chaudhary, N.; Datta, K.; Askin, F.B.; Staab, J.F.; Marr, K.A. Cystic fibrosis transmembrane conductance regulator regulates epithelial cell response to Aspergillus and resultant pulmonary inflammation. Am. J. Respir. Crit. Care Med. 2012, 185, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Bodas, M.; Vij, N. Adapting Proteostasis and Autophagy for Controlling the Pathogenesis of Cystic Fibrosis Lung Disease. Front. Pharmacol. 2019, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.L.; Settembre, C.; Gavina, M.; Raia, V.; Ballabio, A.; Maiuri, L. Cystic fibrosis: A disorder with defective autophagy. Autophagy 2011, 7, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Tosco, A.; Villella, V.R.; Raia, V.; Kroemer, G.; Maiuri, L. Manipulating proteostasis to repair the F508del-CFTR defect in cystic fibrosis. Mol. Cell. Pediatr. 2016, 3, 13. [Google Scholar] [CrossRef]
- Evans, R.J.; Sundaramurthy, V.; Frickel, E.M. The Interplay of Host Autophagy and Eukaryotic Pathogens. Front. Cell Dev. Biol. 2018, 6, 118. [Google Scholar] [CrossRef]
- Rameshwaram, N.R.; Singh, P.; Ghosh, S.; Mukhopadhyay, S. Lipid metabolism and intracellular bacterial virulence: Key to next-generation therapeutics. Future Microbiol. 2018, 13, 1301–1328. [Google Scholar] [CrossRef]
- Walpole, G.F.W.; Grinstein, S.; Westman, J. The role of lipids in host-pathogen interactions. IUBMB Life 2018, 70, 384–392. [Google Scholar] [CrossRef]
- Keyhani, N.O. Lipid biology in fungal stress and virulence: Entomopathogenic fungi. Fungal Biol. 2018, 122, 420–429. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef]
- Ollero, M.; Astarita, G.; Guerrera, I.C.; Sermet-Gaudelus, I.; Trudel, S.; Piomelli, D.; Edelman, A. Plasma lipidomics reveals potential prognostic signatures within a cohort of cystic fibrosis patients. J. Lipid Res. 2011, 52, 1011–1022. [Google Scholar] [CrossRef]
- Ollero, M. Methods for the study of lipid metabolites in cystic fibrosis. J. Cyst. Fibros. Soc. 2004, 3 (Suppl. 2), 97–98. [Google Scholar] [CrossRef] [PubMed]
