The Meaning of Immune Reconstitution after Alemtuzumab Therapy in Multiple Sclerosis


Abstract
1. Introduction
2. Alemtuzumab: From Bench to Bedside
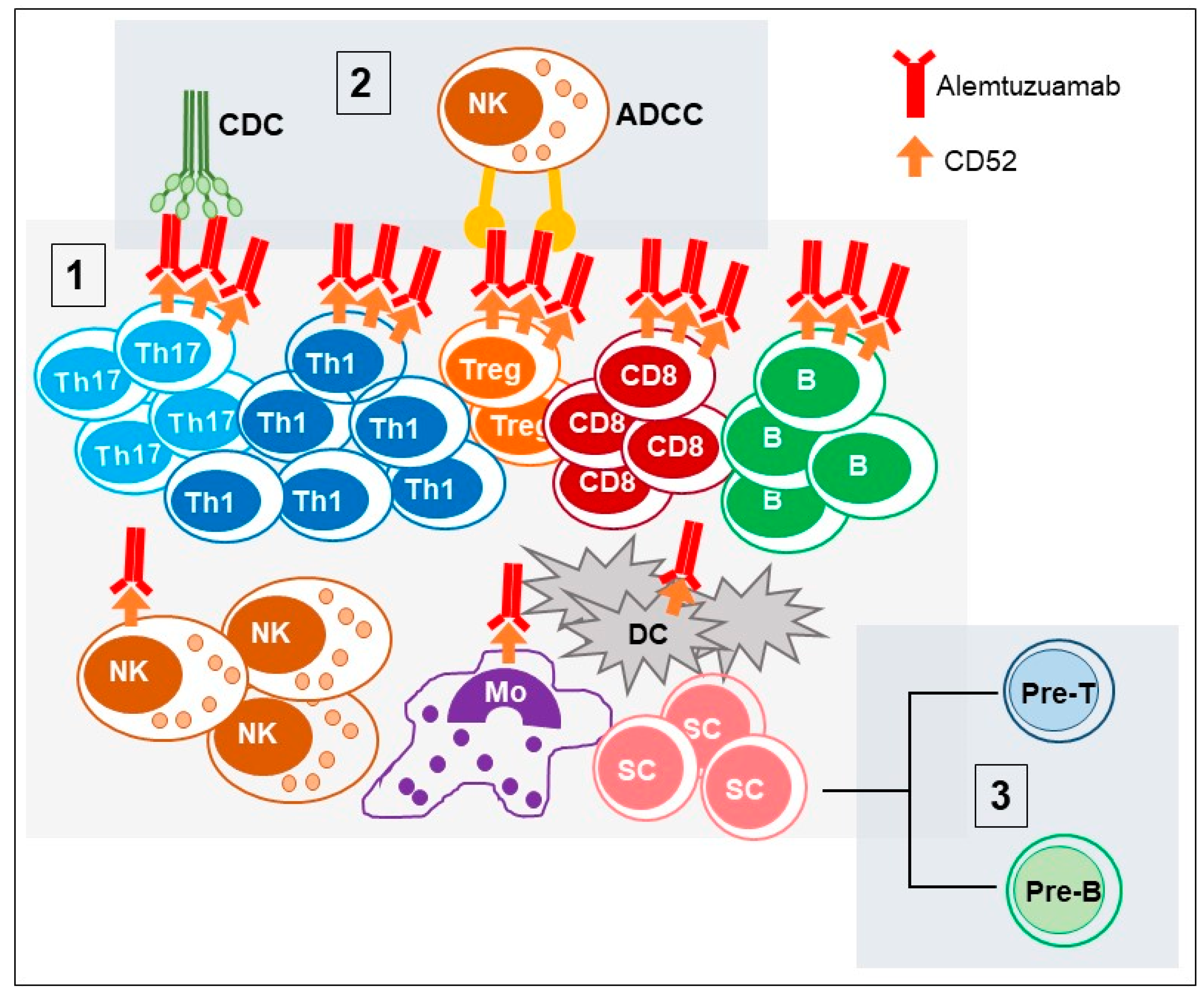
2.1. CD52 Structure and Function (Alemtuzumab Mechanism of Action)
2.2. Alemtuzumab for the Treatment of MS: Preclinical Studies in Transgenic Mice and EAE Models
2.3. Clinical Studies of Alemtuzumab in MS
3. Immune System Reconstitution after Alemtuzumab Treatment
3.1. Reconstitution of T Cells Subsets
3.2. B Cells
3.3. DC/Neutrophils/Macrophages/NK Cells
4. Beyond Depletion and Immune Reconstitution: Alemtuzumab Management
- within 2–6 h after alemtuzumab infusion the so called “cytokine-release syndrome” was observed: The participation of monocytes, macrophages and NK cells to the lysis of lymphocytes mediated by alemtuzumab results in an acute induction of several pro-inflammatory cytokines, such as TNF-α, IL-6 and IFN-γ;
- the depletion of CD8+ could be associated with the increased risk of viral infection;
- the recovery of T cells, as results from homeostatic proliferation rather than thymic reconstitution could be associated with the development of secondary autoimmunity;
- the hyper population of naïve B cells could be responsible for the secondary B cell autoimmunity.
