Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids

 and
and {kind=link}
{kind=link}
Abstract
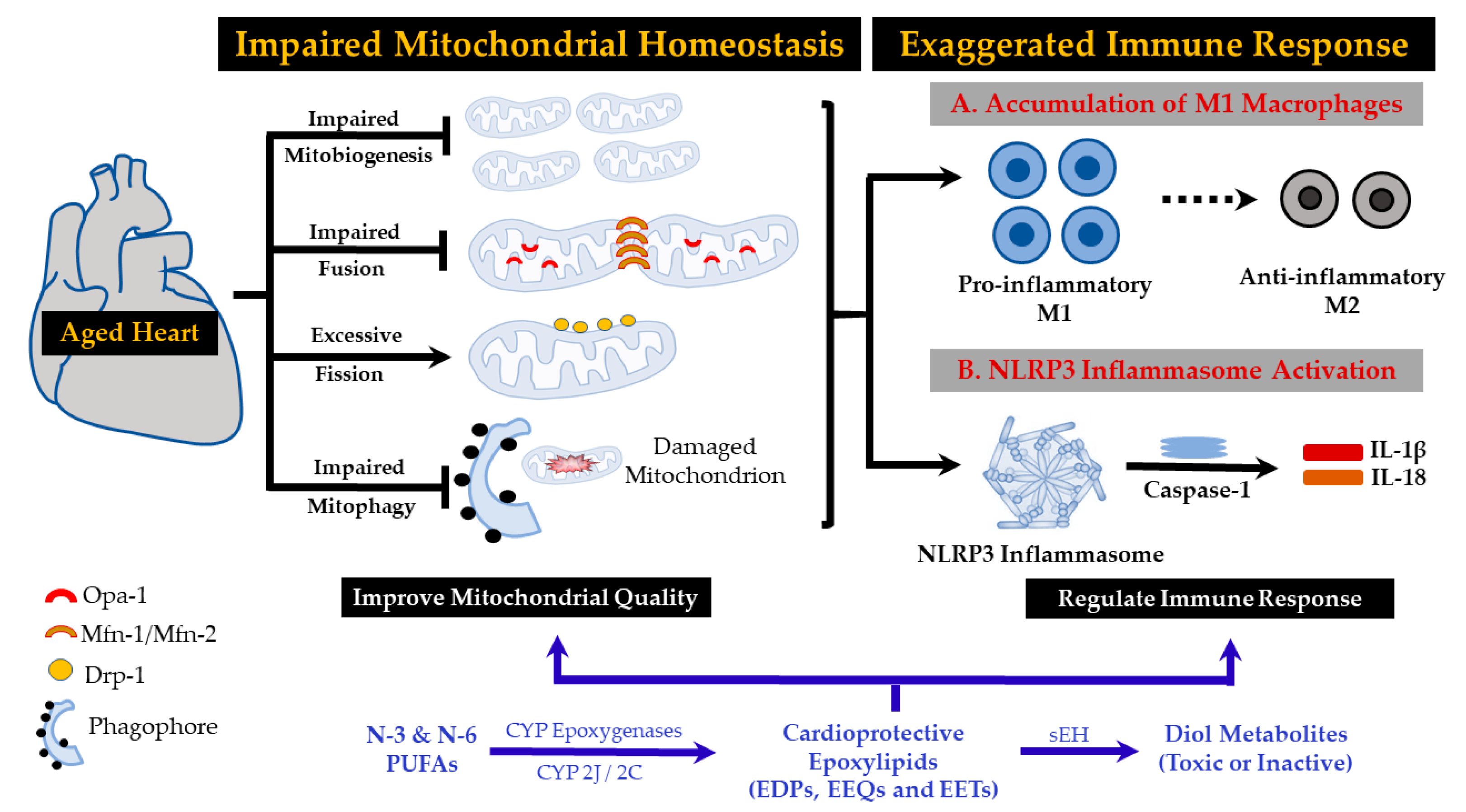
1. Introduction
2. NLRP3 Inflammasomes in Chronic Inflammation and Heart Failure
3. Aging Mitochondria Contribute to NLRP-3 Inflammasome Activation
4. Mitochondria and Oxidative Stress Theory of Aging
5. Impaired Mitophagy and Mitochondrial Dynamics in Cardiac Aging and Heart Failure
6. Macrophages and Chronic Inflammation
7. N-3 and N-6 Polyunsaturated Fatty Acids (PUFAs)
8. Anti-Inflammatory Effects of N-3 and N-6 PUFA-Derived Epoxylipids
9. Mitochondria: Effects of N-3 and N-6 PUFA-Derived Epoxylipids
10. Sex Differences and N-3 and N-6 Polyunsaturated Fatty Acids
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dai, D.-F.; Chiao, Y.-A.; Wessells, R.J.; Bodmer, R.; Szeto, H.H.; Rabinovitch, P.S. Cardiac Aging. In Handbook of the Biology of Aging; Elsevier: Amsterdam, The Netherlands, 2016; pp. 459–494. [Google Scholar]
- Hung, C.-L.; Gonçalves, A.; Shah, A.M.; Cheng, S.; Kitzman, D.; Solomon, S.D. Age-and sex-related influences on left ventricular mechanics in elderly individuals free of prevalent heart failure: The ARIC study (Atherosclerosis Risk in Communities). Circ. Cardiovasc. Imaging 2017, 10, e004510. [Google Scholar] [CrossRef]
- Steenman, M.; Lande, G. Cardiac aging and heart disease in humans. Biophys. Rev. 2017, 9, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Lye, M.; Donnellan, C. Heart disease in the elderly. Heart 2000, 84, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular risks associated with gender and aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 2012, 16, 1492–1526. [Google Scholar] [CrossRef]
- Díez-Villanueva, P.; Alfonso, F. Heart failure in the elderly. J. Geriatr. Cardiol. 2016, 13, 115. [Google Scholar]
- Hilfiker-Kleiner, D.; Landmesser, U.L.F.; Drexler, H. Molecular mechanisms in heart failure: Focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J. Am. Coll. Cardiol. 2006, 48, A56–A66. [Google Scholar] [CrossRef]
- Ezekowitz, J.A.; O’Meara, E.; McDonald, M.A.; Abrams, H.; Chan, M.; Ducharme, A.; Giannetti, N.; Grzeslo, A.; Hamilton, P.G.; Heckman, G.A. 2017 Comprehensive update of the Canadian Cardiovascular Society guidelines for the management of heart failure. Can. J. Cardiol. 2017, 33, 1342–1433. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 128, e240. [Google Scholar] [CrossRef]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef]
- Satomura, H.; Wada, H.; Sakakura, K.; Kubo, N.; Ikeda, N.; Sugawara, Y.; Ako, J.; Momomura, S.-I. Congestive heart failure in the elderly: Comparison between reduced ejection fraction and preserved ejection fraction. J. Cardiol. 2012, 59, 215–219. [Google Scholar] [CrossRef]
- Tsao, C.W.; Lyass, A.; Enserro, D.; Larson, M.G.; Ho, J.E.; Kizer, J.R.; Gottdiener, J.S.; Psaty, B.M.; Vasan, R.S. Temporal trends in the incidence of and mortality associated with heart failure with preserved and reduced ejection fraction. JACC Heart Fail. 2018, 6, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A. Mechanisms of exercise intolerance in heart failure with preserved ejection fraction. Circ. J. 2014, 78, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.; Roger, V.L.; Rodeheffer, R.J.; Bursi, F.; Borlaug, B.A.; Ommen, S.R.; Kass, D.A.; Redfield, M.M. Cardiac structure and ventricular-vascular function in persons with heart failure and preserved ejection fraction from Olmsted County, Minnesota. Circulation 2007, 115, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; Baicu, C.F.; Gaasch, W.H. Diastolic heart failure-abnormalities in active relaxation and passive stiffness of the left ventricle. N. Engl. J. Med. 2004, 350, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Lam, C.S.; Roger, V.L.; Rodeheffer, R.J.; Redfield, M.M. Contractility and ventricular systolic stiffening in hypertensive heart disease insights into the pathogenesis of heart failure with preserved ejection fraction. J. Am. Coll Cardiol. 2009, 54, 410–418. [Google Scholar] [CrossRef]
- Shah, A.M.; Pfeffer, M.A. The many faces of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2012, 9, 555–556. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Danielsen, R.; Thorgeirsson, G.; Einarsson, H.; Ólafsson, Ö.; Aspelund, T.; Harris, T.B.; Launer, L.; Gudnason, V. Prevalence of heart failure in the elderly and future projections: The AGES-Reykjavík study. Scand. Cardiovasc. J. 2017, 51, 183–189. [Google Scholar] [CrossRef]
- Duque, E.R.; Briasoulis, A.; Alvarez, P.A. Heart failure with preserved ejection fraction in the elderly: Pathophysiology, diagnostic and therapeutic approach. J. Geriatr. Cardiol. JGC 2019, 16, 421. [Google Scholar] [PubMed]
- Lazzarini, V.; Mentz, R.J.; Fiuzat, M.; Metra, M.; O’Connor, C.M. Heart failure in elderly patients: Distinctive features and unresolved issues. Eur. J. Heart Fail. 2013, 15, 717–723. [Google Scholar] [CrossRef]
- Upadhya, B.; Kitzman, D.W. Heart failure with preserved ejection fraction in older adults. Heart Fail. Clin. 2017, 13, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart failure with preserved ejection fraction in perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef] [PubMed]
- Valero-Muñoz, M.; Backman, W.; Sam, F. Murine Models of Heart Failure With Preserved Ejection Fraction. JACC Basic Transl. Sci. 2019, 2, 770–789. [Google Scholar] [CrossRef] [PubMed]