- Ziobro, R.; Henry, B.; Edwards, M.J.; Lentsch, A.B.; Gulbins, E. Ceramide mediates lung fibrosis in cystic fibrosis. Biochem. Biophys. Res. Commun. 2013, 434, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; West, R.H.; Manson, M.E.; Ruddy, J.; Jiang, D.; Previs, S.F.; Sonawane, N.D.; Burgess, J.D.; Kelley, T.J. Increased plasma membrane cholesterol in cystic fibrosis cells correlates with CFTR genotype and depends on de novo cholesterol synthesis. Respir. Res. 2010, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Del Ciampo, I.R.; Sawamura, R.; Fernandes, M.I. Cystic fibrosis: From protein-energy malnutrition to obesity with dyslipidemia. Iran. J. Pediatr. 2013, 23, 605–606. [Google Scholar] [PubMed]
- Caretti, A.; Bragonzi, A.; Facchini, M.; De Fino, I.; Riva, C.; Gasco, P.; Musicanti, C.; Casas, J.; Fabrias, G.; Ghidoni, R.; et al. Anti-inflammatory action of lipid nanocarrier-delivered myriocin: Therapeutic potential in cystic fibrosis. Biochim. Biophys. Acta 2014, 1840, 586–594. [Google Scholar] [CrossRef]
- Caretti, A.; Vasso, M.; Bonezzi, F.T.; Gallina, A.; Trinchera, M.; Rossi, A.; Adami, R.; Casas, J.; Falleni, M.; Tosi, D.; et al. Myriocin treatment of CF lung infection and inflammation: Complex analyses for enigmatic lipids. Naunyn-Schmiedeberg Arch. Pharmacol. 2017, 390, 775–790. [Google Scholar] [CrossRef]
- Elkord, E.; Williams, P.E.; Kynaston, H.; Rowbottom, A.W. Human monocyte isolation methods influence cytokine production from in vitro generated dendritic cells. Immunology 2005, 114, 204–212. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, J.G. Bactericidal action of histone. J. Exp. Med. 1958, 108, 925–944. [Google Scholar] [CrossRef]
- Anand, P.; Cermelli, S.; Li, Z.; Kassan, A.; Bosch, M.; Sigua, R.; Huang, L.; Ouellette, A.J.; Pol, A.; Welte, M.A.; et al. A novel role for lipid droplets in the organismal antibacterial response. Elife 2012, 1, e00003. [Google Scholar] [CrossRef]
- Kim, H.S.; Yoon, H.; Minn, I.; Park, C.B.; Lee, W.T.; Zasloff, M.; Kim, S.C. Pepsin-mediated processing of the cytoplasmic histone H2A to strong antimicrobial peptide buforin I. J. Immunol. 2000, 165, 3268–3274. [Google Scholar] [CrossRef]
- Watson, K.; Edwards, R.J.; Shaunak, S.; Parmelee, D.C.; Sarraf, C.; Gooderham, N.J.; Davies, D.S. Extra-nuclear location of histones in activated human peripheral blood lymphocytes and cultured T-cells. Biochem. Pharmacol. 1995, 50, 299–309. [Google Scholar] [CrossRef]
- Zlatanova, J.S.; Srebreva, L.N.; Banchev, T.B.; Tasheva, B.T.; Tsanev, R.G. Cytoplasmic pool of histone H1 in mammalian cells. J. Cell Sci. 1990, 96 Pt 3, 461–468. [Google Scholar]