4.1. Development of Secondary Autoimmunity
4.2. Infection Risk
4.3. Cardiovascolar Risk and New Worning
4.4. Anti-Alemtuzumab Antibodies

{kind=link}
| Cell Type | Alemtuzumab Effect | Involvement in Adverse Events |
|---|---|---|
| CD4+ T cells | 95% depleted in 1 month. Recover in ~32 months [30,31,37,46] Memory CD4+ T cells repopulate faster than naïve CD4+ T cells [49] | Development of secondary autoimmunity conditions was related to “homeostatic proliferation” [49,53] Overproduction of IL-21 by CD4+ T cells could predispose to autoimmunity [53,68,70] |
| Treg cells | CD4+CD25+CD127low Treg cells depleted by 81–86% after infusion [39] Repopulation and restored suppressive function after 24 months [47,55,56,57] 89% of restored Treg cells were memory Treg cellsas result of homeostatic proliferation [47] | The lower amount of Treg cells during the hyper-population of B cells could favor the generation of autoimmunity [49] |
| Th17 cells | Cells and cytokines related to (IL-1β, IL-6, IL-17A, IL-17F, IL-22, IL-23, IL-26, TNF-α, CCL20) are strongly decreased until month 48 [39,47,55] | - |
| Th1 cells | Cells and cytokines related to (IL-12, IFN-γ, CXCL10) are strongly decreased until month 48 [39,47,55] | - |
| Th2 cells | Cells and cytokines related to (IL-4, IL-10 and TGF-β1) increased in 3 months [39] | - |
| CD8+ T cells | 85% depleted in 1 month [30,31,46]. Recover in ~15 months [39] | The depletion of CD8+ were associated with the increased risk of viral infections [85] |
| B cells | Mature CD19+ naïve B lymphocytes decreased (<85%) in one week [49] Hyper-repopulation of immature B cell clones at 3-6 months that convert into mature B cells within 12 months [9,49] Breg cells increase in 5 months [57] | The hyper population of naïve B cells in association with long-lasting depletion of memory B cells could induce secondary B cell autoimmunity [49] Homeostatic proliferation of B cells escaping depletion could contribute to generation of ADA [97] |
| NK cells | Partecipate in the ADCC [93] NK cells are reduced [63] The subset of CD56bright NK cells is expanded [62] | Participate together with monocytes and macrophages to the “cytokine release syndrome” [93] that could bring to cardiovascular and thrombotic adverse reactions [92,94] |
| DC cells | Plasmacitoid DC cells are reduced in 6 months [62] | - |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple Sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef]
- Dos Passos, G.R.; Sato, D.K.; Becker, J.; Fujihara, K. Th17 Cells Pathways in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders: Pathophysiological and Therapeutic Implications. Mediators Inflamm. 2016, 2016, 5314541. [Google Scholar] [CrossRef]
- Salou, M.; Nicol, B.; Garcia, A.; Laplaud, D.-A. Involvement of CD8(+) T Cells in Multiple Sclerosis. Front. Immunol. 2015, 6, 604. [Google Scholar] [CrossRef] [PubMed]
- Zozulya, A.L.; Wiendl, H. The role of regulatory T cells in multiple sclerosis. Nat. Clin. Pract. Neurol. 2008, 4, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Claes, N.; Fraussen, J.; Stinissen, P.; Hupperts, R.; Somers, V. B Cells Are Multifunctional Players in Multiple Sclerosis Pathogenesis: Insights from Therapeutic Interventions. Front. Immunol. 2015, 6, 642. [Google Scholar] [CrossRef]
- González, H.; Pacheco, R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflammation 2014, 11, 201. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Gold, R.; Thompson, A.J.; Otero-Romero, S.; Amato, M.P.; Chandraratna, D.; Clanet, M.; Comi, G.; Derfuss, T.; Fazekas, F.; et al. ECTRIMS/EAN Guideline on the pharmacological treatment of people with multiple sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2018, 24, 96–120. [Google Scholar] [CrossRef] [PubMed]
- Angelis, F.D.; John, N.A.; Brownlee, W.J. Disease-modifying therapies for multiple sclerosis. BMJ 2018, 363, k4674. [Google Scholar] [CrossRef]
- Sellner, J.; Rommer, P.S. Immunological consequences of “immune reconstitution therapy” in multiple sclerosis: A systematic review. Autoimmun. Rev. 2020, 102492. [Google Scholar] [CrossRef]