- Marco Metra, J.R.T. Heart failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. Heart failure with preserved ejection fraction: Present status and future directions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Bellumkonda, L.; Tyrrell, D.; Hummel, S.L.; Goldstein, D.R. Pathophysiology of heart failure and frailty: A common inflammatory origin? Aging Cell 2017, 16, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Komici, K.; Corbi, G.; Pagano, G.; Furgi, G.; Rengo, C.; Femminella, G.D.; Leosco, D.; Bonaduce, D. β-adrenergic receptor responsiveness in aging heart and clinical implications. Front. Physiol. 2014, 4, 396. [Google Scholar] [CrossRef]
- Shioi, T.; Inuzuka, Y. Aging as a substrate of heart failure. J. Cardiol. 2012, 60, 423–428. [Google Scholar] [CrossRef][Green Version]
- Metra, M.; Cotter, G.; El-Khorazaty, J.; Davison, B.A.; Milo, O.; Carubelli, V.; Bourge, R.C.; Cleland, J.G.; Jondeau, G.; Krum, H. Acute heart failure in the elderly: Differences in clinical characteristics, outcomes, and prognostic factors in the VERITAS Study. J. Card. Fail. 2015, 21, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Metra, M.; Mentz, R.J.; Chiswell, K.; Bloomfield, D.M.; Cleland, J.G.F.; Cotter, G.; Davison, B.A.; Dittrich, H.C.; Fiuzat, M.; Givertz, M.M. Acute heart failure in elderly patients: Worse outcomes and differential utility of standard prognostic variables. Insights from the PROTECT trial. Eur. J. Heart Fail. 2015, 17, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Kernaghan, K. Serving seniors: Innovation and public sector service delivery. Innov. J. 2015, 20, 2. [Google Scholar]
- Seals, D.R.; Brunt, V.E.; Rossman, M.J. Keynote lecture: Strategies for optimal cardiovascular aging. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H183–H188. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Cannata, A.; Marcon, G.; Cimmino, G.; Camparini, L.; Ciucci, G.; Sinagra, G.; Loffredo, F.S. Role of circulating factors in cardiac aging. J. Thorac. Dis. 2017, 9, S17. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Li, H.; Hastings, M.H.; Rhee, J.; Trager, L.E.; Roh, J.D.; Rosenzweig, A. Targeting Age-Related Pathways in Heart Failure. Circ. Res. 2020, 126, 533–551. [Google Scholar] [CrossRef]
- Miyamoto, S. Autophagy and cardiac aging. Cell Death Differ. 2019, 26, 653–664. [Google Scholar] [CrossRef]
- Marín-García, J.; Pi, Y.; Goldenthal, M.J. Mitochondrial-nuclear cross-talk in the aging and failing heart. Cardiovasc. Drugs Ther. 2006, 20, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Akhmedov, A.T. Mitochondrial dynamics and cell death in heart failure. Heart Fail. Rev. 2016, 21, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Et. Biophys. Acta Mol. Cell Res. 2011, 1813, 1360–1372. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Ruiz-Meana, M. Not Just the Powerhouse of the Cell: Emerging Roles for Mitochondria in the Heart; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Marin-Garcia, J.; Goldenthal, M.J.; Moe, G.W. Mitochondrial pathology in cardiac failure. Cardiovasc. Res. 2001, 49, 17–26. [Google Scholar] [CrossRef]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.-H.; Chen, C.-H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, oxidative stress and innate immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef]
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16. [Google Scholar] [CrossRef]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Kauppinen, A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases. Cell. Mol. Life Sci. 2012, 69, 2999–3013. [Google Scholar] [CrossRef]
- Gasteiger, G.; D’Osualdo, A.; Schubert, D.A.; Weber, A.; Bruscia, E.M.; Hartl, D. Cellular innate immunity: An old game with new players. J. Innate Immun. 2017, 9, 111–125. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Pérez-Vázquez, D.; Contreras-Castillo, E.; Licona-Limón, P. Innate immune memory, the missing piece of the immunological response. Tip Rev. Espec. En Cienc. Químico-Biológicas 2019, 21, 112–123. [Google Scholar]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMP s and DAMP s: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef]
- Jeannin, P.; Jaillon, S.; Delneste, Y. Pattern recognition receptors in the immune response against dying cells. Curr. Opin. Immunol. 2008, 20, 530–537. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, Y.-J. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 2007, 76, 447–480. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Latz, E.; Duewell, P. NLRP3 Inflammasome Activation in Inflammaging. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2018; pp. 61–73. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Liu, X.-L.; Zhao, R. Induction of pyroptosis and its implications in cancer management. Front. Oncol. 2019, 9, 971. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, R.; Tan, H. Role of pyroptosis in cardiovascular diseases and its therapeutic implications. Int. J. Biol. Sci. 2019, 15, 1345. [Google Scholar] [CrossRef]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Marín-Aguilar, F.; Lechuga-Vieco, A.V.; Alcocer-Gómez, E.; Castejón-Vega, B.; Lucas, J.; Garrido, C.; Peralta-Garcia, A.; Pérez-Pulido, A.J.; Varela-López, A.; Quiles, J.L. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell 2020, 19, e13050. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. The importance of NLRP3 inflammasome in heart failure. J. Card. Fail. 2015, 21, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Wang, L.; Chen, X. Trimethylamine N-oxide exacerbates cardiac fibrosis via activating the NLRP3 inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Pinar, A.A.; Scott, T.E.; Huuskes, B.M.; Cáceres, F.E.T.; Kemp-Harper, B.K.; Samuel, C.S. Targeting the NLRP3 inflammasome to treat cardiovascular fibrosis. Pharm. Ther. 2020, 209, 107511. [Google Scholar] [CrossRef]