- De Lucca, A.J.; Heden, L.O.; Ingber, B.; Bhatnagar, D. Antifungal properties of wheat histones (H1-H4) and purified wheat histone H1. J. Agric. Food Chem. 2011, 59, 6933–6939. [Google Scholar] [CrossRef]
- Hu, B.; Zhang, Y.; Jia, L.; Wu, H.; Fan, C.; Sun, Y.; Ye, C.; Liao, M.; Zhou, J. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 2015, 11, 503–515. [Google Scholar] [CrossRef]
- Negoita, F.; Blomdahl, J.; Wasserstrom, S.; Winberg, M.E.; Osmark, P.; Larsson, S.; Stenkula, K.G.; Ekstedt, M.; Kechagias, S.; Holm, C.; et al. PNPLA3 variant M148 causes resistance to starvation-mediated lipid droplet autophagy in human hepatocytes. J. Cell. Biochem. 2019, 120, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Hu, F.; Shao, L.; Zhang, J.; Zhang, H.; Wen, A.; Zhang, P. Knockdown of ZFAS1 Inhibits Hippocampal Neurons Apoptosis and Autophagy by Activating the PI3K/AKT Pathway via Up-regulating miR-421 in Epilepsy. Neurochem. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Murphy, A.; Zou, F.; Gerard, C.; Klanderman, B.; Schuemann, B.; Lazarus, R.; Garcia, K.C.; Celedon, J.C.; Drumm, M.; et al. IL1B polymorphisms modulate cystic fibrosis lung disease. Pediatr. Pulmonol. 2009, 44, 580–593. [Google Scholar] [CrossRef]
- Gillette, D.D.; Shah, P.A.; Cremer, T.; Gavrilin, M.A.; Besecker, B.Y.; Sarkar, A.; Knoell, D.L.; Cormet-Boyaka, E.; Wewers, M.D.; Butchar, J.P.; et al. Analysis of human bronchial epithelial cell proinflammatory response to Burkholderia cenocepacia infection: Inability to secrete il-1beta. J. Biol. Chem. 2013, 288, 3691–3695. [Google Scholar] [CrossRef] [PubMed]
- Boada-Romero, E.; Letek, M.; Fleischer, A.; Pallauf, K.; Ramon-Barros, C.; Pimentel-Muinos, F.X. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. EMBO J. 2013, 32, 566–582. [Google Scholar] [CrossRef]
- Nowak, J.; Archange, C.; Tardivel-Lacombe, J.; Pontarotti, P.; Pebusque, M.J.; Vaccaro, M.I.; Velasco, G.; Dagorn, J.C.; Iovanna, J.L. The TP53INP2 protein is required for autophagy in mammalian cells. Mol. Biol. Cell 2009, 20, 870–881. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Zaki, M.S.; Al-Gazali, L.; Wang, X.; Rosti, R.O.; Dikoglu, E.; Gelot, A.B.; Rosti, B.; Vaux, K.K.; et al. Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat. Genet. 2015, 47, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Liu, Y.; Datta, S.; Hariri, H.; Seda, M.; Anderson, G.; Peskett, E.; Demetriou, C.; Sousa, S.; Jenkins, D.; et al. SNX14 mutations affect endoplasmic reticulum-associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20. Hum. Mol. Genet. 2018, 27, 1927–1940. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xian, W.J.; Gao, Y.; Jiang, S.; Yu, Q.H.; Zheng, Q.C.; Zhang, Y. Higd1a Protects Cells from Lipotoxicity under High-Fat Exposure. Oxid. Med. Cell. Longev. 2019, 2019, 6051262. [Google Scholar] [CrossRef] [PubMed]
- Idrovo, J.P.; Yang, W.L.; Jacob, A.; Corbo, L.; Nicastro, J.; Coppa, G.F.; Wang, P. Inhibition of lipogenesis reduces inflammation and organ injury in sepsis. J. Surg. Res. 2016, 200, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Mailhot, G.; Ravid, Z.; Barchi, S.; Moreau, A.; Rabasa-Lhoret, R.; Levy, E. CFTR knockdown stimulates lipid synthesis and transport in intestinal Caco-2/15 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1239–G1249. [Google Scholar] [CrossRef]
- Weidberg, H.; Elazar, Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 2011, 4, pe39. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Hansen, M.; Troemel, E. Autophagy and innate immunity: Insights from invertebrate model organisms. Autophagy 2018, 14, 233–242. [Google Scholar] [CrossRef]
- Nozawa, T.; Sano, S.; Minowa-Nozawa, A.; Toh, H.; Nakajima, S.; Murase, K.; Aikawa, C.; Nakagawa, I. TBC1D9 regulates TBK1 activation through Ca2+ signaling in selective autophagy. Nat. Commun. 2020, 11, 770. [Google Scholar] [CrossRef]
- Rhodes, B.; Nash, E.F.; Tullis, E.; Pencharz, P.B.; Brotherwood, M.; Dupuis, A.; Stephenson, A. Prevalence of dyslipidemia in adults with cystic fibrosis. J. Cyst. Fibros. 2010, 9, 24–28. [Google Scholar] [CrossRef]
- Ishimo, M.C.; Belson, L.; Ziai, S.; Levy, E.; Berthiaume, Y.; Coderre, L.; Rabasa-Lhoret, R. Hypertriglyceridemia is associated with insulin levels in adult cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 271–276. [Google Scholar] [CrossRef]
- Freedman, S.D.; Blanco, P.G.; Zaman, M.M.; Shea, J.C.; Ollero, M.; Hopper, I.K.; Weed, D.A.; Gelrud, A.; Regan, M.M.; Laposata, M.; et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N. Engl. J. Med. 2004, 350, 560–569. [Google Scholar] [CrossRef]