- Karussis, D.; Petrou, P. Immune reconstitution therapy (IRT) in multiple sclerosis: The rationale. Immunol. Res. 2018. [Google Scholar] [CrossRef]
- Lunemann, J.D.; Ruck, T.; Muraro, P.A.; Bar-Or, A.; Wiendl, H. Immune reconstitution therapies: Concepts for durable remission in multiple sclerosis. Nat. Rev. Neurol. 2020, 16, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Chatenoud, L. Chapter 81—Treatment of Autoimmune Disease: Biological and Molecular Therapies. In The Autoimmune Diseases, 5th ed.; Rose, N.R., Mackay, I.R., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 1221–1245. ISBN 978-0-12-384929-8. [Google Scholar]
- Ratzinger, G.; Reagan, J.L.; Heller, G.; Busam, K.J.; Young, J.W. Differential CD52 expression by distinct myeloid dendritic cell subsets: Implications for alemtuzumab activity at the level of antigen presentation in allogeneic graft-host interactions in transplantation. Blood 2003, 101, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Buggins, A.G.S.; Mufti, G.J.; Salisbury, J.; Codd, J.; Westwood, N.; Arno, M.; Fishlock, K.; Pagliuca, A.; Devereux, S. Peripheral blood but not tissue dendritic cells express CD52 and are depleted by treatment with alemtuzumab. Blood 2002, 100, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Turner, M.J.; Shields, J.; Gale, M.S.; Hutto, E.; Roberts, B.L.; Siders, W.M.; Kaplan, J.M. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology 2009, 128, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Masuyama, J.; Sohma, Y.; Inazawa, H.; Horie, K.; Kojima, K.; Uemura, Y.; Aoki, Y.; Kaga, S.; Minota, S.; et al. CD52 is a novel costimulatory molecule for induction of CD4+ regulatory T cells. Clin. Immunol. 2006, 120, 247–259. [Google Scholar] [CrossRef]
- Bandala-Sanchez, E.; Zhang, Y.; Reinwald, S.; Dromey, J.A.; Lee, B.-H.; Qian, J.; Böhmer, R.M.; Harrison, L.C. T cell regulation mediated by interaction of soluble CD52 with the inhibitory receptor Siglec-10. Nat. Immunol. 2013, 14, 741–748. [Google Scholar] [CrossRef]
- Bandala-Sanchez, E.; Bediaga, N.G.; Naselli, G.; Neale, A.M.; Harrison, L.C. Siglec-10 expression is up-regulated in activated human CD4+ T cells. Hum. Immunol. 2020, 81, 101–104. [Google Scholar] [CrossRef]
- Xia, M.Q.; Hale, G.; Lifely, M.R.; Ferguson, M.A.; Campbell, D.; Packman, L.; Waldmann, H. Structure of the CAMPATH-1 antigen, a glycosylphosphatidylinositol-anchored glycoprotein which is an exceptionally good target for complement lysis. Biochem. J. 1993, 293, 633–640. [Google Scholar] [CrossRef]
- Gilleece, M.H.; Dexter, T.M. Effect of Campath-1H antibody on human hematopoietic progenitors in vitro. Blood 1993, 82, 807–812. [Google Scholar] [CrossRef]
- Rao, S.P.; Sancho, J.; Campos-Rivera, J.; Boutin, P.M.; Severy, P.B.; Weeden, T.; Shankara, S.; Roberts, B.L.; Kaplan, J.M. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Haile, Y.; Adegoke, A.; Laribi, B.; Lin, J.; Anderson, C.C. Anti-CD52 blocks EAE independent of PD-1 signals and promotes repopulation dominated by double-negative T cells and newly generated T and B cells. Eur. J. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Pang, P.T.; Chretien, N.; Havari, E.; LaMorte, M.J.; Oliver, J.; Pande, N.; Masterjohn, E.; Carter, K.; Reczek, D.; et al. Reduction of inflammation and preservation of neurological function by anti-CD52 therapy in murine experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2015, 285, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Ipek, R.; Homola, G.A.; Rovituso, D.M.; Schampel, A.; Kleinschnitz, C.; Kuerten, S. Anti-CD52 antibody treatment depletes B cell aggregates in the central nervous system in a mouse model of multiple sclerosis. J. Neuroinflammation 2018, 15, 225. [Google Scholar] [CrossRef] [PubMed]
- Ellwardt, E.; Vogelaar, C.F.; Maldet, C.; Schmaul, S.; Bittner, S.; Luchtman, D. Targeting CD52 does not affect murine neuron and microglia function. Eur. J. Pharmacol. 2020, 871, 172923. [Google Scholar] [CrossRef] [PubMed]