- Van Hout, G.P.J.; Bosch, L.; Ellenbroek, G.H.J.M.; De Haan, J.J.; Van Solinge, W.W.; Cooper, M.A.; Arslan, F.; De Jager, S.C.A.; Robertson, A.A.B.; Pasterkamp, G. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef]
- Cañadas-Lozano, D.; Marín-Aguilar, F.; Castejón-Vega, B.; Ryffel, B.; Navarro-Pando, J.M.; Ruiz-Cabello, J.; Alcocer-Gómez, E.; Bullón, P.; Cordero, M.D. Blockade of the NLRP3 inflammasome improves metabolic health and lifespan in obese mice. GeroScience 2020, 42, 715–725. [Google Scholar] [CrossRef]
- Quarles, E.K.; Dai, D.-F.; Tocchi, A.; Basisty, N.; Gitari, L.; Rabinovitch, P.S. Quality control systems in cardiac aging. Ageing Res. Rev. 2015, 23, 101–115. [Google Scholar] [CrossRef]
- Chen, W.; Frangogiannis, N.G. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 415–422. [Google Scholar] [CrossRef]
- Loffredo, F.S.; Nikolova, A.P.; Pancoast, J.R.; Lee, R.T. Heart failure with preserved ejection fraction: Molecular pathways of the aging myocardium. Circ. Res. 2014, 115, 97–107. [Google Scholar] [CrossRef]
- Li, R.; Lu, K.; Wang, Y.; Chen, M.; Zhang, F.; Shen, H.; Yao, D.; Gong, K.; Zhang, Z. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem. Biophys. Res. Commun. 2017, 485, 69–75. [Google Scholar] [CrossRef]
- Bracey, N.A.; Beck, P.L.; Muruve, D.A.; Hirota, S.A.; Guo, J.; Jabagi, H.; Wright, J.R., Jr.; MacDonald, J.A.; Lees-Miller, J.P.; Roach, D. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp. Physiol. 2013, 98, 462–472. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res. 2010, 88, 40–50. [Google Scholar] [CrossRef]
- Zhao, Q.; Sun, Q.; Zhou, L.; Liu, K.; Jiao, K. Complex Regulation of Mitochondrial Function During Cardiac Development. J. Am. Heart Assoc. 2019, 8, e012731. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovasc. Res. 2005, 66, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging; Multidisciplinary Digital Publishing Institute: Basel, Switzerland, 2019. [Google Scholar]
- Fried, L.P.; McNamara, R.L.; Burke, G.L.; Siscovick, D.S. Heart health in older adults. Import of heart disease and opportunities for maintaining cardiac health. West. J. Med. 1997, 167, 240. [Google Scholar] [PubMed]
- Lin, R.; Kerkelä, R. Regulatory Mechanisms of Mitochondrial Function and Cardiac Aging. Int. J. Mol. Sci. 2020, 21, 1359. [Google Scholar] [CrossRef]
- Tocchi, A.; Quarles, E.K.; Basisty, N.; Gitari, L.; Rabinovitch, P.S. Mitochondrial dysfunction in cardiac aging. Biochim. Et Biophys. Acta Bioenerg. 2015, 1847, 1424–1433. [Google Scholar] [CrossRef]
- Linton, P.-J.; Gurney, M.; Sengstock, D.; Mentzer, R.M., Jr.; Gottlieb, R.A. This old heart: Cardiac aging and autophagy. J. Mol. Cell. Cardiol. 2015, 83, 44–54. [Google Scholar] [CrossRef]
- Volt, H.; García, J.A.; Doerrier, C.; Díaz-Casado, M.E.; Guerra-Librero, A.; López, L.C.; Escames, G.; Tresguerres, J.A.; Acuña-Castroviejo, D. Same molecule but different expression: Aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J. Pineal Res. 2016, 60, 193–205. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757. [Google Scholar] [CrossRef]
- Andriollo-Sanchez, M.; Hininger-Favier, I.; Meunier, N.; Venneria, E.; O’Connor, J.M.; Maiani, G.; Coudray, C.; Roussel, A.M. Age-related oxidative stress and antioxidant parameters in middle-aged and older European subjects: The ZENITH study. Eur. J. Clin. Nutr. 2005, 59, S58–S62. [Google Scholar] [CrossRef]
- Naregal, G.V.; Devaranavadagi, B.B.; Patil, S.G.; Aski, B.S. Elevation of oxidative stress and decline in endogenous antioxidant defense in elderly individuals with hypertension. J. Clin. Diagn. Res. 2017, 11, BC09. [Google Scholar] [CrossRef]
- Nuttall, S.L.; Dunne, F.; Kendall, M.J.; Martin, U. Age-independent oxidative stress in elderly patients with non-insulin-dependent diabetes mellitus. QJM 1999, 92, 33–38. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rababa’h, A.M.; Guillory, A.N.; Mustafa, R.; Hijjawi, T. Oxidative stress and cardiac remodeling: An updated edge. Curr. Cardiol. Rev. 2018, 14, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, A.; Hamuro, J.; Nakamura, H.; Kondo, N.; Hirabayashi, Y.; Ishizaki-Koizumi, S.; Hirakawa, T.; Inoue, T.; Yodoi, J. Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxid. Redox Signal. 2002, 4, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Bullone, M.; Lavoie, J.-P. The contribution of oxidative stress and inflamm-aging in human and equine asthma. Int. J. Mol. Sci. 2017, 18, 2612. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.B.; Colucci, W.S. Mitochondrial Oxidative Stress in Heart Failure: “Oxygen Wastage” Revisited; Am Heart Assoc: Dallas, TX, USA, 2000. [Google Scholar]
- Han, Y.; Chen, J.Z. Oxidative stress induces mitochondrial DNA damage and cytotoxicity through independent mechanisms in human cancer cells. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef]
- Dai, D.-F.; Rabinovitch, P.S. Cardiac aging in mice and humans: The role of mitochondrial oxidative stress. Trends Cardiovasc. Med. 2009, 19, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-j.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Kanneganti, T.-D.; Vandanmagsar, B.; Zhu, X.; Ravussin, A.; Adijiang, A.; Owen, J.S.; Thomas, M.J.; Francis, J.; Parks, J.S. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 2012, 1, 56–68. [Google Scholar] [CrossRef]
- Hamilton, C.; Anand, P.K. Right place, right time: Localisation and assembly of the NLRP3 inflammasome. F1000Research. 2019, 8, 676. [Google Scholar] [CrossRef]