- Olveira, G.; Dorado, A.; Olveira, C.; Padilla, A.; Rojo-Martinez, G.; Garcia-Escobar, E.; Gaspar, I.; Gonzalo, M.; Soriguer, F. Serum phospholipid fatty acid profile and dietary intake in an adult Mediterranean population with cystic fibrosis. Br. J. Nutr. 2006, 96, 343–349. [Google Scholar] [CrossRef]
- Peretti, N.; Roy, C.C.; Drouin, E.; Seidman, E.; Brochu, P.; Casimir, G.; Levy, E. Abnormal intracellular lipid processing contributes to fat malabsorption in cystic fibrosis patients. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G609–G615. [Google Scholar] [CrossRef] [PubMed]
- Dogliotti, E.; Vezzoli, G.; Nouvenne, A.; Meschi, T.; Terranegra, A.; Mingione, A.; Brasacchio, C.; Raspini, B.; Cusi, D.; Soldati, L. Nutrition in calcium nephrolithiasis. J. Transl. Med. 2013, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- White, N.M.; Jiang, D.; Burgess, J.D.; Bederman, I.R.; Previs, S.F.; Kelley, T.J. Altered cholesterol homeostasis in cultured and in vivo models of cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L476–L486. [Google Scholar] [CrossRef] [PubMed]
- Desbenoit, N.; Saussereau, E.; Bich, C.; Bourderioux, M.; Fritsch, J.; Edelman, A.; Brunelle, A.; Ollero, M. Localized lipidomics in cystic fibrosis: TOF-SIMS imaging of lungs from Pseudomonas aeruginosa-infected mice. Int. J. Biochem. Cell Biol. 2014, 52, 77–82. [Google Scholar] [CrossRef]
- Gentzsch, M.; Choudhury, A.; Chang, X.B.; Pagano, R.E.; Riordan, J.R. Misassembled mutant DeltaF508 CFTR in the distal secretory pathway alters cellular lipid trafficking. J. Cell Sci. 2007, 120, 447–455. [Google Scholar] [CrossRef]
- Gentzsch, M.; Chang, X.B.; Cui, L.; Wu, Y.; Ozols, V.V.; Choudhury, A.; Pagano, R.E.; Riordan, J.R. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Chillappagari, S.; Venkatesan, S.; Garapati, V.; Mahavadi, P.; Munder, A.; Seubert, A.; Sarode, G.; Guenther, A.; Schmeck, B.T.; Tummler, B.; et al. Impaired TLR4 and HIF expression in cystic fibrosis bronchial epithelial cells downregulates hemeoxygenase-1 and alters iron homeostasis in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L791–L799. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Agi, E. Heat shock proteins in infection. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 498, 90–100. [Google Scholar] [CrossRef]
