The Role of MicroRNAs in Repair Processes in Multiple Sclerosis


Abstract
1. Introduction
1.1. Overview
1.2. Remyelination
1.3. MicroRNAs
2. Contribution of miRNAs to Remyelination in CNS Cells
2.1. OPC Intrinsic miRNAs
2.2. Microglia
2.3. Astrocytes
2.4. Neurons
3. Peripheral Immune miRNAs
3.1. Leukocytes
3.2. Monocytes
4. miRNAs as Therapeutic Targets in Multiple Sclerosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, W.J.; Chen, W.W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Gran, B. The essential role of t cells in multiple sclerosis: A reappraisal. Biomed. J. 2014, 37, 34–40. [Google Scholar] [CrossRef]
- Nikic, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Bruck, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef] [PubMed]
- University of California; San Francisco MS-EPIC Team; Cree, B.A.; Gourraud, P.A.; Oksenberg, J.R.; Bevan, C.; Crabtree-Hartman, E.; Gelfand, J.M.; Goodin, D.S.; Graves, J.; et al. Long-term evolution of multiple sclerosis disability in the treatment ERA. Ann. Neurol. 2016, 80, 499–510. [Google Scholar] [PubMed]
- Miller, D.H.; Leary, S.M. Primary-progressive multiple sclerosis. Lancet Neurol. 2007, 6, 903–912. [Google Scholar] [CrossRef]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Miron, V.E.; Kuhlmann, T.; Antel, J.P. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim. Biophys. Acta 2011, 1812, 184–193. [Google Scholar] [CrossRef]
- Smith, P.M.; Jeffery, N.D. Histological and ultrastructural analysis of white matter damage after naturally-occurring spinal cord injury. Brain Pathol. 2006, 16, 99–109. [Google Scholar] [CrossRef]
- Gupta, R.; Rowshan, K.; Chao, T.; Mozaffar, T.; Steward, O. Chronic nerve compression induces local demyelination and remyelination in a rat model of carpal tunnel syndrome. Exp. Neurol. 2004, 187, 500–508. [Google Scholar] [CrossRef]
- Glezer, I.; Lapointe, A.; Rivest, S. Innate immunity triggers oligodendrocyte progenitor reactivity and confines damages to brain injuries. FASEB J. 2006, 20, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, M.; Franklin, R.J. Myelin regeneration in demyelinating disorders: New developments in biology and clinical pathology. Curr. Opin. Neurol. 2007, 20, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Gensert, J.M.; Goldman, J.E. Endogenous progenitors remyelinate demyelinated axons in the adult cns. Neuron 1997, 19, 197–203. [Google Scholar] [CrossRef]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef]
- Kotter, M.R.; Li, W.W.; Zhao, C.; Franklin, R.J. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in development, myelin generation and beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Molina-Gonzalez, I.; Miron, V.E. Astrocytes in myelination and remyelination. Neurosci. Lett. 2019, 713, 134532. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Davies, C.L.; Holloway, R.K.; Labrak, Y.; Ireland, G.; Carradori, D.; Dillenburg, A.; Borger, E.; Soong, D.; Richardson, J.C.; et al. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 2019, 22, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The role of astrocytes in multiple sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.; Kroner, A.; David, S. Iron efflux from astrocytes plays a role in remyelination. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 4841–4847. [Google Scholar] [CrossRef]
- Tress, O.; Maglione, M.; May, D.; Pivneva, T.; Richter, N.; Seyfarth, J.; Binder, S.; Zlomuzica, A.; Seifert, G.; Theis, M.; et al. Panglial gap junctional communication is essential for maintenance of myelin in the cns. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 7499–7518. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-astrocyte crosstalk: An intimate molecular conversation. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Holley, J.E.; Gveric, D.; Newcombe, J.; Cuzner, M.L.; Gutowski, N.J. Astrocyte characterization in the multiple sclerosis glial scar. Neuropathol. Appl. Neurobiol. 2003, 29, 434–444. [Google Scholar] [CrossRef]
- Bjelobaba, I.; Begovic-Kupresanin, V.; Pekovic, S.; Lavrnja, I. Animal models of multiple sclerosis: Focus on experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2018, 96, 1021–1042. [Google Scholar] [CrossRef]
- Procaccini, C.; De Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of multiple sclerosis. Eur. J. Pharm. 2015, 759, 182–191. [Google Scholar] [CrossRef]
- Blakemore, W.F.; Franklin, R.J. Remyelination in experimental models of toxin-induced demyelination. Curr. Top. Microbiol. Immunol. 2008, 318, 193–212. [Google Scholar]