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 2017, 171, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- De la Roche, M.; Hamilton, C.; Mortensen, R.; Jeyaprakash, A.A.; Ghosh, S.; Anand, P.K. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J. Cell Biol. 2018, 217, 3560–3576. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P.; Sulaiman Rahman, H. Antioxidant and oxidative stress: A mutual interplay in age-related diseases. Front. Pharm. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Y.; Hu, Y.J.; Zhao, X.Y.; Zhong, Y.; Zeng, L.L.; Chen, X.B.; Yuan, J.; Wu, J.; Sun, Y.; Kong, W. Age-related changes in mitochondrial antioxidant enzyme Trx2 and TXNIP–Trx2–ASK 1 signal pathways in the auditory cortex of a mimetic aging rat model: Changes to Trx2 in the auditory cortex. FEBS J. 2015, 282, 2758–2774. [Google Scholar] [CrossRef]
- Huy, H.; Song, H.Y.; Kim, M.J.; Kim, W.S.; Kim, D.O.; Byun, J.E.; Lee, J.; Park, Y.J.; Kim, T.D.; Yoon, S.R. TXNIP regulates AKT-mediated cellular senescence by direct interaction under glucose-mediated metabolic stress. Aging Cell 2018, 17, e12836. [Google Scholar] [CrossRef]
- Lane, T.; Flam, B.; Lockey, R.; Kolliputi, N. TXNIP shuttling: Missing link between oxidative stress and inflammasome activation. Front. Physiol. 2013, 4, 50. [Google Scholar] [CrossRef]
- Oberacker, T.; Bajorat, J.; Ziola, S.; Schroeder, A.; Röth, D.; Kastl, L.; Edgar, B.A.; Wagner, W.; Gülow, K.; Krammer, P.H. Enhanced expression of thioredoxin-interacting-protein regulates oxidative DNA damage and aging. FEBS Lett. 2018, 592, 2297–2307. [Google Scholar] [CrossRef]
- Sverdlov, A.L.; Chan, W.P.A.; Procter, N.E.K.; Chirkov, Y.Y.; Ngo, D.T.M.; Horowitz, J.D. Reciprocal regulation of NO signaling and TXNIP expression in humans: Impact of aging and ramipril therapy. Int. J. Cardiol. 2013, 168, 4624–4630. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xu, X.; Tang, C.; Gao, P.; Chen, X.; Xiong, X.; Yang, M.; Yang, S.; Zhu, X.; Yuan, S. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018, 16, 32–46. [Google Scholar] [CrossRef]
- Kong, X.; Lu, A.-L.; Yao, X.-M.; Hua, Q.; Li, X.-Y.; Qin, L.; Zhang, H.-M.; Meng, G.-X.; Su, Q. Activation of NLRP3 inflammasome by advanced glycation end products promotes pancreatic islet damage. Oxidative Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.P.; Fu, J.; Liu, L.H.; Wu, K.F.; Liu, H.X.; Qi, B.M.; Liu, Y.; Qi, B.L. Honokiol antagonizes doxorubicin-induced cardiomyocyte senescence by inhibiting TXNIP-mediated NLRP3 inflammasome activation. Int. J. Mol. Med. 2020, 45, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn Ii, G.W. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Seo, A.Y.; Joseph, A.-M.; Dutta, D.; Hwang, J.C.Y.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, Mitochondrial Dynamics, and Homeostasis in Cardiovascular Aging. Oxidative Med. Cell. Longev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.-P.; Picard, M.; Pelletier, F.S.-J.; Sgarioto, N.; Auger, M.-J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Ishii, T.; Suda, H.; Akatsuka, A.; Hartman, P.S.; Goto, S.; Miyazawa, M.; Ishii, N. Age-related changes of mitochondrial structure and function in Caenorhabditis elegans. Mech. Ageing Dev. 2006, 127, 763–770. [Google Scholar] [CrossRef]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Sabbah, H.N. Targeting mitochondrial dysfunction in the treatment of heart failure. Expert Rev. Cardiovasc. Ther. 2016, 14, 1305–1313. [Google Scholar] [CrossRef]
- Dorn Ii, G.W. Mitochondrial dynamics in heart disease. Biochim. Et Biophys. Acta Mol. Cell Res. 2013, 1833, 233–241. [Google Scholar] [CrossRef]
- Park, S.; Won, J.-H.; Hwang, I.; Hong, S.; Lee, H.K.; Yu, J.-W. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 2015, 5, 15489. [Google Scholar] [CrossRef]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef]
- Miller, C.; Ferrero, M.; Bers, D.M.; Dedkova, E.N. Mitochondrial quality control in aging and heart failure: Influence of ketone bodies. Biophys. J. 2018, 114, 661a–662a. [Google Scholar] [CrossRef]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.Z.; Macleod, K.F. In Brief: Mitophagy: Mechanisms and role in human disease. J. Pathol. 2016, 240, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Shires, S.E.; Gustafsson, Å.B. Mitophagy and heart failure. J. Mol. Med. 2015, 93, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef]
- Kubli, D.A.; Quinsay, M.N.; Gustafsson, Å.B. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun. Integr. Biol. 2013, 6, e24511. [Google Scholar] [CrossRef]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and autophagy in the heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.-H.; Chen, C.; Wang, D.W. AMPKα2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.J.; Gustafsson, Å.B. The Aging Heart: Mitophagy at the Center of Rejuvenation. Front. Cardiovasc. Med. 2020, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-dit-Félix, A.A.; Williams, E.G.; Jha, P.; Sasso, G.L.; Huzard, D. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Yoon, J.-H.; Ryu, J.-H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Li, Z.; Meng, C.; Wu, J.; Zhao, Y.; Zhao, J. Parkin-Dependent Mitophagy Is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats. Cells 2019, 8, 897. [Google Scholar] [CrossRef] [PubMed]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K. Mitochondria and cardiovascular diseases—from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Zhou, R. Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol. 2014, 3, 63–71. [Google Scholar] [CrossRef]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 2017, 21, 455–466. [Google Scholar] [CrossRef]
- Biernacka, A.; Frangogiannis, N.G. Aging and cardiac fibrosis. Aging Dis. 2011, 2, 158. [Google Scholar]