- MacConmara, M.; Nwadei, I.; Kirk, A.D. Chapter 96—Immunosuppressive Biologic Agents. In Transplantation of the Liver, 3rd ed.; Busuttil, R.W., Klintmalm, G.B.G., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 1343–1353. ISBN 978-1-4557-0268-8. [Google Scholar]
- Ruck, T.; Bittner, S.; Wiendl, H.; Meuth, S.G. Alemtuzumab in Multiple Sclerosis: Mechanism of Action and Beyond. Int. J. Mol. Sci. 2015, 16, 16414–16439. [Google Scholar] [CrossRef] [PubMed]
- Hale, G.; Rebello, P.; Brettman, L.R.; Fegan, C.; Kennedy, B.; Kimby, E.; Leach, M.; Lundin, J.; Mellstedt, H.; Moreton, P.; et al. Blood concentrations of alemtuzumab and antiglobulin responses in patients with chronic lymphocytic leukemia following intravenous or subcutaneous routes of administration. Blood 2004, 104. [Google Scholar] [CrossRef]
- CAMMS223 Trial Investigators; Coles, A.J.; Compston, D.A.S.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Norris, K.; Tandon, P.K. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801. [Google Scholar] [CrossRef]
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet Lond. Engl. 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet Lond. Engl. 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Havrdova, E.; Arnold, D.L.; Cohen, J.A.; Hartung, H.-P.; Fox, E.J.; Giovannoni, G.; Schippling, S.; Selmaj, K.W.; Traboulsee, A.; Compston, D.A.S.; et al. Alemtuzumab CARE-MS I 5-year follow-up. Neurology 2017, 89, 1107–1116. [Google Scholar] [CrossRef]
- Coles, A.J.; Cohen, J.A.; Fox, E.J.; Giovannoni, G.; Hartung, H.P.; Havrdova, E.; Schippling, S.; Selmaj, K.W.; Traboulsee, A.; Compston, D.A.S.; et al. Alemtuzumab CARE-MS II 5-year follow-up: Efficacy and safety findings. Neurology 2017, 89, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Wijmeersch, B.V.; Singer, B.A.; Boster, A.; Broadley, S.; Fernández, Ó.; Freedman, M.S.; Izquierdo, G.; Lycke, J.; Pozzilli, C.; Sharrack, B.; et al. Efficacy of alemtuzumab over 6 years in relapsing–remitting multiple sclerosis patients who relapsed between courses 1 and 2: Post hoc analysis of the CARE-MS studies. Mult. Scler. J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bertolotto, A.; Arroyo, R.; Celius, E.G.; Comi, G.; Havrdova, E.K.; Honeycutt, W.D.; Hunter, S.F.; Izquierdo, G.; Kornek, B.; Miller, T.; et al. Quality of Life Improves with Alemtuzumab Over 6 Years in Relapsing-Remitting Multiple Sclerosis Patients with or without Autoimmune Thyroid Adverse Events: Post Hoc Analysis of the CARE-MS Studies. Neurol. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.L.; Tuohy, O.; Compston, D.A.S.; Kumararatne, D.S.; Coles, A.J.; Jones, J.L. Immune competence after alemtuzumab treatment of multiple sclerosis. Neurology 2013, 81, 872–876. [Google Scholar] [CrossRef]
- Kousin-Ezewu, O.; Azzopardi, L.; Parker, R.A.; Tuohy, O.; Compston, A.; Coles, A.; Jones, J. Accelerated lymphocyte recovery after alemtuzumab does not predict multiple sclerosis activity. Neurology 2014, 82, 2158–2164. [Google Scholar] [CrossRef]
- Hill-Cawthorne, G.A.; Button, T.; Tuohy, O.; Jones, J.L.; May, K.; Somerfield, J.; Green, A.; Giovannoni, G.; Compston, D.A.S.; Fahey, M.T.; et al. Long term lymphocyte reconstitution after alemtuzumab treatment of multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 298–304. [Google Scholar] [CrossRef]
- Zhang, X.; Tao, Y.; Chopra, M.; Ahn, M.; Marcus, K.L.; Choudhary, N.; Zhu, H.; Markovic-Plese, S. Differential Reconstitution of T Cell Subsets following Immunodepleting Treatment with Alemtuzumab (Anti-CD52 Monoclonal Antibody) in Patients with Relapsing-Remitting Multiple Sclerosis. J. Immunol. 2013, 191, 5867–5874. [Google Scholar] [CrossRef]
- Cucci, A.; Barbero, P.; Clerico, M.; Ferrero, B.; Versino, E.; Contessa, G.; Demercanti, S.; Viglietta, E.; Di Liberto, A.; Vai, A.G.; et al. Pro-inflammatory cytokine and chemokine mRNA blood level in multiple sclerosis is related to treatment response and interferon-beta dose. J. Neuroimmunol. 2010, 226, 150–157. [Google Scholar] [CrossRef]
- Stampanoni Bassi, M.; Iezzi, E.; Landi, D.; Monteleone, F.; Gilio, L.; Simonelli, I.; Musella, A.; Mandolesi, G.; De Vito, F.; Furlan, R.; et al. Delayed treatment of MS is associated with high CSF levels of IL-6 and IL-8 and worse future disease course. J. Neurol. 2018, 265, 2540–2547. [Google Scholar] [CrossRef]