- Stewart, G.R.; Young, D.B. Heat-shock proteins and the host-pathogen interaction during bacterial infection. Curr. Opin. Immunol. 2004, 16, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Osterloh, A.; Breloer, M. Heat shock proteins: Linking danger and pathogen recognition. Med. Microbiol. Immunol. 2008, 197, 1–8. [Google Scholar] [CrossRef]
- Dokladny, K.; Myers, O.B.; Moseley, P.L. Heat shock response and autophagy--cooperation and control. Autophagy 2015, 11, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Tardelli, M.; Bruschi, F.V.; Trauner, M. The role of metabolic lipases in the pathogenesis and management of liver disease. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Bonezzi, F.; Piccoli, M.; Dei Cas, M.; Paroni, R.; Mingione, A.; Monasky, M.M.; Caretti, A.; Riganti, C.; Ghidoni, R.; Pappone, C.; et al. Sphingolipid Synthesis Inhibition by Myriocin Administration Enhances Lipid Consumption and Ameliorates Lipid Response to Myocardial Ischemia Reperfusion Injury. Front. Physiol. 2019, 10, 986. [Google Scholar] [CrossRef]
- Sasset, L.; Zhang, Y.; Dunn, T.M.; Di Lorenzo, A. Sphingolipid De Novo Biosynthesis: A Rheostat of Cardiovascular Homeostasis. Trends Endocrinol. Metab. TEM 2016, 27, 807–819. [Google Scholar] [CrossRef]
- Cai, L.; Oyeniran, C.; Biswas, D.D.; Allegood, J.; Milstien, S.; Kordula, T.; Maceyka, M.; Spiegel, S. ORMDL proteins regulate ceramide levels during sterile inflammation. J. Lipid Res. 2016, 57, 1412–1422. [Google Scholar] [CrossRef]
- Chen, L.; Duan, Y.; Wei, H.; Ning, H.; Bi, C.; Zhao, Y.; Qin, Y.; Li, Y. Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors. Expert Opin. Investig. Drugs 2019, 28, 917–930. [Google Scholar] [CrossRef]
- Ammanathan, V.; Mishra, P.; Chavalmane, A.K.; Muthusamy, S.; Jadhav, V.; Siddamadappa, C.; Manjithaya, R. Restriction of intracellular Salmonella replication by restoring TFEB-mediated xenophagy. Autophagy 2019, 1–14. [Google Scholar] [CrossRef]
- Khoury, T.; Asombang, A.W.; Berzin, T.M.; Cohen, J.; Pleskow, D.K.; Mizrahi, M. The Clinical Implications of Fatty Pancreas: A Concise Review. Dig. Dis. Sci. 2017, 62, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Tham, R.T.; Heyerman, H.G.; Falke, T.H.; Zwinderman, A.H.; Bloem, J.L.; Bakker, W.; Lamers, C.B. Cystic fibrosis: MR imaging of the pancreas. Radiology 1991, 179, 183–186. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