- Dewan, S.K.; Zheng, S.-B.; Xia, S.-J.; Kalionis, B. Senescent remodeling of the immune system and its contribution to the predisposition of the elderly to infections. Chin. Med. J. 2012, 125, 3325–3331. [Google Scholar]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Deleidi, M.; Jäggle, M.; Rubino, G. Immune aging, dysmetabolism, and inflammation in neurological diseases. Front. Neurosci. 2015, 9, 172. [Google Scholar] [CrossRef]
- Ramos, G.C.; Van Den Berg, A.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abeßer, M.; Heinze, K.G. Myocardial aging as a T-cell–mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef] [PubMed]
- Harari, E.; Guo, L.; Smith, S.L.; Braumann, R.E.; Virmani, R.; Finn, A.V. Heart-resident macrophages: Are they involved in the rhythm of every beat? J. Thorac. Dis. 2017, 9, 2264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oishi, Y.; Manabe, I. Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech. Dis. 2016, 2, 1–8. [Google Scholar] [CrossRef]
- Honold, L.; Nahrendorf, M. Resident and monocyte-derived macrophages in cardiovascular disease. Circ. Res. 2018, 122, 113–127. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, S.; Dunne, A.; Monaghan, M.G. The role of macrophages in the infarcted myocardium: Orchestrators of ECM remodelling. Front. Cardiovasc. Med. 2019, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Koenig, A.L.; Lavine, K.J.; Apte, R.S. Macrophage Plasticity and Function in the Eye and Heart. Trends Immunol. 2019, 40, 825–841. [Google Scholar] [CrossRef]
- Yap, J.; Cabrera-Fuentes, H.A.; Irei, J.; Hausenloy, D.J.; Boisvert, W.A. Role of Macrophages in Cardioprotection. Int. J. Mol. Sci. 2019, 20, 2474. [Google Scholar] [CrossRef]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- DeBerge, M.; Shah, S.J.; Wilsbacher, L.; Thorp, E. Macrophages in heart failure with reduced versus preserved ejection fraction. Trends Mol. Med. 2019, 25, 328–340. [Google Scholar] [CrossRef]
- Pinto, A.R.; Godwin, J.W.; Chandran, A.; Hersey, L.; Ilinykh, A.; Debuque, R.; Wang, L.; Rosenthal, N.A. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging 2014, 6, 399. [Google Scholar] [CrossRef]
- Toba, H.; Cannon, P.L.; Yabluchanskiy, A.; Iyer, R.P.; D’Armiento, J.; Lindsey, M.L. Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H375–H383. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xie, Y.; Sun, X.; Zeh Iii, H.J.; Kang, R.; Lotze, M.T.; Tang, D. DAMPs, ageing, and cancer: The ‘DAMP Hypothesis’. Ageing Res. Rev. 2015, 24, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.L.; Dixit, V.D. Drivers of age-related inflammation and strategies for healthspan extension. Immunol. Rev. 2015, 265, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhang, H.; Zhao, Y.; Wang, Y.; Wang, W.; He, Y.; Zhang, W.; Zhang, W.; Zhu, X.; Zhou, Y. Telomere dysfunction disturbs macrophage mitochondrial metabolism and the NLRP3 inflammasome through the PGC-1α/TNFAIP3 axis. Cell Rep. 2018, 22, 3493–3506. [Google Scholar] [CrossRef]
- Van Beek, A.A.; Van den Bossche, J.; Mastroberardino, P.G.; de Winther, M.P.J.; Leenen, P.J.M. Metabolic alterations in aging macrophages: Ingredients for inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef]
- Woodcock, K.J.; Kierdorf, K.; Pouchelon, C.A.; Vivancos, V.; Dionne, M.S.; Geissmann, F. Macrophage-derived upd3 cytokine causes impaired glucose homeostasis and reduced lifespan in Drosophila fed a lipid-rich diet. Immunity 2015, 42, 133–144. [Google Scholar] [CrossRef]
- Spadaro, O.; Goldberg, E.L.; Camell, C.D.; Youm, Y.-H.; Kopchick, J.J.; Nguyen, K.Y.; Bartke, A.; Sun, L.Y.; Dixit, V.D. Growth hormone receptor deficiency protects against age-related NLRP3 inflammasome activation and immune senescence. Cell Rep. 2016, 14, 1571–1580. [Google Scholar] [CrossRef]
- Sokoła-Wysoczańska, E.; Wysoczański, T.; Wagner, J.; Czyż, K.; Bodkowski, R.; Lochyński, S.; Patkowska-Sokoła, B. Polyunsaturated fatty acids and their potential therapeutic role in cardiovascular system disorders—A review. Nutrients 2018, 10, 1561. [Google Scholar] [CrossRef]
- Rodríguez, M.G.; Rebollar, P.; Mattioli, S.; Castellini, C. n-3 PUFA sources (precursor/products): A review of current knowledge on rabbit. Animals 2019, 9, 806. [Google Scholar]
- Ander, B.P.; Dupasquier, C.M.C.; Prociuk, M.A.; Pierce, G.N. Polyunsaturated fatty acids and their effects on cardiovascular disease. Exp. Clin. Cardiol. 2003, 8, 164. [Google Scholar]
- Liu, J.J.; Green, P.; Mann, J.J.; Rapoport, S.I.; Sublette, M.E. Pathways of polyunsaturated fatty acid utilization: Implications for brain function in neuropsychiatric health and disease. Brain Res. 2015, 1597, 220–246. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.C. Interplay between n-3 and n-6 long-chain polyunsaturated fatty acids and the endocannabinoid system in brain protection and repair. Lipids 2017, 52, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Fisslthaler, B.; Popp, R.; Kiss, L.; Potente, M.; Harder, D.R.; Fleming, I.; Busse, R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999, 401, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Westphal, C.; Konkel, A.; Schunck, W.-H. Cytochrome p450 Enzymes in the Bioactivation of Polyunsaturated Fatty Acids and Their Role in Cardiovascular Disease. In Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450; Springer: Berlin/Heidelberg, Germany, 2015; pp. 151–187. [Google Scholar]
- Schunck, W.-H.; Konkel, A.; Fischer, R.; Weylandt, K.-H. Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases. Pharm. Ther. 2018, 183, 177–204. [Google Scholar] [CrossRef]