- Bărcuţean, L.I.; Romaniuc, A.; Maier, S.; Bajko, Z.; Moţăţăianu, A.; Adina, H.; Simu, I.; Andone, S.; Bălaşa, R. Clinical and Serological Biomarkers of Treatment’s Response in Multiple Sclerosis Patients Treated Continuously with Interferonβ-1b for More than a Decade. CNS Neurol. Disord. Drug Targets 2018, 17, 780–792. [Google Scholar] [CrossRef]
- Balasa, R.; Maier, S.; Voidazan, S.; Hutanu, A.; Bajko, Z.; Motataianu, A.; Tilea, B.; Tiu, C. Assessment of Interleukin-17A, Interleukin-10 and Transforming Growth Factor-Beta1 Serum Titers in Relapsing Remitting Multiple Sclerosis Patients Treated with Avonex, Possible Biomarkers for Treatment Response. CNS Neurol. Disord. Drug Targets 2017, 16, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Khademi, M.; Bornsen, L.; Rafatnia, F.; Andersson, M.; Brundin, L.; Piehl, F.; Sellebjerg, F.; Olsson, T. The effects of natalizumab on inflammatory mediators in multiple sclerosis: Prospects for treatment-sensitive biomarkers. Eur. J. Neurol. 2009, 16, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Ganji, A.; Monfared, M.E.; Shapoori, S.; Nourbakhsh, P.; Ghazavi, A.; Ghasami, K.; Mosayebi, G. Effects of interferon and glatiramer acetate on cytokine patterns in multiple sclerosis patients. Cytokine 2020, 126, 154911. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J.; Fox, E.; Vladic, A.; Gazda, S.K.; Brinar, V.; Selmaj, K.W.; Skoromets, A.; Stolyarov, I.; Bass, A.; Sullivan, H.; et al. Alemtuzumab more effective than interferon β-1a at 5-year follow-up of CAMMS223 Clinical Trial. Neurology 2012, 78, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- De Mercanti, S.; Rolla, S.; Cucci, A.; Bardina, V.; Cocco, E.; Vladic, A.; Soldo-Butkovic, S.; Habek, M.; Adamec, I.; Horakova, D.; et al. Alemtuzumab long-term immunologic effect: Treg suppressor function increases up to 24 months. Neurol. Neuroimmunol. Neuroinflammation 2016, 3, e194. [Google Scholar] [CrossRef] [PubMed]
- Rolla, S.; De Mercanti, S.F.; Bardina, V.; Horakova, D.; Habek, M.; Adamec, I.; Cocco, E.; Annovazzi, P.; Vladic, A.; Novelli, F.; et al. Lack of CD4 + T cell percent decrease in alemtuzumab-treated multiple sclerosis patients with persistent relapses. J. Neuroimmunol. 2017, 313. [Google Scholar] [CrossRef]
- Baker, D.; Herrod, S.S.; Alvarez-Gonzalez, C.; Giovannoni, G.; Schmierer, K. Interpreting lymphocyte reconstitution data from the pivotal phase 3 trials of alemtuzumab. JAMA Neurol. 2017, 74, 961–969. [Google Scholar] [CrossRef]
- Jones, J.L.; Thompson, S.A.J.; Loh, P.; Davies, J.L.; Tuohy, O.C.; Curry, A.J.; Azzopardi, L.; Hill-Cawthorne, G.; Fahey, M.T.; Compston, A.; et al. Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc. Natl. Acad. Sci. USA 2013, 110, 20200–20205. [Google Scholar] [CrossRef]
- Baccala, R.; Theofilopoulos, A.N. The new paradigm of T-cell homeostatic proliferation-induced autoimmunity. Trends Immunol. 2005, 26, 5–8. [Google Scholar] [CrossRef]
- Krupica, T.; Fry, T.J.; Mackall, C.L. Autoimmunity during lymphopenia: A two-hit model. Clin. Immunol. Orlando Fla. 2006, 120, 121–128. [Google Scholar] [CrossRef]
- Costelloe, L.; Jones, J.; Coles, A. Secondary autoimmune diseases following alemtuzumab therapy for multiple sclerosis. Expert Rev. Neurother. 2012, 12, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L.; Thompson, S.A.J.; Jones, J.L.; Robertson, V.H.; Hale, G.; Waldmann, H.; Compston, D.A.S.; Coles, A.J. Lymphocyte homeostasis following therapeutic lymphocyte depletion in multiple sclerosis. Eur. J. Immunol. 2005, 35, 3332–3342. [Google Scholar] [CrossRef] [PubMed]
- Clerico, M.; Rolla, S.; De Mercanti, S.; Bardina, V.; Cucci, A.; Taverna, D.; Cocco, E.; Vladic, A.; Habek, M.; Adamec, I.; et al. Six year prospective immunological study of Alemtuzumab treated patients: Focus on CD4+ T cell subsets (P5.360). Neurology 2018, 90, P5.360. [Google Scholar]