- Madill, M.; Fitzgerald, D.; O’Connell, K.E.; Dev, K.K.; Shen, S.; FitzGerald, U. In vitro and ex vivo models of multiple sclerosis. Drug Discov. Today 2016, 21, 1504–1511. [Google Scholar] [CrossRef]
- Tagliafierro, L.; Bonawitz, K.; Glenn, O.C.; Chiba-Falek, O. Gene expression analysis of neurons and astrocytes isolated by laser capture microdissection from frozen human brain tissues. Front. Mol. Neurosci. 2016, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.T.S.; Fuh, S.C.; Karamchandani, J.R.; Woulfe, J.M.J.; Munoz, D.G.; Ellezam, B.; Blain, M.; Ho, M.K.; Bedell, B.J.; Antel, J.P.; et al. Astrocytes in the pathogenesis of multiple sclerosis: An in situ microRNA study. J. Neuropathol. Exp. Neurol. 2019, 78, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Honce, J.M. Gray matter pathology in ms: Neuroimaging and clinical correlations. Mult. Scler. Int. 2013, 2013, 627870. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.R.; Button, T.; Schmierer, K.; Hunter, K.; Tozer, D.J.; Wheeler-Kingshott, C.A.; Coles, A.; Miller, D.H. Sample sizes for lesion magnetisation transfer ratio outcomes in remyelination trials for multiple sclerosis. Mult. Scler. Relat. Disord. 2014, 3, 237–243. [Google Scholar] [CrossRef][Green Version]
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Levy, R.J.; Huguenard, J.R.; Pasca, S.P. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef]
- Mehta, A.; Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016, 16, 279–294. [Google Scholar] [CrossRef]
- Cao, D.D.; Li, L.; Chan, W.Y. MicroRNAs: Key regulators in the central nervous system and their implication in neurological diseases. Int. J. Mol. Sci. 2016, 17, 842. [Google Scholar] [CrossRef]
- Ghadiri, N.; Emamnia, N.; Ganjalikhani-Hakemi, M.; Ghaedi, K.; Etemadifar, M.; Salehi, M.; Shirzad, H.; Nasr-Esfahani, M.H. Analysis of the expression of miR-34a, miR-199a, miR-30c and miR-19a in peripheral blood CD4+T lymphocytes of relapsing-remitting multiple sclerosis patients. Gene 2018, 659, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.R.; Mackenzie, J.; Chaplin, G.; Jablonski, N.G.; Griffiths, L. Circulating microRNAs involved in multiple sclerosis. Mol. Biol. Rep. 2012, 39, 6219–6225. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Piket, E.; Khademi, M.; James, T.; Brundin, L.; Olsson, T.; Piehl, F.; Jagodic, M. Circulating miR-150 in CSF is a novel candidate biomarker for multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e219. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, H.B.; Hesse, D.; Krakauer, M.; Sorensen, P.S.; Sellebjerg, F. Differential microRNA expression in blood in multiple sclerosis. Mult. Scler. 2013, 19, 1849–1857. [Google Scholar] [CrossRef]
- Fenoglio, C.; Cantoni, C.; De Riz, M.; Ridolfi, E.; Cortini, F.; Serpente, M.; Villa, C.; Comi, C.; Monaco, F.; Mellesi, L.; et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci. Lett. 2011, 504, 9–12. [Google Scholar] [CrossRef]
- Selmaj, I.; Cichalewska, M.; Namiecinska, M.; Galazka, G.; Horzelski, W.; Selmaj, K.W.; Mycko, M.P. Global exosome transcriptome profiling reveals biomarkers for multiple sclerosis. Ann. Neurol. 2017, 81, 703–717. [Google Scholar] [CrossRef]
- Vistbakka, J.; Elovaara, I.; Lehtimaki, T.; Hagman, S. Circulating microRNAs as biomarkers in progressive multiple sclerosis. Mult. Scler. 2017, 23, 403–412. [Google Scholar] [CrossRef]
- Ingwersen, J.; Menge, T.; Wingerath, B.; Kaya, D.; Graf, J.; Prozorovski, T.; Keller, A.; Backes, C.; Beier, M.; Scheffler, M.; et al. Natalizumab restores aberrant miRNA expression profile in multiple sclerosis and reveals a critical role for miR-20b. Ann. Clin. Transl. Neurol. 2015, 2, 43–55. [Google Scholar] [CrossRef]
- McCoy, C.E. MiR-155 dysregulation and therapeutic intervention in multiple sclerosis. Adv. Exp. Med. Biol. 2017, 1024, 111–131. [Google Scholar]
- Dugas, J.C.; Cuellar, T.L.; Scholze, A.; Ason, B.; Ibrahim, A.; Emery, B.; Zamanian, J.L.; Foo, L.C.; McManus, M.T.; Barres, B.A. Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron 2010, 65, 597–611. [Google Scholar] [CrossRef]
- Zhao, X.; He, X.; Han, X.; Yu, Y.; Ye, F.; Chen, Y.; Hoang, T.; Xu, X.; Mi, Q.S.; Xin, M.; et al. MicroRNA-mediated control of oligodendrocyte differentiation. Neuron 2010, 65, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Shin, J.Y.; McManus, M.T.; Ptacek, L.J.; Fu, Y.H. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann. Neurol. 2009, 66, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Aung, L.L.; Balashov, K.E. Decreased dicer expression is linked to increased expression of co-stimulatory molecule CD80 on B cells in multiple sclerosis. Mult. Scler. 2015, 21, 1131–1138. [Google Scholar] [CrossRef]
