NeuroHeal Reduces Muscle Atrophy and Modulates Associated Autophagy

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Denervation Model
2.2. Hindlimb Immobilization Model
2.3. Drug Treatment
2.4. Grip Test
2.5. Histology
2.6. Immunoblotting
2.7. In Vitro Experiments
2.8. Statistics
3. Results
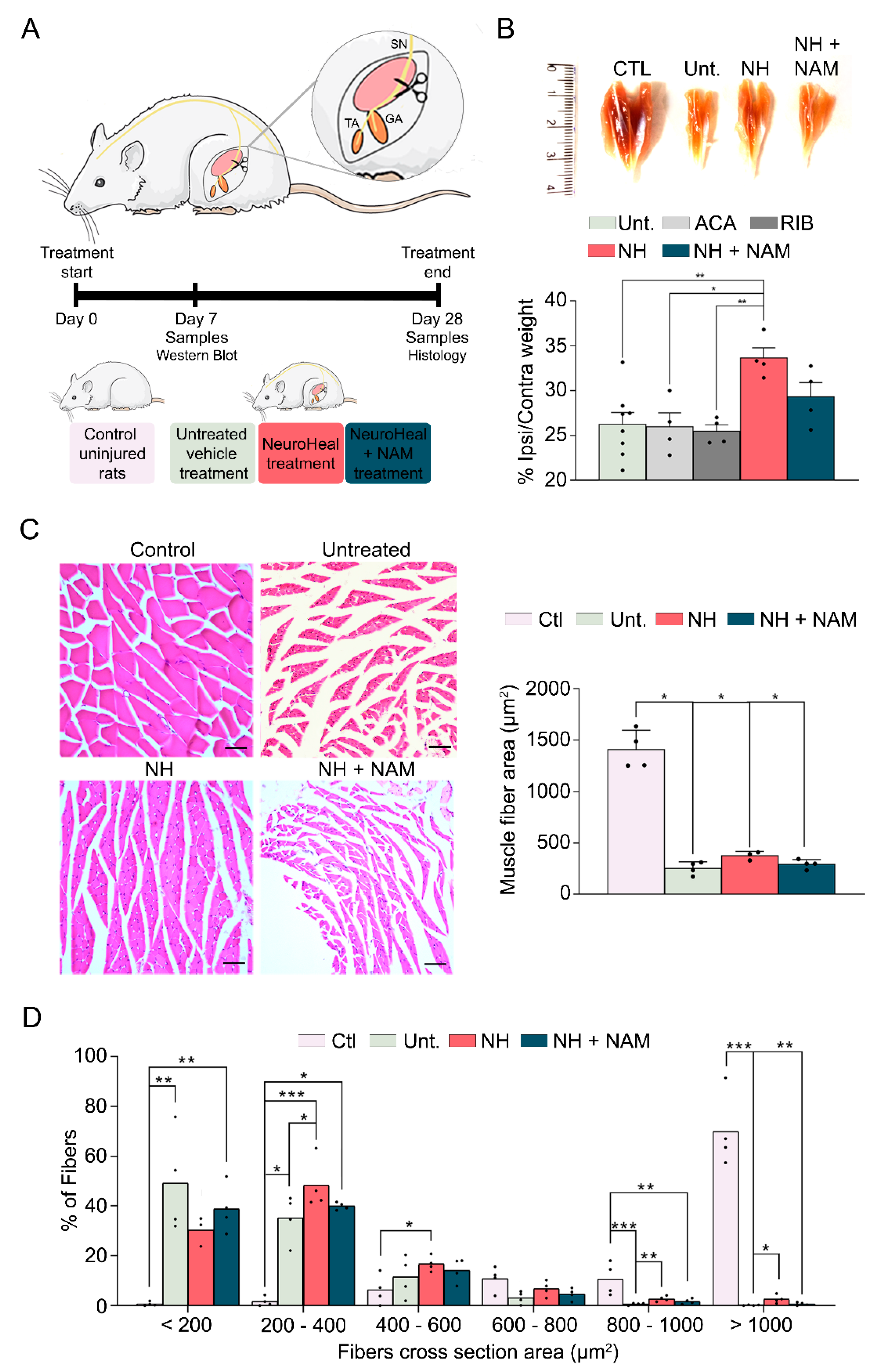
3.1. NeuroHeal Reduces Atrophy-Related Histological Parameters
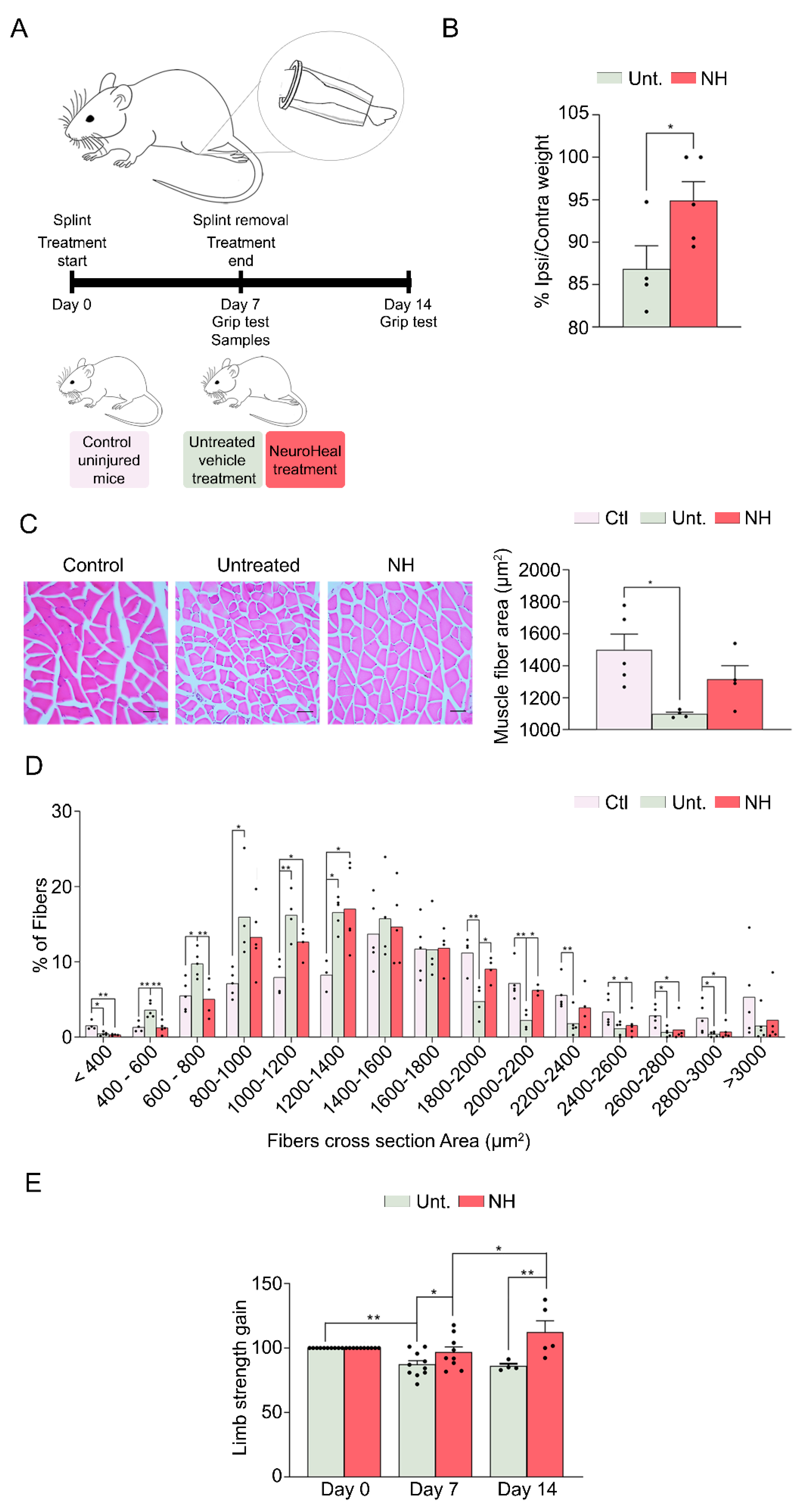
3.2. NeuroHeal Reduces Muscle Atrophy in a Model of Mechanical Unloading
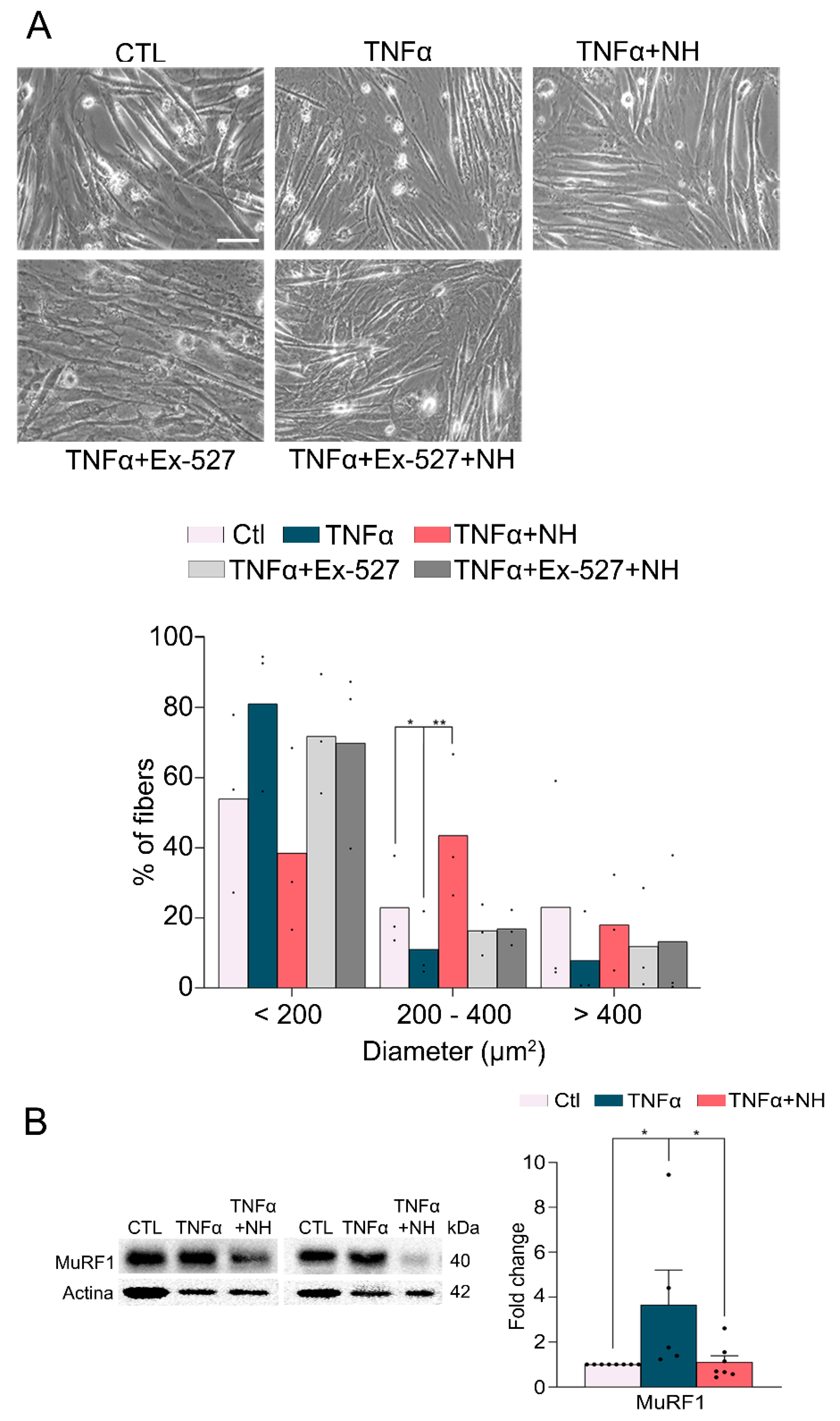
3.3. NeuroHeal Preserved Fiber Diameter in Atrophied Myotubes Cells In Vitro
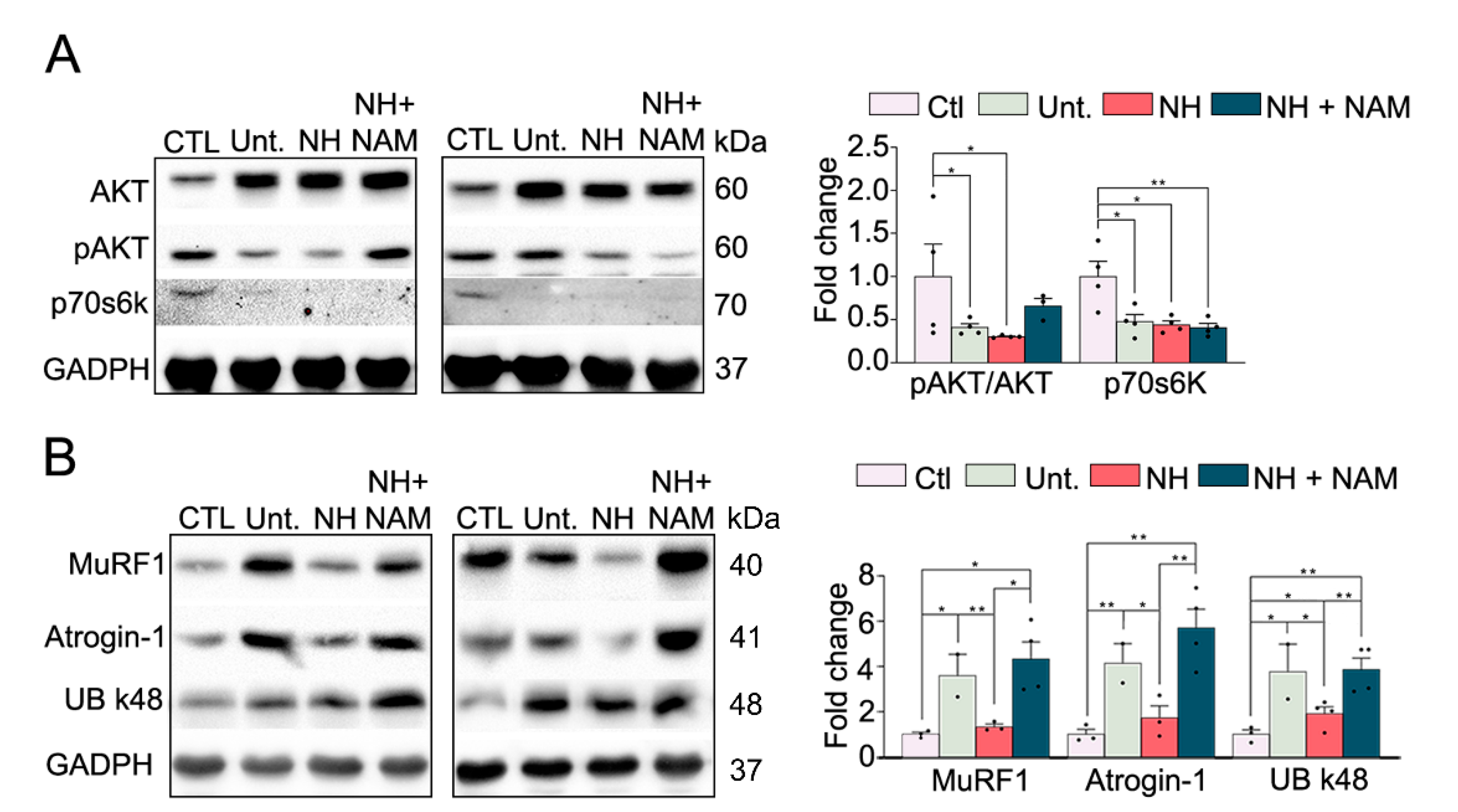
3.4. NeuroHeal Reduced Proteasome Markers
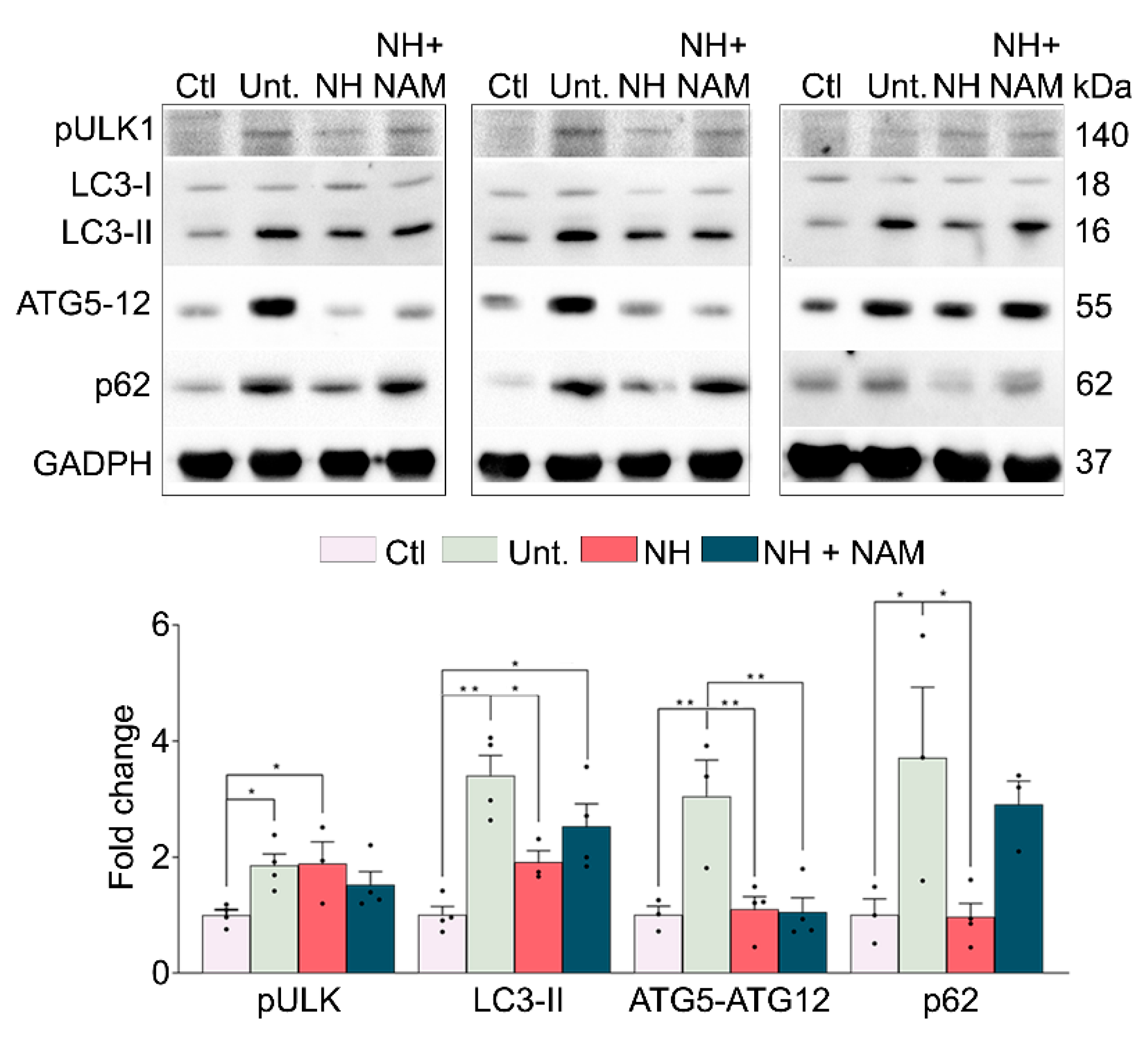
3.5. NeuroHeal Modulates Atrophy Associated Autophagy
4. Discussion
5. Study Limitations and Future Research
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cohen, I.R.; Lambris, J.D. Advances in Experimental Medicine and Biology. Springer Singapore 2018, 1088, 1434–1436. [Google Scholar]
- Legrand, D.; Vaes, B.; Matheï, C.; Adriaensen, W.; Van Pottelbergh, G.; Degryse, J.-M. Muscle Strength and Physical Performance as Predictors of Mortality, Hospitalization, and Disability in the Oldest Old. J. Am. Geriatr. Soc. 2014, 62, 1030–1038. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.a.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Romeo-Guitart, D.; Forés, J.; Herrando-Grabulosa, M.; Valls, R.; Leiva-Rodríguez, T.; Galea, E.; González-Pérez, F.; Navarro, X.; Petegnief, V.; Bosch, A.; et al. Neuroprotective Drug for Nerve Trauma Revealed Using Artificial Intelligence. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Romeo-Guitart, D.; Forés, J.; Navarro, X.; Casas, C. Boosted Regeneration and Reduced Denervated Muscle Atrophy by NeuroHeal in a Pre-clinical Model of Lumbar Root Avulsion with Delayed Reimplantation. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Vargas, R.; Lang, C.H. Alcohol Accelerates Loss of Muscle and Impairs Recovery of Muscle Mass Resulting From Disuse Atrophy. Alcohol. Clin. Exp. Res. 2007, 32, 128–137. [Google Scholar] [CrossRef]
- Magne, H.; Savary-Auzeloux, I.; Vazeille, E.; Claustre, A.; Attaix, D.; Anne, L.; Véronique, S.-L.; Philippe, G.; Dardevet, D.; Combaret, L. Lack of muscle recovery after immobilization in old rats does not result from a defect in normalization of the ubiquitin-proteasome and the caspase-dependent apoptotic pathways. J. Physiol. 2011, 589, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.M.; Kazi, A.A.; Hong-Brown, L.; Lang, C.H. Delayed Recovery of Skeletal Muscle Mass following Hindlimb Immobilization in mTOR Heterozygous Mice. PLoS ONE 2012, 7, e38910. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Marín-Corral, J.; Sanchez, F.; Mielgo, V.; Alvarez, F.J.; Gáldiz, J.B.; Gea, J. Reference values of respiratory and peripheral muscle function in rats. J. Anim. Physiol. Anim. Nutr. 2010, 94, e393–e401x. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Bustamante, V.; Cejudo, P.; Gáldiz, J.B.; Gea, J.; de Lucas, P.; Martínez-Llorens, J.; Ortega, F.; Puente-Maestu, L.; Roca, J.; et al. Normativa SEPAR sobre disfunción muscular de los pacientes con enfermedad pulmonar obstructiva crónica. Arch. Bronconeumol. 2015, 51, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Cabrera, A.; Fermoselle, C.; Urtreger, A.J.; Mateu-Jimenez, M.; Diament, M.J.; de Kier Joffé, E.D.B.; Sandri, M.; Barreiro, E. Pharmacological Strategies in Lung Cancer-Induced Cachexia: Effects on Muscle Proteolysis, Autophagy, Structure, and Weakness. J. Cell. Physiol. 2014, 229, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Cabrera, A.; Fermoselle, C.; Salmela, I.; Yelamos, J.; Barreiro, E. MicroRNA expression and protein acetylation pattern in respiratory and limb muscles of Parp-1−/− and Parp-2−/− mice with lung cancer cachexia. Biochim. Biophys. Acta 2015, 1850, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.T.; Yin, Y.; Yang, Y.J.; Lv, P.J.; Shi, Y.; Lu, L.; Wei, L.B. Resveratrol prevents TNF-α-induced muscle atrophy via regulation of Akt/mTOR/FoxO1 signaling in C2C12 myotubes. Int. Immunopharmacol. 2014, 19, 206–213. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Lagirand-Cantaloube, J.; Offner, N.; Csibi, A.; Leibovitch, M.P.; Batonnet-Pichon, S.; Tintignac, L.A.; Segura, C.T.; Leibovitch, S.A. The initiation factor eIF3-f is a major target for Atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008, 27, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Marcos-DeJuana, C.; Marmolejo-Martínez-Artesero, S.; Navarro, X.; Casas, C. Novel neuroprotective therapy with NeuroHeal by autophagy induction for damaged neonatal motoneurons. Theranostics 2020, 10, 5154–5168. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 Coordinately Activates Protein Degradation by the Autophagic/Lysosomal and Proteasomal Pathways in Atrophying Muscle Cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Goldberg, A.L. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J. Biol. Chem. 2013, 288, 30515–30526. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Molecular mechanisms modulating muscle mass. Trends Mol. Med. 2003, 9, 344–350. [Google Scholar] [CrossRef]
- Glass, D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 2005, 37, 1974–19848. [Google Scholar] [CrossRef]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef]
- Heszele, M.F.C.; Price, S.R. Insulin-Like Growth Factor I: The Yin and Yang of Muscle Atrophy. Endocrinology 2004, 145, 4803–4805. [Google Scholar] [CrossRef][Green Version]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Labeit, S.; Kohl, C.H.; Witt, C.C.; Labeit, D.; Jung, J.; Granzier, H. Modulation of Muscle Atrophy, Fatigue and MLC Phosphorylation by MuRF1 as Indicated by Hindlimb Suspension Studies on MuRF1-KO Mice. J. Biomed. Biotechnol. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Eddins, M.J.; Marblestone, J.G.; Suresh Kumar, K.G.; Leach, C.A.; Sterner, D.E.; Mattern, M.R.; Nicholson, B. Targeting the Ubiquitin E3 Ligase MuRF1 to Inhibit Muscle Atrophy. Cell Biochem. Biophys. 