- Katragadda, D.; Batchu, S.N.; Cho, W.J.; Chaudhary, K.R.; Falck, J.R.; Seubert, J.M. Epoxyeicosatrienoic acids limit damage to mitochondrial function following stress in cardiac cells. J. Mol. Cell. Cardiol. 2009, 46, 867–875. [Google Scholar] [CrossRef]
- Nayeem, M.A. Role of oxylipins in cardiovascular diseases. Acta Pharm. Sin. 2018, 39, 1142–1154. [Google Scholar] [CrossRef]
- Samokhvalov, V.; Alsaleh, N.; El-Sikhry, H.E.; Jamieson, K.L.; Chen, C.B.; Lopaschuk, D.G.; Carter, C.; Light, P.E.; Manne, R.; Falck, J.R. Epoxyeicosatrienoic acids protect cardiac cells during starvation by modulating an autophagic response. Cell Death Dis. 2013, 4, e885. [Google Scholar] [CrossRef] [PubMed]
- Ulu, A.; Lee, K.S.S.; Miyabe, C.; Yang, J.; Hammock, B.G.; Dong, H.; Hammock, B.D. An omega-3 epoxide of docosahexaenoic acid lowers blood pressure in angiotensin-II dependent hypertension. J. Cardiovasc. Pharm. 2014, 64, 87. [Google Scholar] [CrossRef]
- Wang, R.-X.; Chai, Q.; Lu, T.; Lee, H.-C. Activation of vascular BK channels by docosahexaenoic acid is dependent on cytochrome P450 epoxygenase activity. Cardiovasc. Res. 2011, 90, 344–352. [Google Scholar] [CrossRef]
- Thomson, S.J.; Askari, A.; Bishop-Bailey, D. Anti-inflammatory effects of epoxyeicosatrienoic acids. Int. J. Vasc. Med. 2012, 2012. [Google Scholar] [CrossRef]
- Jamieson, K.L.; Endo, T.; Darwesh, A.M.; Samokhvalov, V.; Seubert, J.M. Cytochrome P450-derived eicosanoids and heart function. Pharm. Ther. 2017, 179, 47–83. [Google Scholar] [CrossRef] [PubMed]
- Hrdlicka, J.; Neckář, J.; Papousek, F.; Huskova, Z.; Kikerlova, S.; Vanourkova, Z.; Vernerova, Z.; Vasinova, J.; Hammock, B.D.; Hwang, S.H. Epoxyeicosatrienoic acid-based therapy attenuate the progression of postischemic heart failure in normotensive Sprague-Dawley but not in hypertensive Ren-2 transgenic rats. Front. Pharm. 2019, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D. Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharm. Ther. 2018, 192, 1–19. [Google Scholar] [CrossRef]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Tsenovoy, P.L.; Thompson, E.A.; Falck, J.R.; Touchon, R.; Sodhi, K.; Rezzani, R.; Shapiro, J.I.; Abraham, N.G. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins Other Lipid Mediat. 2015, 116, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Neckář, J.; Hye Khan, M.A.; Gross, G.J.; Cyprová, M.; Hrdlička, J.; Kvasilová, A.; Falck, J.R.; Campbell, W.B.; Sedláková, L.; Škutová, Š. Epoxyeicosatrienoic acid analog EET-B attenuates post-myocardial infarction remodeling in spontaneously hypertensive rats. Clin. Sci. 2019, 133, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Li, N.; He, Y.; Timofeyev, V.; Lu, L.; Tsai, H.-J.; Kim, I.-H.; Tuteja, D.; Mateo, R.K.P.; Singapuri, A. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 18733–18738. [Google Scholar] [CrossRef]
- Westphal, C.; Spallek, B.; Konkel, A.; Marko, L.; Qadri, F.; DeGraff, L.M.; Schubert, C.; Bradbury, J.A.; Regitz-Zagrosek, V.; Falck, J.R. CYP2J2 overexpression protects against arrhythmia susceptibility in cardiac hypertrophy. PLoS ONE 2013, 8, e73490. [Google Scholar] [CrossRef]
- He, Z.; Zhang, X.; Chen, C.; Wen, Z.; Hoopes, S.L.; Zeldin, D.C.; Wang, D.W. Cardiomyocyte-specific expression of CYP2J2 prevents development of cardiac remodelling induced by angiotensin II. Cardiovasc. Res. 2015, 105, 304–317. [Google Scholar] [CrossRef]
- O’Shea, K.M.; Chess, D.J.; Khairallah, R.J.; Hecker, P.A.; Lei, B.; Walsh, K.; Des Rosiers, C.; Stanley, W.C. ω-3 Polyunsaturated fatty acids prevent pressure overload-induced ventricular dilation and decrease in mitochondrial enzymes despite no change in adiponectin. Lipids Health Dis. 2010, 9, 95. [Google Scholar] [CrossRef]
- Darwesh, A.M.; Sosnowski, D.K.; Lee, T.Y.T.; Keshavarz-Bahaghighat, H.; Seubert, J.M. Insights into the cardioprotective properties of n-3 PUFAs against ischemic heart disease via modulation of the innate immune system. Chem. Biol. Interact. 2019, 308, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Romagna, E. Effect of n-3 Polyunsaturated Fatty Acids in Patients with Chronic Heart Failure (the GISSI-HF Trial): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2008, 372, 1223–1230. [Google Scholar]
- Kromhout, D.; Giltay, E.J.; Geleijnse, J.M.; Alpha Omega Trial, G. n-3 fatty acids and cardiovascular events after myocardial infarction. N. Engl. J. Med. 2010, 363, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Block, R.C.; Liu, L.; Herrington, D.M.; Huang, S.; Tsai, M.Y.; O’Connell, T.D.; Shearer, G.C. Predicting risk for incident heart failure with omega-3 fatty acids: From MESA. JACC Heart Fail. 2019, 7, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, J.; Jędrzejewski, T.; Pawlikowska, M.; Pacuła, A.J.; Ścianowski, J.; Kozak, W. The weakening effect of soluble epoxide hydrolase inhibitor AUDA on febrile response to lipopolysaccharide and turpentine in rat. J. Physiol. Biochem. 2017, 73, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Ai, D.; Pang, W.; Li, N.; Xu, M.; Jones, P.D.; Yang, J.; Zhang, Y.; Chiamvimonvat, N.; Shyy, J.Y.J.; Hammock, B.D. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 564–569. [Google Scholar] [CrossRef]
- Akhnokh, M.K.; Yang, F.H.; Samokhvalov, V.; Jamieson, K.L.; Cho, W.J.; Wagg, C.; Takawale, A.; Wang, X.; Lopaschuk, G.D.; Hammock, B.D. Inhibition of soluble epoxide hydrolase limits mitochondrial damage and preserves function following ischemic injury. Front. Pharm. 2016, 7, 133. [Google Scholar] [CrossRef]
- Darwesh, A.M.; Keshavarz-Bahaghighat, H.; Jamieson, K.L.; Seubert, J.M. Genetic Deletion or Pharmacological Inhibition of Soluble Epoxide Hydrolase Ameliorates Cardiac Ischemia/Reperfusion Injury by Attenuating NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2019, 20, 3502. [Google Scholar] [CrossRef]