- Lewkowicz, P.; Cwiklinska, H.; Mycko, M.P.; Cichalewska, M.; Domowicz, M.; Lewkowicz, N.; Jurewicz, A.; Selmaj, K.W. Dysregulated RNA-induced silencing complex (RISC) assembly within CNS corresponds with abnormal miRNA expression during autoimmune demyelination. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 7521–7537. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ren, C.; Qu, X.; Wu, X.; Dong, F.; Chand, Y.K.; Fan, H.; Yao, R.; Geng, D. MiR-219 attenuates demyelination in cuprizone-induced demyelinated mice by regulating monocarboxylate transporter 1. Eur. J. Neurosci. 2017, 45, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Moyano, A.L.; Ma, Z.; Deng, Y.; Lin, Y.; Zhao, C.; Zhang, L.; Jiang, M.; He, X.; Ma, Z.; et al. MiR-219 cooperates with miR-338 in myelination and promotes myelin repair in the CNS. Dev. Cell 2017, 40, 566–582.e565. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Volsko, C.; Datta, U.; Regev, K.; Dutta, R. Expression of disease-related miRNAs in white-matter lesions of progressive multiple sclerosis brains. Ann. Clin. Transl. Neurol. 2019, 6, 854–862. [Google Scholar] [CrossRef]
- Reijerkerk, A.; Lopez-Ramirez, M.A.; van Het Hof, B.; Drexhage, J.A.; Kamphuis, W.W.; Kooij, G.; Vos, J.B.; van der Pouw Kraan, T.C.; van Zonneveld, A.J.; Horrevoets, A.J.; et al. MicroRNAs regulate human brain endothelial cell-barrier function in inflammation: Implications for multiple sclerosis. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 6857–6863. [Google Scholar] [CrossRef]
- Long, H.C.; Wu, R.; Liu, C.F.; Xiong, F.L.; Xu, Z.; He, D.; Zhang, Y.F.; Shao, B.; Zhang, P.A.; Xu, G.Y.; et al. MiR-125a-5p regulates vitamin d receptor expression in a mouse model of experimental autoimmune encephalomyelitis. Neurosci. Bull. 2020, 36, 110–120. [Google Scholar] [CrossRef]
- Tripathi, A.; Volsko, C.; Garcia, J.P.; Agirre, E.; Allan, K.C.; Tesar, P.J.; Trapp, B.D.; Castelo-Branco, G.; Sim, F.J.; Dutta, R. Oligodendrocyte intrinsic miR-27a controls myelination and remyelination. Cell Rep. 2019, 29, 904–919.e909. [Google Scholar] [CrossRef]
- Morquette, B.; Juzwik, C.A.; Drake, S.S.; Charabati, M.; Zhang, Y.; Lecuyer, M.A.; Galloway, D.A.; Dumas, A.; de Faria Junior, O.; Paradis-Isler, N.; et al. MicroRNA-223 protects neurons from degeneration in experimental autoimmune encephalomyelitis. Brain J. Neurol. 2019, 142, 2979–2995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.G.; Lu, M.; Wang, X.; Shang, X.; Elias, S.B.; Chopp, M. MiR-146a promotes remyelination in a cuprizone model of demyelinating injury. Neuroscience 2017, 348, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Saba, R.; Gushue, S.; Huzarewich, R.L.; Manguiat, K.; Medina, S.; Robertson, C.; Booth, S.A. microRNA 146a (miR-146a) is over-expressed during prion disease and modulates the innate immune response and the microglial activation state. PLoS ONE 2012, 7, e30832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.G.; Lu, M.; Zhang, Y.; Shang, X.; Chopp, M. MiR-146a promotes oligodendrocyte progenitor cell differentiation and enhances remyelination in a model of experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2019, 125, 154–162. [Google Scholar] [CrossRef]
- Galloway, D.A.; Williams, J.B.; Moore, C.S. Effects of fumarates on inflammatory human astrocyte responses and oligodendrocyte differentiation. Ann. Clin. Transl. Neurol. 2017, 4, 381–391. [Google Scholar] [CrossRef]
- Li, B.; Wang, X.; Choi, I.Y.; Wang, Y.C.; Liu, S.; Pham, A.T.; Moon, H.; Smith, D.J.; Rao, D.S.; Boldin, M.P.; et al. MiR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J. Clin. Investig. 2017, 127, 3702–3716. [Google Scholar] [CrossRef]
- Budde, H.; Schmitt, S.; Fitzner, D.; Opitz, L.; Salinas-Riester, G.; Simons, M. Control of oligodendroglial cell number by the miR-17-92 cluster. Development 2010, 137, 2127–2132. [Google Scholar] [CrossRef]
- Kuypers, N.J.; Bankston, A.N.; Howard, R.M.; Beare, J.E.; Whittemore, S.R. Remyelinating oligodendrocyte precursor cell miRNAs from the Sfmbt2 cluster promote cell cycle arrest and differentiation. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 1698–1710. [Google Scholar] [CrossRef]
- Buller, B.; Chopp, M.; Ueno, Y.; Zhang, L.; Zhang, R.L.; Morris, D.; Zhang, Y.; Zhang, Z.G. Regulation of serum response factor by miRNA-200 and miRNA-9 modulates oligodendrocyte progenitor cell differentiation. Glia 2012, 60, 1906–1914. [Google Scholar] [CrossRef]