- Haas, J.; Würthwein, C.; Korporal-Kuhnke, M.; Viehoever, A.; Jarius, S.; Ruck, T.; Pfeuffer, S.; Meuth, S.G.; Wildemann, B. Alemtuzumab in Multiple Sclerosis: Short- and Long-Term Effects of Immunodepletion on the Peripheral Treg Compartment. Front. Immunol. 2019, 10, 1204. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, W.; Lund, B.T.; Traboulsee, A.; Javed, A.; Dunn, J.; Weiner, L.P.; Li, P.; Kelland, E.E.; Levy, A. Leukocyte repopulation following alemtuzumab treatment in relapsing-remitting MS contains multiple regulatory immune cell types. In Proceedings of the Multiple Sclerosis Journal; Sage Publications Ltd.: London, UK, 2017; Volume 23, pp. 506–507. [Google Scholar]
- Thompson, S.A.J.; Jones, J.L.; Cox, A.L.; Compston, D.A.S.; Coles, A.J. B-Cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J. Clin. Immunol. 2010, 30, 99–105. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, G.; Shin, H.-J.; Hyun, J.-W.; Kim, S.-H.; Lee, E.; Kim, H.J. Restoration of regulatory B cell deficiency following alemtuzumab therapy in patients with relapsing multiple sclerosis. J. Neuroinflammation 2018, 15, 300. [Google Scholar] [CrossRef]
- Turner, M.J.; LaMorte, M.J.; Chretien, N.; Havari, E.; Roberts, B.L.; Kaplan, J.M.; Siders, W.M. Immune status following alemtuzumab treatment in human CD52 transgenic mice. J. Neuroimmunol. 2013, 261, 29–36. [Google Scholar] [CrossRef]
- Thomas, K.; Eisele, J.; Rodriguez-Leal, F.A.; Hainke, U.; Ziemssen, T. Acute effects of alemtuzumab infusion in patients with active relapsing-remitting MS. Neurol. Neuroimmunol. Neuroinflammation 2016, 3, e228. [Google Scholar] [CrossRef]
- Gross, C.C.; Ahmetspahic, D.; Ruck, T.; Schulte-Mecklenbeck, A.; Schwarte, K.; Jorgens, S.; Scheu, S.; Windhagen, S.; Graefe, B.; Melzer, N.; et al. Alemtuzumab treatment alters circulating innate immune cells in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2016, 3, e289. [Google Scholar] [CrossRef]
- Li, Z.; Richards, S.; Surks, H.K.; Jacobs, A.; Panzara, M.A. Clinical pharmacology of alemtuzumab, an anti-CD52 immunomodulator, in multiple sclerosis. Clin. Exp. Immunol. 2018, 194, 295–314. [Google Scholar] [CrossRef]
- Kasper, L.H.; Arnold, D.L.; Coles, A.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Palmer, J.; Margolin, D.H.; Panzara, M.A.; Compston, D.A.S. Lymphocyte subset dynamics following alemtuzumab treatment in patients who relapsed on a prior therapy. J. Neuroimmunol. 2014, 275, 63–64. [Google Scholar] [CrossRef]
- Freedman, M.; Kaplan, J.; Markovic-Plese, S. Insights into the mechanisms of the therapeutic efficacy of alemtuzumab in multiple sclerosis. J Clin Cell Immunol 2013, 4, 1–18. [Google Scholar] [CrossRef]
- Hartung, H.-P.; Aktas, O.; Boyko, A.N. Alemtuzumab: A new therapy for active relapsing-remitting multiple sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tuohy, O.; Costelloe, L.; Hill-Cawthorne, G.; Bjornson, I.; Harding, K.; Robertson, N.; May, K.; Button, T.; Azzopardi, L.; Kousin-Ezewu, O.; et al. Alemtuzumab treatment of multiple sclerosis: Long-term safety and efficacy. J. Neurol. Neurosurg. Psychiatry 2015, 86, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.L.; Phuah, C.L.; Cox, A.L.; Thompson, S.A.; Ban, M.; Shawcross, J.; Walton, A.; Sawcer, S.J.; Compston, A.; Coles, A.J. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J. Clin. Investig. 2009, 119, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Kuchen, S.; Robbins, R.; Sims, G.P.; Sheng, C.; Phillips, T.M.; Lipsky, P.E.; Ettinger, R. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J. Immunol. Baltim. MD 2007, 179, 5886–5896. [Google Scholar] [CrossRef]
- Sakuraba, K.; Oyamada, A.; Fujimura, K.; Spolski, R.; Iwamoto, Y.; Leonard, W.J.; Yoshikai, Y.; Yamada, H. Interleukin-21 signaling in B cells, but not in T cells, is indispensable for the development of collagen-induced arthritis in mice. Arthritis Res. Ther. 2016, 18, 188. [Google Scholar] [CrossRef]
- Pariani, N.; Willis, M.; Muller, I.; Healy, S.; Nasser, T.; McGowan, A.; Lyons, G.; Jones, J.; Chatterjee, K.; Dayan, C.; et al. Alemtuzumab-Induced Thyroid Dysfunction Exhibits Distinctive Clinical and Immunological Features. J. Clin. Endocrinol. Metab. 2018, 103, 3010–3018. [Google Scholar] [CrossRef]