- Imig, J.D.; Zhao, X.; Capdevila, J.H.; Morisseau, C.; Hammock, B.D. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 2002, 39, 690–694. [Google Scholar] [CrossRef]
- Jamieson, K.L.; Samokhvalov, V.; Akhnokh, M.K.; Lee, K.; Cho, W.J.; Takawale, A.; Wang, X.; Kassiri, Z.; Seubert, J.M. Genetic deletion of soluble epoxide hydrolase provides cardioprotective responses following myocardial infarction in aged mice. Prostaglandins Other Lipid Mediat. 2017, 132, 47–58. [Google Scholar] [CrossRef]
- Ulu, A.; Davis, B.B.; Tsai, H.-J.; Kim, I.-H.; Morisseau, C.; Inceoglu, B.; Fiehn, O.; Hammock, B.D.; Weiss, R.H. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J. Cardiovasc. Pharm. 2008, 52, 314. [Google Scholar] [CrossRef] [PubMed]
- Merabet, N.; Bellien, J.; Glevarec, E.; Nicol, L.; Lucas, D.; Remy-Jouet, I.; Bounoure, F.; Dreano, Y.; Wecker, D.; Thuillez, C. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J. Mol. Cell. Cardiol. 2012, 52, 660–666. [Google Scholar] [CrossRef]
- Stevenson, M.D.; Canugovi, C.; Vendrov, A.E.; Hayami, T.; Bowles, D.E.; Krause, K.-H.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates inflammation in ischemic heart failure: Role of soluble epoxide hydrolase. Antioxid. Redox Signal. 2019, 31, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Vacková, Š.; Kopkan, L.; Kikerlová, S.; Husková, Z.; Sadowski, J.; Kompanowska-Jezierska, E.; Hammock, B.D.; Imig, J.D.; Táborský, M.; Melenovský, V. Pharmacological blockade of soluble epoxide hydrolase attenuates the progression of congestive heart failure combined with chronic kidney disease: Insights from studies with Fawn-hooded hypertensive rats. Front. Pharm. 2019, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, Y.; Liu, X.; Chen, J.; Cai, Q.; Wang, J.; Huang, H. Apocynin improving cardiac remodeling in chronic renal failure disease is associated with up-regulation of epoxyeicosatrienoic acids. Oncotarget 2015, 6, 24699. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, V.; Jamieson, K.L.; Darwesh, A.M.; Keshavarz-Bahaghighat, H.; Lee, T.Y.T.; Edin, M.; Lih, F.; Zeldin, D.C.; Seubert, J.M. Deficiency of Soluble Epoxide Hydrolase Protects Cardiac Function Impaired by LPS-Induced Acute Inflammation. Front. Pharm. 2019, 9, 1572. [Google Scholar] [CrossRef]
- Bannehr, M.; Löhr, L.; Gelep, J.; Haverkamp, W.; Schunck, W.-H.; Gollasch, M.; Wutzler, A. Linoleic acid metabolite DiHOME decreases post-ischemic cardiac recovery in murine hearts. Cardiovasc. Toxicol. 2019, 19, 365–371. [Google Scholar] [CrossRef]
- Ha, J.; Dobretsov, M.; Kurten, R.C.; Grant, D.F.; Stimers, J.R. Effect of linoleic acid metabolites on Na+/K+ pump current in N20. 1 oligodendrocytes: Role of membrane fluidity. Toxicol. Appl. Pharm. 2002, 182, 76–83. [Google Scholar] [CrossRef]
- Harrell, M.D.; Stimers, J.R. Differential effects of linoleic acid metabolites on cardiac sodium current. J. Pharm. Exp. Ther. 2002, 303, 347–355. [Google Scholar] [CrossRef]
- Stimers, J.R.; Dobretsov, M.; Hastings, S.L.; Jude, A.R.; Grant, D.F. Effects of linoleic acid metabolites on electrical activity in adult rat ventricular myocytes. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 1999, 1438, 359–368. [Google Scholar] [CrossRef]
- Chaudhary, K.R.; Zordoky, B.N.M.; Edin, M.L.; Alsaleh, N.; El-Kadi, A.O.S.; Zeldin, D.C.; Seubert, J.M. Differential effects of soluble epoxide hydrolase inhibition and CYP2J2 overexpression on postischemic cardiac function in aged mice. Prostaglandins Other Lipid Mediat. 2013, 104, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Edin, M.L.; Wang, Z.; Bradbury, J.A.; Graves, J.P.; Lih, F.B.; DeGraff, L.M.; Foley, J.F.; Torphy, R.; Ronnekleiv, O.K.; Tomer, K.B. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 2011, 25, 3436–3447. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef]
- Shoieb, S.M.; El-Ghiaty, M.A.; Alqahtani, M.A.; El-Kadi, A.O.S. Cytochrome P450-derived eicosanoids and inflammation in liver diseases. Prostaglandins Other Lipid Mediat. 2020, 147, 106400. [Google Scholar] [CrossRef]
- Li, X.; Zhu, F.; Meng, W.; Zhang, F.; Hong, J.; Zhang, G.; Wang, F. CYP2J2/EET reduces vulnerability to atrial fibrillation in chronic pressure overload mice. J. Cell. Mol. Med. 2020, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Darwesh, A.M.; Jamieson, K.L.; Wang, C.; Samokhvalov, V.; Seubert, J.M. Cardioprotective effects of CYP-derived epoxy metabolites of docosahexaenoic acid involve limiting NLRP3 inflammasome activation (1). Can. J. Physiol Pharm. 2019, 97, 544–556. [Google Scholar] [CrossRef]
- Capozzi, M.E.; Hammer, S.S.; McCollum, G.W.; Penn, J.S. Epoxygenated fatty acids inhibit retinal vascular inflammation. Sci. Rep. 2016, 6, 39211. [Google Scholar] [CrossRef]
- Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar]
- Ramos-Campo, D.J.; Ávila-Gandía, V.; López-Román, F.J.; Miñarro, J.; Contreras, C.; Soto-Méndez, F.; Domingo Pedrol, J.C.; Luque-Rubia, A.J. Supplementation of Re-Esterified Docosahexaenoic and Eicosapentaenoic Acids Reduce Inflammatory and Muscle Damage Markers after Exercise in Endurance Athletes: A Randomized, Controlled Crossover Trial. Nutrients 2020, 12, 719. [Google Scholar] [CrossRef]
- Ulu, A.; Harris, T.R.; Morisseau, C.; Miyabe, C.; Inoue, H.; Schuster, G.; Dong, H.; Iosif, A.-M.; Liu, J.-Y.; Weiss, R.H. Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II dependent hypertension. J. Cardiovasc. Pharm. 2013, 62, 285. [Google Scholar] [CrossRef]