- Lau, P.; Verrier, J.D.; Nielsen, J.A.; Johnson, K.R.; Notterpek, L.; Hudson, L.D. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 11720–11730. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.T.; Fu, Y.H. MiR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis. Model Mech. 2009, 2, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Afrang, N.; Tavakoli, R.; Tasharrofi, N.; Alian, A.; Naderi Sohi, A.; Kabiri, M.; Fathi-Roudsari, M.; Soufizomorrod, M.; Rajaei, F.; Soleimani, M.; et al. A critical role for miR-184 in the fate determination of oligodendrocytes. Stem Cell Res. Ther. 2019, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, J.; Zheng, M.; Gao, F.; Ju, G. Specification and maintenance of oligodendrocyte precursor cells from neural progenitor cells: Involvement of microrna-7a. Mol. Biol. Cell 2012, 23, 2867–2878. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef]
- Dutta, R.; Chomyk, A.M.; Chang, A.; Ribaudo, M.V.; Deckard, S.A.; Doud, M.K.; Edberg, D.D.; Bai, B.; Li, M.; Baranzini, S.E.; et al. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann. Neurol. 2013, 73, 637–645. [Google Scholar] [CrossRef]
- Galloway, D.A.; Blandford, S.N.; Berry, T.; Williams, J.B.; Stefanelli, M.; Ploughman, M.; Moore, C.S. MiR-223 promotes regenerative myeloid cell phenotype and function in the demyelinated central nervous system. Glia 2019, 67, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, D.; Ren, Y.; Zhang, Z.; Guo, X.; Ma, M.; Xue, Z.; Lv, J.; Liu, H.; Xi, Q.; et al. Mir223 restrains autophagy and promotes CNS inflammation by targeting ATG16L1. Autophagy 2019, 15, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.M.; Perron, G.; Won, S.Y.; Michell-Robinson, M.A.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-Or, A.; Antel, J.P. Mertk is a functional regulator of myelin phagocytosis by human myeloid cells. J. Immunol. 2016, 196, 3375–3384. [Google Scholar] [CrossRef]
- Moore, C.S.; Rao, V.T.; Durafourt, B.A.; Bedell, B.J.; Ludwin, S.K.; Bar-Or, A.; Antel, J.P. MiR-155 as a multiple sclerosis-relevant regulator of myeloid cell polarization. Ann. Neurol. 2013, 74, 709–720. [Google Scholar] [CrossRef]
- Junker, A.; Krumbholz, M.; Eisele, S.; Mohan, H.; Augstein, F.; Bittner, R.; Lassmann, H.; Wekerle, H.; Hohlfeld, R.; Meinl, E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain J. Neurol. 2009, 132, 3342–3352. [Google Scholar] [CrossRef]
- Han, S.R.; Kang, Y.H.; Jeon, H.; Lee, S.; Park, S.J.; Song, D.Y.; Min, S.S.; Yoo, S.M.; Lee, M.S.; Lee, S.H. Differential expression of miRNAs and behavioral change in the cuprizone-induced demyelination mouse model. Int. J. Mol. Sci. 2020, 21, 646. [Google Scholar] [CrossRef]
- Patel, J.R.; Klein, R.S. Mediators of oligodendrocyte differentiation during remyelination. FEBS Lett. 2011, 585, 3730–3737. [Google Scholar] [CrossRef]
- Fang, X.; Sun, D.; Wang, Z.; Yu, Z.; Liu, W.; Pu, Y.; Wang, D.; Huang, A.; Liu, M.; Xiang, Z.; et al. MiR-30a positively regulates the inflammatory response of microglia in experimental autoimmune encephalomyelitis. Neurosci. Bull. 2017, 33, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Freilich, R.W.; Woodbury, M.E.; Ikezu, T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS ONE 2013, 8, e79416. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, S.; Talebi, F.; Chan, W.F.; Masoumi, F.; Vojgani, M.; Power, C.; Noorbakhsh, F. MicroRNA-181 variants regulate t cell phenotype in the context of autoimmune neuroinflammation. Front. Immunol. 2017, 8, 758. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.O.; Hashemifar, S.S.; Wu, C.J.; Cho, S.; Nguyen, D.T.; Lin, L.L.; Khan, A.A.; Lu, L.F. Excessive expression of miR-27 impairs Treg-mediated immunological tolerance. J. Clin. Investig. 2017, 127, 530–542. [Google Scholar] [CrossRef]
- Nakahama, T.; Hanieh, H.; Nguyen, N.T.; Chinen, I.; Ripley, B.; Millrine, D.; Lee, S.; Nyati, K.K.; Dubey, P.K.; Chowdhury, K.; et al. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17-producing T-helper cell differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 11964–11969. [Google Scholar] [CrossRef]
- De Santis, G.; Ferracin, M.; Biondani, A.; Caniatti, L.; Rosaria Tola, M.; Castellazzi, M.; Zagatti, B.; Battistini, L.; Borsellino, G.; Fainardi, E.; et al. Altered miRNA expression in T regulatory cells in course of multiple sclerosis. J. Neuroimmunol. 2010, 226, 165–171. [Google Scholar] [CrossRef]