- Cossburn, M.; Pace, A.A.; Jones, J.; Ali, R.; Ingram, G.; Baker, K.; Hirst, C.; Zajicek, J.; Scolding, N.; Boggild, M.; et al. Autoimmune disease after alemtuzumab treatment for multiple sclerosis in a multicenter cohort. Neurology 2011, 77, 573–579. [Google Scholar] [CrossRef]
- Coles, A.J.; Wing, M.; Smith, S.; Coraddu, F.; Greer, S.; Taylor, C.; Weetman, A.; Hale, G.; Chatterjee, V.K.; Waldmann, H.; et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet Lond. Engl. 1999, 354, 1691–1695. [Google Scholar] [CrossRef]
- Havla, J.; Warnke, C.; Derfuss, T.; Kappos, L.; Hartung, H.-P.; Hohlfeld, R. Interdisciplinary Risk Management in the Treatment of Multiple Sclerosis. Dtsch. Arzteblatt Int. 2016, 113, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Wray, S.; Havrdova, E.; Snydman, D.R.; Arnold, D.L.; Cohen, J.A.; Coles, A.J.; Hartung, H.-P.; Selmaj, K.W.; Weiner, H.L.; Daizadeh, N.; et al. Infection risk with alemtuzumab decreases over time: Pooled analysis of 6-year data from the CAMMS223, CARE-MS I, and CARE-MS II studies and the CAMMS03409 extension study. Mult. Scler. Houndmills Basingstoke Engl. 2019, 25, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Watanabe, R.; Teague, J.E.; Schlapbach, C.; Tawa, M.C.; Adams, N.; Dorosario, A.A.; Chaney, K.S.; Cutler, C.S.; Leboeuf, N.R.; et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci. Transl. Med. 2012, 4, 117ra7. [Google Scholar] [CrossRef]
- Turner, M.J.; Havari, E.; Dodge, J.; Treleaven, C.; Shihabuddin, L.; Roberts, B.; Kaplan, J.; Siders, W.M. Preservation of lymphocyte migratory ability following anti-CD52 therapy. In Proceedings of the Multiple Sclerosis Journal; Sage Publications Ltd.: London, UK, 2013; Volume 19, p. 573. [Google Scholar]
- Buonomo, A.R.; Zappulo, E.; Viceconte, G.; Scotto, R.; Borgia, G.; Gentile, I. Risk of opportunistic infections in patients treated with alemtuzumab for multiple sclerosis. Expert Opin. Drug Saf. 2018, 17, 709–717. [Google Scholar] [CrossRef]
- Yann, K.; Jackson, F.; Sharaf, N.; Mihalova, T.; Talbot, P.; Rog, D.; Pace, A. Acute respiratory distress syndrome following alemtuzumab therapy for relapsing multiple sclerosis. Mult. Scler. Relat. Disord. 2017, 14, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Penkert, H.; Delbridge, C.; Wantia, N.; Wiestler, B.; Korn, T. Fulminant Central Nervous System Nocardiosis in a Patient Treated With Alemtuzumab for Relapsing-Remitting Multiple Sclerosis. JAMA Neurol. 2016, 73, 757–759. [Google Scholar] [CrossRef]
- Sheikh-Taha, M.; Corman, L.C. Pulmonary Nocardia beijingensis infection associated with the use of alemtuzumab in a patient with multiple sclerosis. Mult. Scler. J. 2017. [Google Scholar] [CrossRef]
- Canham, L.J.W.; Manara, A.; Fawcett, J.; Rolinski, M.; Mortimer, A.; Inglis, K.E.A.; Cottrell, D.A. Mortality from Listeria monocytogenes meningoencephalitis following escalation to alemtuzumab therapy for relapsing-remitting Multiple Sclerosis. Mult. Scler. Relat. Disord. 2018, 24, 38–41. [Google Scholar] [CrossRef]
- Pappolla, A.; Midaglia, L.; Boix Rodríguez, C.P.; Puig, A.A.; Lung, M.; Camps, I.R.; Castilló, J.; Mulero, P.; Vidal-Jordana, A.; Arrambide, G.; et al. Simultaneous CMV and Listeria infection following alemtuzumab treatment for multiple sclerosis. Neurology 2019, 92, 296–298. [Google Scholar] [CrossRef]
- Rau, D.; Lang, M.; Harth, A.; Naumann, M.; Weber, F.; Tumani, H.; Bayas, A. Listeria Meningitis Complicating Alemtuzumab Treatment in Multiple Sclerosis--Report of Two Cases. Int. J. Mol. Sci. 2015, 16, 14669–14676. [Google Scholar] [CrossRef]
- Winkelmann, A.; Loebermann, M.; Reisinger, E.C.; Hartung, H.-P.; Zettl, U.K. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R.; Kümpfel, T. Alemtuzumab and Multiple Sclerosis: Another Note of Caution. JAMA Neurol. 2016, 73, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Clerico, M.; De Mercanti, S.; Artusi, C.A.; Durelli, L.; Naismith, R.T. Active CMV infection in two patients with multiple sclerosis treated with alemtuzumab. Mult. Scler. 2017, 23. [Google Scholar] [CrossRef]