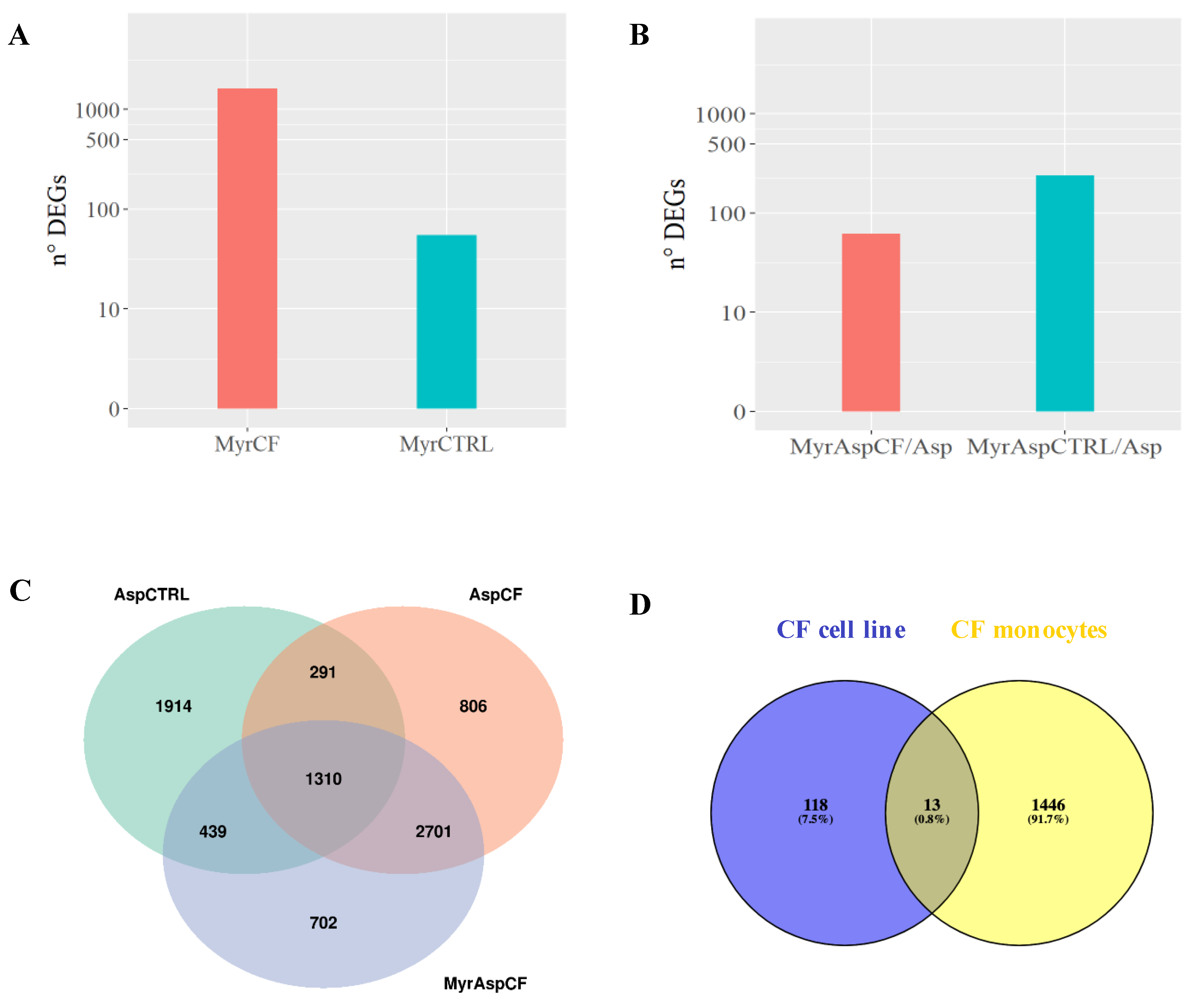{kind=link}
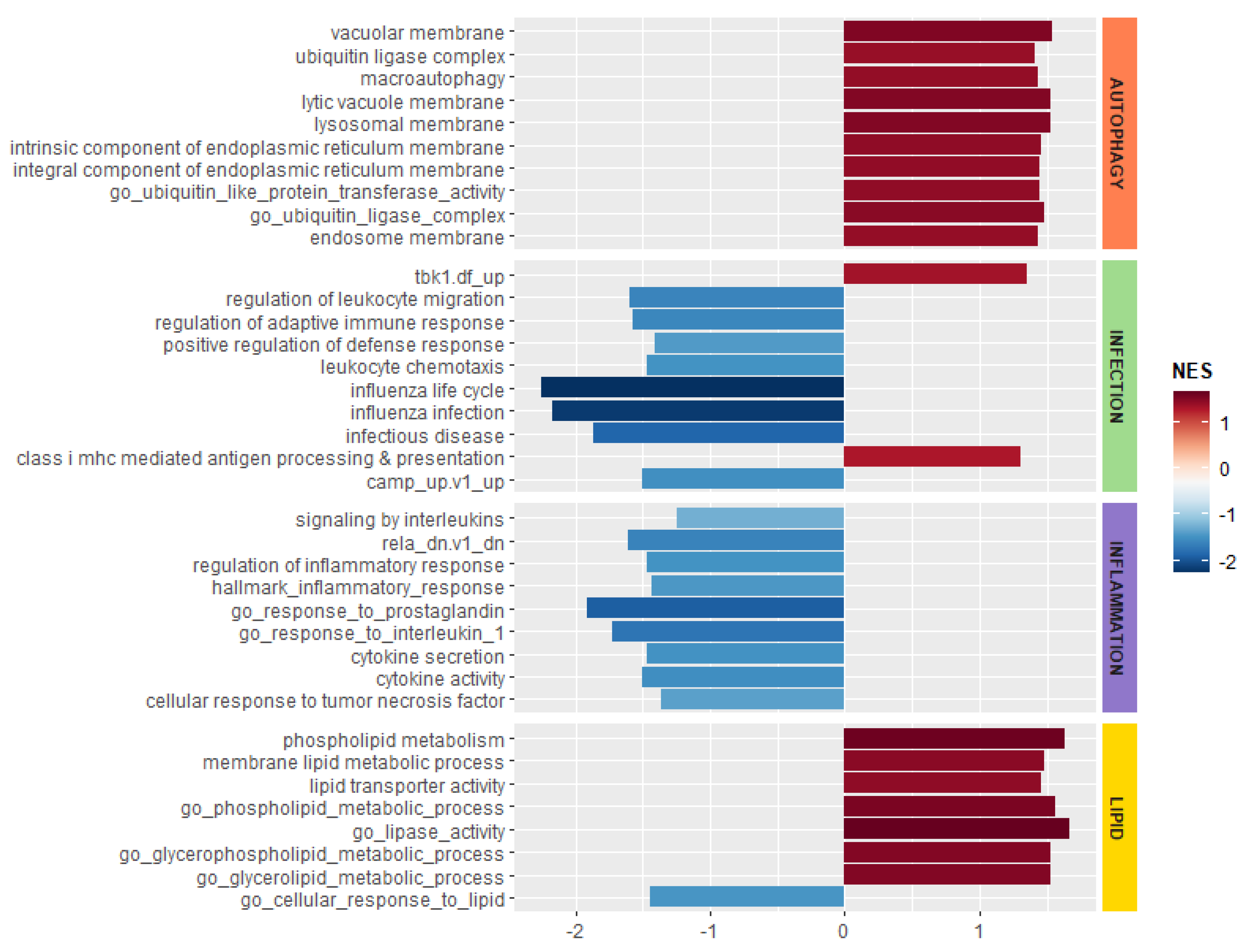{kind=link}
{kind=link}
{kind=link}
| Symbol | Log2FoldChange | Padj | Description | Ensembl |
|---|---|---|---|---|
| HIST1H2BH | 1.86 | 8.07 × 10−5 | histone cluster 1 H2B family member h | ENSG00000275713 |
| HIST1H2BG | 1.56 | 0.001394033 | histone cluster 1 H2B family member g | ENSG00000273802 |
| HIST1H2BO | 1.54 | 0.021985759 | histone cluster 1 H2B family member o | ENSG00000274641 |
| HIST1H2BE | 1.53 | 0.007600047 | histone cluster 1 H2B family member e | ENSG00000274290 |
| HIST1H2BJ | 1.29 | 0.002758228 | histone cluster 1 H2B family member j | ENSG00000124635 |
| HIST1H2BD | 1.21 | 0.001620018 | histone cluster 1 H2B family member d | ENSG00000158373 |
| HIST1H4C | 1.14 | 0.004057245 | histone cluster 1 H4 family member c | ENSG00000197061 |
| H2AFX | 1.04 | 0.009827979 | H2A histone family member X | ENSG00000188486 |
| HIST1H3A | 1.02 | 0.05944192 | histone cluster 1 H3 family member a | ENSG00000275714 |
| HIST2H2BE | 0.88 | 0.028428384 | histone cluster 2 H2B family member e | ENSG00000184678 |
| H2AFZ | 0.63 | 0.044623207 | H2A histone family member Z | ENSG00000164032 |
| HIST1H3J | 0.93 | 0.204175587 | histone cluster 1 H3 family member j | ENSG00000197153 |
| HIST1H4I | 0.72 | 0.065534653 | histone cluster 1 H4 family member i | ENSG00000276180 |
| HIST1H2AC | 0.47 | 0.318228769 | histone cluster 1 H2A family member c | ENSG00000180573 |
| Symbol | Log2FoldChange | Padj | Description | Ensembl |
|---|---|---|---|---|
| HSPA6 | 3.41 | 2.65 × 10−9 | heat shock protein family A (Hsp70) member 6 | ENSG00000173110 |
| DNAJA4 | 2.71 | 2.25 × 10−16 | DnaJ heat shock protein family (Hsp40) member A4 | ENSG00000140403 |
| HSPA1B | 2.71 | 1.19 × 10−10 | heat shock protein family A (Hsp70) member 1B | ENSG00000204388 |
| HSPA1A | 2.64 | 3.02 × 10−13 | heat shock protein family A (Hsp70) member 1A | ENSG00000204389 |
| DNAJB1 | 2.47 | 8.60 × 10−13 | DnaJ heat shock protein family (Hsp40) member B1 | ENSG00000132002 |
| BAG3 | 2.14 | 1.79 × 10−11 | BCL2 associated athanogene 3 | ENSG00000151929 |