- Piatek, P.; Namiecinska, M.; Domowicz, M.; Przygodzka, P.; Wieczorek, M.; Michlewska, S.; Lewkowicz, N.; Tarkowski, M.; Lewkowicz, P. MS CD49d(+)CD154(+) lymphocytes reprogram oligodendrocytes into immune reactive cells affecting CNS regeneration. Cells 2019, 8, 1508. [Google Scholar] [CrossRef]
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to oligodendrocyte: An epigenetic journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.Y.; Rao, V.T.; Bin, J.M.; Gris, P.; Sangaralingam, M.; Kennedy, T.E.; Antel, J.P. Heterogeneity of oligodendrocyte progenitor cells in adult human brain. Ann. Clin. Transl. Neurol. 2014, 1, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.A.; Molnar, V.; Szilagyi, G.T.; Elkjaer, M.L.; Nawrocki, A.; Okarmus, J.; Wlodarczyk, A.; Thygesen, E.K.; Palkovits, M.; Gallyas, F., Jr.; et al. Experimental demyelination and axonal loss are reduced in microRNA-146a deficient mice. Front. Immunol. 2018, 9, 490. [Google Scholar] [CrossRef] [PubMed]
- Marangon, D.; Boda, E.; Parolisi, R.; Negri, C.; Giorgi, C.; Montarolo, F.; Perga, S.; Bertolotto, A.; Buffo, A.; Abbracchio, M.P.; et al. In vivo silencing of miR-125a-3p promotes myelin repair in models of white matter demyelination. Glia 2020. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Marangon, D.; Coppolino, G.T.; Mendez, A.M.; Finardi, A.; Costa, G.D.; Martinelli, V.; Furlan, R.; Abbracchio, M.P. MiR-125a-3p timely inhibits oligodendroglial maturation and is pathologically up-regulated in human multiple sclerosis. Sci. Rep. 2016, 6, 34503. [Google Scholar] [CrossRef]
- Letzen, B.S.; Liu, C.; Thakor, N.V.; Gearhart, J.D.; All, A.H.; Kerr, C.L. MicroRNA expression profiling of oligodendrocyte differentiation from human embryonic stem cells. PLoS ONE 2010, 5, e10480. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Veremeyko, T.; Starossom, S.C.; Weiner, H.L.; Ponomarev, E.D. Detection of microRNAs in microglia by real-time PCR in normal CNS and during neuroinflammation. J. Vis. Exp. JOVE 2012, 23, 4097. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Yang, T.; Dai, Y.; Chen, G.; Cui, S. Dissecting the dual role of the glial scar and scar-forming astrocytes in spinal cord injury. Front. Cell. Neurosci. 2020, 14, 78. [Google Scholar] [CrossRef]
- Haindl, M.T.; Kock, U.; Zeitelhofer-Adzemovic, M.; Fazekas, F.; Hochmeister, S. The formation of a glial scar does not prohibit remyelination in an animal model of multiple sclerosis. Glia 2019, 67, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Chang, A.; Doud, M.K.; Kidd, G.J.; Ribaudo, M.V.; Young, E.A.; Fox, R.J.; Staugaitis, S.M.; Trapp, B.D. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 2011, 69, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, B.G. Epidemiology of multiple sclerosis. Neurol. Clin. 1996, 14, 291–308. [Google Scholar] [CrossRef]
- Gautier, H.O.; Evans, K.A.; Volbracht, K.; James, R.; Sitnikov, S.; Lundgaard, I.; James, F.; Lao-Peregrin, C.; Reynolds, R.; Franklin, R.J.; et al. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat. Commun. 2015, 6, 8518. [Google Scholar] [CrossRef]
- Juzwik, C.A.; Drake, S.; Lecuyer, M.A.; Johnson, R.M.; Morquette, B.; Zhang, Y.; Charabati, M.; Sagan, S.M.; Bar-Or, A.; Prat, A.; et al. Neuronal microRNA regulation in experimental autoimmune encephalomyelitis. Sci. Rep. 2018, 8, 13437. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple sclerosis: Mechanisms and immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- De la Vega Gallardo, N.; Dittmer, M.; Dombrowski, Y.; Fitzgerald, D.C. Regenerating CNS myelin: Emerging roles of regulatory T cells and ccn proteins. Neurochem. Int. 2019, 130, 104349. [Google Scholar] [CrossRef]
- Miron, V.E. Beyond immunomodulation: The regenerative role for regulatory T cells in central nervous system remyelination. J. Cell Commun. Signal. 2017, 11, 191–192. [Google Scholar] [CrossRef]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef]
- Raposo, C.; Graubardt, N.; Cohen, M.; Eitan, C.; London, A.; Berkutzki, T.; Schwartz, M. CNS repair requires both effector and regulatory T cells with distinct temporal and spatial profiles. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 10141–10155. [Google Scholar] [CrossRef] [PubMed]