2011, 60, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Quy, P.N.; Kuma, A.; Pierre, P.; Mizushima, N. Proteasome-dependent Activation of Mammalian Target of Rapamycin Complex 1 (mTORC1) Is Essential for Autophagy Suppression and Muscle Remodeling Following Denervation. J. Biol. Chem. 2013, 288, 1125–1134. [Google Scholar] [CrossRef]
- Sala, D.; Ivanova, S.; Plana, N.; Ribas, V.; Duran, J.; Bach, D.; Turkseven, S.; Laville, M.; Vidal, H.; Karczewska-Kupczewska, M.; et al. Autophagy-regulating TP53INP2 mediates muscle wasting and is repressed in diabetes. J. Clin. Invest. 2014, 124, 1914–1927. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Won, K.-J.; Lee, H.M.; Hwang, B.-Y.; Bae, Y.-M.; Choi, W.S.; Song, H.; Lim, K.W.; Lee, C.-K.; Kim, B. p38 MAPK Participates in Muscle-Specific RING Finger 1-Mediated Atrophy in Cast-Immobilized Rat Gastrocnemius Muscle. Korean J. Physiol. Pharmacol. 2009, 13, 491. [Google Scholar] [CrossRef]
- Xia, H.-G.; Zhang, L.; Chen, G.; Zhang, T.; Liu, J.; Jin, M.; Ma, X.; Ma, D.; Yuan, J. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy 2010, 6, 61–66. [Google Scholar] [CrossRef]
- Sandri, M.; Coletto, L.; Grumati, P.; Bonaldo, P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [CrossRef]
- Odeh, M.; Tamir-Livne, Y.; Haas, T.; Bengal, E. P38α MAPK coordinates the activities of several metabolic pathways that together induce atrophy of denervated muscles. FEBS J. 2020, 287, 73–93. [Google Scholar] [CrossRef]