| HSPA2 | 2.00 | 1.54 × 10−5 | heat shock protein family A (Hsp70) member 2 | ENSG00000126803 |
| SERPINH1 | 1.94 | 3.78 × 10−9 | serpin family H member 1 | ENSG00000149257 |
| HSPD1 | 1.68 | 3.78 × 10−9 | heat shock protein family D (Hsp60) member 1 | ENSG00000144381 |
| ZFAND2A | 1.60 | 1.29 × 10−9 | zinc finger AN1-type containing 2A | ENSG00000178381 |
| HSPE1 | 1.40 | 7.08 × 10−5 | heat shock protein family E (Hsp10) member 1 | ENSG00000115541 |
| HSP90AA1 | 1.35 | 1.13 × 10−6 | heat shock protein 90 alpha family class A member 1 | ENSG00000080824 |
| DNAJB4 | 1.13 | 5.80 × 10−7 | DnaJ heat shock protein family (Hsp40) member B4 | ENSG00000162616 |
| HSPA1L | 1.13 | 8.07 × 10−5 | heat shock protein family A (Hsp70) member 1 like | ENSG00000204390 |
| DNAJA1 | 0.90 | 6.78 × 10−5 | DnaJ heat shock protein family (Hsp40) member A1 | ENSG00000086061 |
| HSP90AB1 | 0.82 | 0.002280106 | heat shock protein 90 alpha family class B member 1 | ENSG00000096384 |
| HSPA8 | 0.53 | 0.058998158 | heat shock protein family A (Hsp70) member 8 | ENSG00000109971 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mingione, A.; Ottaviano, E.; Barcella, M.; Merelli, I.; Rosso, L.; Armeni, T.; Cirilli, N.; Ghidoni, R.; Borghi, E.; Signorelli, P. Cystic Fibrosis Defective Response to Infection Involves Autophagy and Lipid Metabolism. Cells 2020, 9, 1845. https://doi.org/10.3390/cells9081845
Mingione A, Ottaviano E, Barcella M, Merelli I, Rosso L, Armeni T, Cirilli N, Ghidoni R, Borghi E, Signorelli P. Cystic Fibrosis Defective Response to Infection Involves Autophagy and Lipid Metabolism. Cells. 2020; 9(8):1845. https://doi.org/10.3390/cells9081845
Chicago/Turabian StyleMingione, Alessandra, Emerenziana Ottaviano, Matteo Barcella, Ivan Merelli, Lorenzo Rosso, Tatiana Armeni, Natalia Cirilli, Riccardo Ghidoni, Elisa Borghi, and Paola Signorelli. 2020. "Cystic Fibrosis Defective Response to Infection Involves Autophagy and Lipid Metabolism" Cells 9, no. 8: 1845. https://doi.org/10.3390/cells9081845
APA StyleMingione, A., Ottaviano, E., Barcella, M., Merelli, I., Rosso, L., Armeni, T., Cirilli, N., Ghidoni, R., Borghi, E., & Signorelli, P. (2020). Cystic Fibrosis Defective Response to Infection Involves Autophagy and Lipid Metabolism. Cells, 9(8), 1845. https://doi.org/10.3390/cells9081845