- Koeniger, T.; Kuerten, S. Splitting the “unsplittable”: Dissecting resident and infiltrating macrophages in experimental autoimmune encephalomyelitis. Int. J. Mol. Sci. 2017, 18, 2072. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, G.; Theodorou, D.; Kendirli, A.; Jordao, M.J.C.; Staszewski, O.; Phulphagar, K.; Cantuti-Castelvetri, L.; Dagkalis, A.; Bessis, A.; Simons, M.; et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat. Neurosci. 2018, 21, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Nikolic, I.; Yang, J.; Castillo, L.; Deng, N.; Chan, C.L.; Yeung, N.K.; Dodson, E.; Elsworth, B.; Spielman, C.; et al. microRNAs as potential therapeutics to enhance chemosensitivity in advanced prostate cancer. Sci. Rep. 2018, 8, 7820. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Chang, S. Development of novel therapeutic agents by inhibition of oncogenic micrornas. Int. J. Mol. Sci. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing miRNA-LncRNA interactions. Methods Mol. Biol. 2016, 1402, 271–286. [Google Scholar]
- Louloupi, A.; Orom, U.A.V. Inhibiting Pri-miRNA processing with target site blockers. Methods Mol. Biol. 2018, 1823, 63–68. [Google Scholar]
- Lu, Q.; Wu, R.; Zhao, M.; Garcia-Gomez, A.; Ballestar, E. miRNAs as therapeutic targets in inflammatory disease. Trends Pharm. Sci. 2019, 40, 853–865. [Google Scholar] [CrossRef]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’Arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550. [Google Scholar] [CrossRef]
- Mai, H.; Fan, W.; Wang, Y.; Cai, Y.; Li, X.; Chen, F.; Chen, X.; Yang, J.; Tang, P.; Chen, H.; et al. Intranasal administration of miR-146a agomir rescued the pathological process and cognitive impairment in an AD mouse model. Mol. Nucleic Acids 2019, 18, 681–695. [Google Scholar] [CrossRef]
- Nally, F.K.; De Santi, C.; McCoy, C.E. Nanomodulation of macrophages in multiple sclerosis. Cells 2019, 8, 543. [Google Scholar] [CrossRef]
- Osorio-Querejeta, I.; Alberro, A.; Munoz-Culla, M.; Mager, I.; Otaegui, D. Therapeutic potential of extracellular vesicles for demyelinating diseases; challenges and opportunities. Front. Mol. Neurosci. 2018, 11, 434. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Cuesta, M.; Osorio-Querejeta, I.; Otaegui, D. Extracellular vesicles in multiple sclerosis: What are they telling us? Front. Cell. Neurosci. 2014, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Prabhakar, S.; Balaj, L.; Lai, C.P.; Cerione, R.A.; Breakefield, X.O. Delivery of therapeutic proteins via extracellular vesicles: Review and potential treatments for parkinson’s disease, glioma, and schwannoma. Cell. Mol. Neurobiol. 2016, 36, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Querejeta, I.; Carregal-Romero, S.; Ayerdi-Izquierdo, A.; Mager, I.; Nash, A.L.; Wood, M.; Egimendia, A.; Betanzos, M.; Alberro, A.; Iparraguirre, L.; et al. MiR-219a-5p enriched extracellular vesicles induce OPC differentiation and EAE improvement more efficiently than liposomes and polymeric nanoparticles. Pharmaceutics 2020, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Pusic, A.D.; Pusic, K.M.; Kraig, R.P. What are exosomes and how can they be used in multiple sclerosis therapy? Expert Rev. Neurother. 2014, 14, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Pusic, A.D.; Kraig, R.P. Phasic treatment with interferon gamma stimulates release of exosomes that protect against spreading depression. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2015, 35, 795–807. [Google Scholar] [CrossRef]
- Pusic, K.M.; Pusic, A.D.; Kraig, R.P. Environmental enrichment stimulates immune cell secretion of exosomes that promote CNS myelination and may regulate inflammation. Cell. Mol. Neurobiol. 2016, 36, 313–325. [Google Scholar] [CrossRef]
- Haney, M.J.; Zhao, Y.; Harrison, E.B.; Mahajan, V.; Ahmed, S.; He, Z.; Suresh, P.; Hingtgen, S.D.; Klyachko, N.L.; Mosley, R.L.; et al. Specific transfection of inflamed brain by macrophages: A new therapeutic strategy for neurodegenerative diseases. PLoS ONE 2013, 8, e61852. [Google Scholar] [CrossRef]
- Li, S.P.; Lin, Z.X.; Jiang, X.Y.; Yu, X.Y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharm. Sin. 2018, 39, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Plemel, J.R.; Keough, M.B.; Duncan, G.J.; Sparling, J.S.; Yong, V.W.; Stys, P.K.; Tetzlaff, W. Remyelination after spinal cord injury: Is it a target for repair? Prog. Neurobiol. 2014, 117, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Diao, H.J.; Low, W.C.; Milbreta, U.; Lu, Q.R.; Chew, S.Y. Nanofiber-mediated microRNA delivery to enhance differentiation and maturation of oligodendroglial precursor cells. J. Control. Release Off. J. Control. Release Soc. 2015, 208, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Diao, H.J.; Low, W.C.; Lu, Q.R.; Chew, S.Y. Topographical effects on fiber-mediated microRNA delivery to control oligodendroglial precursor cells development. Biomaterials 2015, 70, 105–114. [Google Scholar] [CrossRef]