- Brownlee, W.J.; Chataway, J. Opportunistic infections after alemtuzumab: New cases of norcardial infection and cytomegalovirus syndrome. Mult. Scler. J. 2017. [Google Scholar] [CrossRef]
- Barone, S.; Scannapieco, S.; Torti, C.; Filippelli, E.; Pisani, V.; Granata, A.; Console, D.; Demonte, G.; Tallarico, T.; Polidoro, S.; et al. Hepatic microabscesses during CMV reactivation in a multiple sclerosis patient after alemtuzumab treatment. Mult. Scler. Relat. Disord. 2018, 20, 6–8. [Google Scholar] [CrossRef]
- McCall, B. Alemtuzumab to be restricted pending review, says EMA. Lancet Lond. Engl. 2019. [Google Scholar] [CrossRef]
- Azevedo, C.J.; Kutz, C.; Dix, A.; Boster, A.; Sanossian, N.; Kaplan, J. Intracerebral haemorrhage during alemtuzumab administration. Lancet Neurol. 2019, 18, 329–331. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Maggi, E.; Vultaggio, A.; Matucci, A. Acute infusion reactions induced by monoclonal antibody therapy. Expert Rev. Clin. Immunol. 2011, 7, 55–63. [Google Scholar] [CrossRef]
- Libertinova, J.; Meluzinova, E.; Nema, E.; Rockova, P.; Elisak, M.; Petrzalka, M.; Mojzisova, H.; Hammer, J.; Tomek, A.; Marusic, P. Elevated D-dimer as an immediate response to alemtuzumab treatment. Mult. Scler. Houndmills Basingstoke Engl. 2020, 1352458520904277. [Google Scholar] [CrossRef]
- Mercanti, S.F.D.; Rolla, S.; Matta, M.; Iudicello, M.; Franchin, E.; Clerico, M. D-dimer Increasing After First Alemtuzumab Administration in a Multiple Sclerosis Patient. Int. J. Clin. Exp. Med. Sci. 2019, 5, 67. [Google Scholar] [CrossRef]
- Ali, L.; Saxena, G.; Jones, M.; Leisegang, G.R.; Gammon, L.; Gnanapavan, S.; Giovannoni, G.; Schmierer, K.; Baker, D.; Kang, A.S. A cell-based assay for the detection of neutralizing antibodies against alemtuzumab. BioTechniques 2020. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Ali, L.; Saxena, G.; Pryce, G.; Jones, M.; Schmierer, K.; Giovannoni, G.; Gnanapavan, S.; Munger, K.C.; Samkoff, L.; et al. The Irony of Humanization: Alemtuzumab, the First, But One of the Most Immunogenic, Humanized Monoclonal Antibodies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, N.; Baker, D.; Kang, A.S.; Pryce, G.; Marta, M.; Visser, L.H.; Hofmann, W.E.; Gnanapavan, S.; Giovannoni, G.; Schmierer, K. Alemtuzumab depletion failure can occur in multiple sclerosis. Immunology 2018, 154, 253–260. [Google Scholar] [CrossRef]
- Eggers, C. Abrogation of the lymphocyte depleting action of Alemtuzumab by neutralizing antibodies—A case report P1231. In Proceedings of the Multiple Sclerosis Journal; Sage Publications Ltd.: London, UK, 2017; Volume 23, p. 655. [Google Scholar]
- Akgün, K.; Kretschmann, N.; Haase, R.; Proschmann, U.; Kitzler, H.H.; Reichmann, H.; Ziemssen, T. Profiling individual clinical responses by high-frequency serum neurofilament assessment in MS. Neurol. Neuroimmunol. Neuroinflammation 2019, 6, e555. [Google Scholar] [CrossRef]
- Bierhansl, L.; Ruck, T.; Pfeuffer, S.; Gross, C.C.; Wiendl, H.; Meuth, S.G. Signatures of immune reprogramming in anti-CD52 therapy of MS: Markers for risk stratification and treatment response. Neurol. Res. Pract. 2019, 1, 40. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rolla, S.; Maglione, A.; De Mercanti, S.F.; Clerico, M. The Meaning of Immune Reconstitution after Alemtuzumab Therapy in Multiple Sclerosis. Cells 2020, 9, 1396. https://doi.org/10.3390/cells9061396
Rolla S, Maglione A, De Mercanti SF, Clerico M. The Meaning of Immune Reconstitution after Alemtuzumab Therapy in Multiple Sclerosis. Cells. 2020; 9(6):1396. https://doi.org/10.3390/cells9061396
Chicago/Turabian StyleRolla, Simona, Alessandro Maglione, Stefania Federica De Mercanti, and Marinella Clerico. 2020. "The Meaning of Immune Reconstitution after Alemtuzumab Therapy in Multiple Sclerosis" Cells 9, no. 6: 1396. https://doi.org/10.3390/cells9061396
APA StyleRolla, S., Maglione, A., De Mercanti, S. F., & Clerico, M. (2020). The Meaning of Immune Reconstitution after Alemtuzumab Therapy in Multiple Sclerosis. Cells, 9(6), 1396. https://doi.org/10.3390/cells9061396