- Vergne, I.; Roberts, E.; Elmaoued, R.A.; Tosch, V.; Delgado, M.A.; Proikas-Cezanne, T.; Laporte, J.; Deretic, V. Control of autophagy initiation by phosphoinositide 3-phosphatase jumpy. EMBO J. 2009, 28, 2244–2258. [Google Scholar] [CrossRef]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar]
- Rusmini, P.; Polanco, M.J.; Cristofani, R.; Cicardi, M.E.; Meroni, M.; Galbiati, M.; Piccolella, M.; Messi, E.; Giorgetti, E.; Lieberman, A.P.; et al. Aberrant Autophagic Response in The Muscle of A Knock-in Mouse Model of Spinal and Bulbar Muscular Atrophy. Sci. Rep. 2015, 5, 15174. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef]
- Waite, K.A.; De-La Mota-Peynado, A.; Vontz, G.; Roelofs, J. Starvation Induces Proteasome Autophagy with Different Pathways for Core and Regulatory Particles. J. Biol. Chem. 2016, 291, 3239–3253. [Google Scholar] [CrossRef]
- Kageyama, S.; Sou, Y.; Uemura, T.; Kametaka, S.; Saito, T.; Ishimura, R.; Kouno, T.; Bedford, L.; Mayer, R.J.; Lee, M.-S.; et al. Proteasome Dysfunction Activates Autophagy and the Keap1-Nrf2 Pathway. J. Biol. Chem. 2014, 289, 24944–24955. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Demishtein, A.; Fraiberg, M.; Berko, D.; Tirosh, B.; Elazar, Z.; Navon, A. SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy 2017, 13, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Wakatsuki, S.; Walters, K.J. Ubiquitin-binding domains — from structures to functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Yau, R.G.; Doerner, K.; Castellanos, E.R.; Haakonsen, D.L.; Werner, A.; Wang, N.; Yang, X.W.; Martinez-Martin, N.; Matsumoto, M.L.; Dixit, V.M.; et al. Assembly and Function of Heterotypic Ubiquitin Chains in Cell-Cycle and Protein Quality Control. Cell 2017, 171, 918–933.e20. [Google Scholar] [CrossRef]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Kaiser, S.E.; Riley, B.E.; Shaler, T.A.; Trevino, R.S.; Becker, C.H.; Schulman, H.; Kopito, R.R. Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods 2011, 8, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Savova, A.; Danieli, A.; Romanov, J.; Tremel, S.; Ebner, M.; Peterbauer, T.; Sztacho, M.; Trapannone, R.; Tarafder, A.K.; et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018, 37, 1–21. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Jiang, W.-X.; Qin, L.-Y.; Gong, Z.; Wan, W.; Li, J.; Wang, Y.; Zhang, H.; Peng, C.; Zhou, T.; et al. Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat. Commun. 2019, 10, 5792. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Ling, H.; Yuan, Z.; Fang, B.; Bloom, G.; Fukasawa, K.; Koomen, J.; Chen, J.; Lane, W.S.; Seto, E. SIRT1 Negatively Regulates the Activities, Functions, and Protein Levels of hMOF and TIP60. Mol. Cell. Biol. 2012, 32, 2823–2836. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Kitabayashi, I. Sirt1 physically interacts with Tip60 and negatively regulates Tip60-mediated acetylation of H2AX. Biochem. Biophys. Res. Commun. 2009, 390, 1355–1360. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Leiva-Rodriguez, T.; Forés, J.; Casas, C. Improved Motor Nerve Regeneration by SIRT1/Hif1a-Mediated Autophagy. Cells 2019, 8, 1354. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Casas, C. NeuroHeal Treatment Alleviates Neuropathic Pain and Enhances Sensory Axon Regeneration. Cells 2020, 9, 808. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Casas, C. Network-centric medicine for peripheral nerve injury: Treating the whole to boost endogenous mechanisms of neuroprotection and regeneration. Neural Regen. Res. 2019, 14, 1122. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marmolejo-Martínez-Artesero, S.; Romeo-Guitart, D.; Mañas-García, L.; Barreiro, E.; Casas, C. NeuroHeal Reduces Muscle Atrophy and Modulates Associated Autophagy. Cells 2020, 9, 1575. https://doi.org/10.3390/cells9071575
Marmolejo-Martínez-Artesero S, Romeo-Guitart D, Mañas-García L, Barreiro E, Casas C. NeuroHeal Reduces Muscle Atrophy and Modulates Associated Autophagy. Cells. 2020; 9(7):1575. https://doi.org/10.3390/cells9071575
Chicago/Turabian StyleMarmolejo-Martínez-Artesero, Sara, David Romeo-Guitart, Laura Mañas-García, Esther Barreiro, and Caty Casas. 2020. "NeuroHeal Reduces Muscle Atrophy and Modulates Associated Autophagy" Cells 9, no. 7: 1575. https://doi.org/10.3390/cells9071575
APA StyleMarmolejo-Martínez-Artesero, S., Romeo-Guitart, D., Mañas-García, L., Barreiro, E., & Casas, C. (2020). NeuroHeal Reduces Muscle Atrophy and Modulates Associated Autophagy. Cells, 9(7), 1575. https://doi.org/10.3390/cells9071575