- Milbreta, U.; Lin, J.; Pinese, C.; Ong, W.; Chin, J.S.; Shirahama, H.; Mi, R.; Williams, A.; Bechler, M.E.; Wang, J.; et al. Scaffold-mediated sustained, non-viral delivery of miR-219/miR-338 promotes CNS remyelination. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Ong, W.; Wang, K.; Wang, M.; Nizetic, D.; Chew, S.Y. Effects of miR-219/miR-338 on microglia and astrocyte behaviors and astrocyte-oligodendrocyte precursor cell interactions. Neural Regen. Res. 2020, 15, 739–747. [Google Scholar]
- Jalali Monfared, M.; Nasirinezhad, F.; Ebrahimi-Barough, S.; Hasanzade, G.; Saberi, H.; Tavangar, S.M.; Asadpour, S.; Aryan, L.; Barabadi, Z.; Ai, J. Transplantation of miR-219 overexpressed human endometrial stem cells encapsulated in fibrin hydrogel in spinal cord injury. J. Cell. Physiol. 2019, 234, 18887–18896. [Google Scholar] [CrossRef]
- Song, J.; Li, X.; Li, Y.; Che, J.; Li, X.; Zhao, X.; Chen, Y.; Zheng, X.; Yuan, W. Biodegradable and biocompatible cationic polymer delivering microRNA-221/222 promotes nerve regeneration after sciatic nerve crush. Int. J. Nanomed. 2017, 12, 4195–4208. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Yue, P.; Jing, L.; Zhao, X.; Zhu, H.; Teng, J. Down-regulation of taurine-up-regulated gene 1 attenuates inflammation by sponging miR-9-5p via targeting NF-kappaB1/p50 in multiple sclerosis. Life Sci. 2019, 233, 116731. [Google Scholar] [CrossRef]
- Ghasemi-Kasman, M.; Zare, L.; Baharvand, H.; Javan, M. In vivo conversion of astrocytes to myelinating cells by miR-302/367 and valproate to enhance myelin repair. J. Tissue Eng. Regen. Med. 2018, 12, e462–e472. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.B.; Chen, L.X.; Qu, X.B.; Ren, C.L.; Wu, X.X.; Dong, F.X.; Zhang, B.L.; Gao, D.S.; Yao, R.Q. Transplanted miR-219-overexpressing oligodendrocyte precursor cells promoted remyelination and improved functional recovery in a chronic demyelinated model. Sci. Rep. 2017, 7, 41407. [Google Scholar] [CrossRef] [PubMed]
- Horga, A.; Montalban, X. FTY720 (fingolimod) for relapsing multiple sclerosis. Expert Rev. Neurother. 2008, 8, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.; Baharvand, H.; Javan, M. Enhanced remyelination following lysolecithin-induced demyelination in mice under treatment with fingolimod (FTY720). Neuroscience 2015, 311, 34–44. [Google Scholar] [CrossRef]
- Miron, V.E.; Ludwin, S.K.; Darlington, P.J.; Jarjour, A.A.; Soliven, B.; Kennedy, T.E.; Antel, J.P. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am. J. Pathol. 2010, 176, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Cuesta, M.; Alberro, A.; Munoz-Culla, M.; Osorio-Querejeta, I.; Fernandez-Mercado, M.; Lopetegui, I.; Tainta, M.; Prada, A.; Castillo-Trivino, T.; Falcon-Perez, J.M.; et al. The first dose of fingolimod affects circulating extracellular vesicles in multiple sclerosis patients. Int. J. Mol. Sci. 2018, 19, 2448. [Google Scholar] [CrossRef]
- Potenza, N.; Mosca, N.; Mondola, P.; Damiano, S.; Russo, A.; De Felice, B. Human miR-26a-5p regulates the glutamate transporter SLC1A1 (EAAT3) expression. Relevance in multiple sclerosis. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 317–323. [Google Scholar] [CrossRef]
- Waschbisch, A.; Atiya, M.; Linker, R.A.; Potapov, S.; Schwab, S.; Derfuss, T. Glatiramer acetate treatment normalizes deregulated microRNA expression in relapsing remitting multiple sclerosis. PLoS ONE 2011, 6, e24604. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Ellestad, K.K.; Maingat, F.; Warren, K.G.; Han, M.H.; Steinman, L.; Baker, G.B.; Power, C. Impaired neurosteroid synthesis in multiple sclerosis. Brain J. Neurol. 2011, 134, 2703–2721. [Google Scholar] [CrossRef]
- Duan, C.; Liu, Y.; Li, Y.; Chen, H.; Liu, X.; Chen, X.; Yue, J.; Zhou, X.; Yang, J. Sulfasalazine alters microglia phenotype by competing endogenous RNA effect of miR-136-5p and long non-coding RNA HOTAIR in cuprizone-induced demyelination. Biochem. Pharmacol. 2018, 155, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Li, Z.; Cheng, L.; Zhang, L.; Zha, X.; Jing, J. Low frequency pulsed electromagnetic field promotes differentiation of oligodendrocyte precursor cells through upregulation of miR-219-5p in vitro. Life Sci. 2019, 223, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.L.; Syed, I.; Smith, T.L.; Harrison, B.S. The regenerative effects of electromagnetic field on spinal cord injury. Electromagn. Biol. Med. 2017, 36, 74–87. [Google Scholar] [CrossRef] [PubMed]


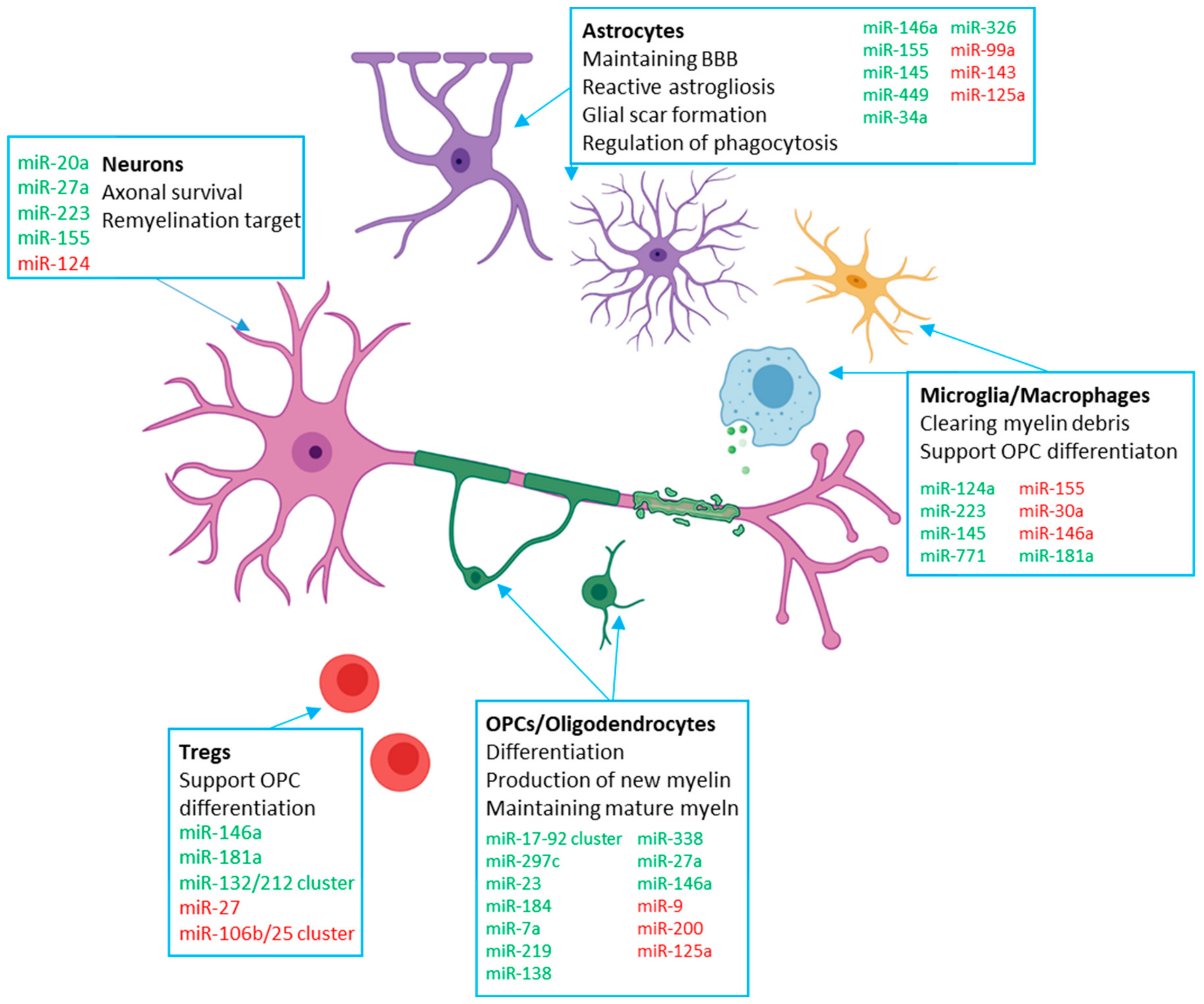{kind=link}
| miRNA | Cell Type | Role in Remyelination |
|---|---|---|
| miR-219 | Oligodendrocytes | - Promotes early and late stages of OPC differentiation [50,51,55,56] - Maintenance of mature myelin sheath [52] |
| miR-138 | Oligodendrocytes | - Promotes early and inhibits late OPC differentiation [50] |
| miR-338 | Oligodendrocytes | - Supports miR-219 in promoting OPC differentiation [56,57] |
| miR-125a | Oligodendrocytes Astrocytes Neurons | - Impairs OPC differentiation - Promotes astrogliosis [32] - Supports BBB maintenance [58] - Reduction is neuroprotective [59] |
| miR-27a | Oligodendrocytes Neurons | - Steady-state expression needed for OPC specification, proliferation and differentiation [60] - Increase is neuroprotective [61] |
| miR-146a | Oligodendrocytes Microglia Astrocytes T cells | - Promotes OPC differentiation [62] -Tempers pro-inflammatory microglia activation and promotes anti-inflammatory activation [63,64] - Reduction associated with astrocyte withdrawal from inflammatory activation [32,65] - Inhibits Th17 differentiation while supporting Treg differentiation [66] |
| miR-17-92 cluster | Oligodendrocytes | - Promotes OPC proliferation [67] |
| miR-297c | Oligodendrocytes | - Promotes G0/1 cell cycle arrest and OPC differentiation [68] |
| miR-9 | Oligodendrocytes | - Overexpression impairs differentiation [69] - Suppresses expression of peripheral myelin protein [70] |
| miR-200 | Oligodendrocytes | - Overexpression impairs differentiation [69] |
| miR-23 | Oligodendrocytes | - Inhibits lamin B1, supporting myelin maintenance [71] |
| miR-184 | Oligodendrocytes | - Commits NPCs to OPC lineage, expression enhances myelination [72] |
| miR-7a | Oligodendrocytes | - Promotes generation of OPCs, inhibits maturation [73] |
| miR-124 | Microglia Neurons | - Promotes quiescent state [74] - Upregulation in demyelinated hippocampal axons associated with memory dysfunction [74,75] |
| miR-223 | Microglia Neurons Macrophages | - Required for efficient anti-inflammatory activation and phagocytosis of myelin debris [76] - Increase is neuroprotective [61] - Overall deficiency ameliorates EAE progression and neuroinflammation [77] |
| miR-155 | Microglia Astrocytes Neurons | - Promotes pro-inflammatory activation of microglia and impairs myelin phagocytosis [78,79] - Expression in astrocytes releases microglia of inhibitory control of phagocytosis [80] - May be neuroprotective via the Nogo pathway [81,82] |
| miR-30a | Microglia | - Overexpression promotes release of factors that induce OPC apoptosis [83] |
| miR-145 | Microglia Astrocytes | - Strongly associated with anti-inflammatory microglia activation [84] - Inhibits astrogliosis [32] |
| miR-771 | Microglia | - Strongly associated with anti-inflammatory microglia activation [84] |
| miR-99a | Astrocytes | - Decrease increases astrocyte proliferation [32] |
| miR-143 | Astrocytes | - Decrease increases astrocyte proliferation [32] |
| miR-449 | Astrocytes | - Attenuates glial scar formation [32] |
| miR-34a | Astrocytes | - Expression in astrocytes releases microglia of inhibitory control of phagocytosis [80] |
| miR-326 | Astrocytes | - Expression in astrocytes releases microglia of inhibitory control of phagocytosis [80] |
| miR-20a | Neurons | - May be neuroprotective via the Nogo pathway [81] |
| miR-181a | T cells Macrophages | - Promotes Treg differentiation [85] - Inhibits Th1 differentiation and pro-inflammatory macrophage polarisation [85] |
| miR-27 | T cells | - Impairs Treg differentiation and Treg immunosuppressive activity [86] |
| miR-132/212 cluster | T cells | - Suppression inhibits Th1/Th17 differentiation without interfering with Treg differentiation [87] |
| miR-106b/25 cluster | T cells | - Possibly impairs Treg suppressor function by interfering with the TGF-β pathway [88] |
| miR-665 | Oligodendrocytes | - Impairs OPC differentiation when induced by CD49d+CD154+ lymphocytes [89] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duffy, C.P.; McCoy, C.E. The Role of MicroRNAs in Repair Processes in Multiple Sclerosis. Cells 2020, 9, 1711. https://doi.org/10.3390/cells9071711
Duffy CP, McCoy CE. The Role of MicroRNAs in Repair Processes in Multiple Sclerosis. Cells. 2020; 9(7):1711. https://doi.org/10.3390/cells9071711
Chicago/Turabian StyleDuffy, Conor P., and Claire E. McCoy. 2020. "The Role of MicroRNAs in Repair Processes in Multiple Sclerosis" Cells 9, no. 7: 1711. https://doi.org/10.3390/cells9071711
APA StyleDuffy, C. P., & McCoy, C. E. (2020). The Role of MicroRNAs in Repair Processes in Multiple Sclerosis. Cells, 9(7), 1711. https://doi.org/10.3390/cells9071711