- Napimoga, M.H.; Rocha, E.P.; Trindade-da-Silva, C.A.; Demasi, A.P.D.; Martinez, E.F.; Macedo, C.G.; Abdalla, H.B.; Bettaieb, A.; Haj, F.G.; Clemente-Napimoga, J.T. Soluble epoxide hydrolase inhibitor promotes immunomodulation to inhibit bone resorption. J. Periodontal Res. 2018, 53, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.M.; McReynolds, C.B.; Schmidt, W.K.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharm. Ther. 2017, 180, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bratt, J.; Franzi, L.; Liu, J.-Y.; Zhang, G.; Zeki, A.A.; Vogel, C.F.A.; Williams, K.; Dong, H.; Lin, Y. Soluble epoxide hydrolase inhibitor attenuates inflammation and airway hyperresponsiveness in mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, K.R.; Kubala, L.; Newman, J.W.; Kim, I.H.; Eiserich, J.P.; Hammock, B.D. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl Acad Sci. USA 2005, 102, 9772–9777. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, A.L.; Liao, J.; Li, H.; Dong, H.; Chung, Y.T.; Bai, H.; Matkowskyj, K.A.; Hammock, B.D.; Yang, G.-Y. Soluble epoxide hydrolase gene deficiency or inhibition attenuates chronic active inflammatory bowel disease in IL-10 (−/−) mice. Dig. Dis. Sci. 2012, 57, 2580–2591. [Google Scholar] [CrossRef]
- Yeh, C.-F.; Chuang, T.Y.; Hung, Y.-W.; Lan, M.-Y.; Tsai, C.-H.; Huang, H.-X.; Lin, Y.-Y. Soluble epoxide hydrolase inhibition enhances anti-inflammatory and antioxidative processes, modulates microglia polarization, and promotes recovery after ischemic stroke. Neuropsychiatr. Dis. Treat. 2019, 15, 2927. [Google Scholar] [CrossRef]
- Yang, H.-H.; Duan, J.-X.; Liu, S.-K.; Xiong, J.-B.; Guan, X.-X.; Zhong, W.-J.; Sun, C.-C.; Zhang, C.-Y.; Luo, X.-Q.; Zhang, Y.-F. A COX-2/sEH dual inhibitor PTUPB alleviates lipopolysaccharide-induced acute lung injury in mice by inhibiting NLRP3 inflammasome activation. Theranostics 2020, 10, 4749. [Google Scholar] [CrossRef]
- Zimmer, B.; Angioni, C.; Osthues, T.; Toewe, A.; Thomas, D.; Pierre, S.C.; Geisslinger, G.; Scholich, K.; Sisignano, M. The oxidized linoleic acid metabolite 12, 13-DiHOME mediates thermal hyperalgesia during inflammatory pain. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 669–678. [Google Scholar] [CrossRef]
- Levan, S.R. Neonatal gut-microbiome-derived 12, 13 DiHOME impedes tolerance and promotes childhood atopy and asthma. Biorxiv 2018, e311704. [Google Scholar] [CrossRef]
- Levan, S.R.; Stamnes, K.A.; Lin, D.L.; Panzer, A.R.; Fukui, E.; McCauley, K.; Fujimura, K.E.; McKean, M.; Ownby, D.R.; Zoratti, E.M. Elevated faecal 12, 13-diHOME concentration in neonates at high risk for asthma is produced by gut bacteria and impedes immune tolerance. Nat. Microbiol. 2019, 4, 1851–1861. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef] [PubMed]
- Batchu, S.N.; Lee, S.B.; Samokhvalov, V.; Chaudhary, K.R.; El-Sikhry, H.; Weldon, S.M.; Seubert, J.M. Novel soluble epoxide hydrolase inhibitor protects mitochondrial function following stress. Can. J. Physiol. Pharm. 2012, 90, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.R.; Cho, W.J.; Yang, F.; Samokhvalov, V.; El-Sikhry, H.E.; Daniel, E.E.; Seubert, J.M. Effect of ischemia reperfusion injury and epoxyeicosatrienoic acids on caveolin expression in mouse myocardium. J. Cardiovasc. Pharm. 2013, 61, 258–263. [Google Scholar] [CrossRef] [PubMed]
- El-Sikhry, H.E.; Alsaleh, N.; Dakarapu, R.; Falck, J.R.; Seubert, J.M. Novel roles of epoxyeicosanoids in regulating cardiac mitochondria. PloS ONE 2016, 11, e0160380. [Google Scholar] [CrossRef]
- Oni-Orisan, A.; Alsaleh, N.; Lee, C.R.; Seubert, J.M. Epoxyeicosatrienoic acids and cardioprotection: The road to translation. J. Mol. Cell. Cardiol. 2014, 74, 199–208. [Google Scholar] [CrossRef]
- Batchu, S.N.; Lee, S.B.; Qadhi, R.S.; Chaudhary, K.R.; El-Sikhry, H.; Kodela, R.; Falck, J.R.; Seubert, J.M. Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. Br. J. Pharm. 2011, 162, 897–907. [Google Scholar] [CrossRef]
- Sarkar, P.; Zaja, I.; Bienengraeber, M.; Rarick, K.R.; Terashvili, M.; Canfield, S.; Falck, J.R.; Harder, D.R. Epoxyeicosatrienoic acids pretreatment improves amyloid β-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H475–H484. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, G.; Yang, S.; Ping, W.; Fu, X.; Zhang, N.; Wang, D.W.; Wang, J. CYP2J2 and EETs protect against oxidative stress and apoptosis in vivo and in vitro following lung ischemia/reperfusion. Cell. Physiol. Biochem. 2014, 33, 1663–1680. [Google Scholar] [CrossRef]
- Cao, J.; Singh, S.P.; McClung, J.A.; Joseph, G.; Vanella, L.; Barbagallo, I.; Jiang, H.; Falck, J.R.; Arad, M.; Shapiro, J.I. EET intervention on Wnt1, NOV, and HO-1 signaling prevents obesity-induced cardiomyopathy in obese mice. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H368–H380. [Google Scholar] [CrossRef]
- Liu, L.; Puri, N.; Raffaele, M.; Schragenheim, J.; Singh, S.P.; Bradbury, J.A.; Bellner, L.; Vanella, L.; Zeldin, D.C.; Cao, J. Ablation of soluble epoxide hydrolase reprogram white fat to beige-like fat through an increase in mitochondrial integrity, HO-1-adiponectin in vitro and in vivo. Prostaglandins Other Lipid Mediat. 2018, 138, 1–8. [Google Scholar] [CrossRef]
- Galvao, T.F.; Khairallah, R.J.; Dabkowski, E.R.; Brown, B.H.; Hecker, P.A.; O’Connell, K.A.; O’Shea, K.M.; Sabbah, H.N.; Rastogi, S.; Daneault, C. Marine n3 polyunsaturated fatty acids enhance resistance to mitochondrial permeability transition in heart failure but do not improve survival. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H12–H21. [Google Scholar] [CrossRef]
- Stanley, W.C.; Khairallah, R.J.; Dabkowski, E.R. Update on lipids and mitochondrial function: Impact of dietary n-3 polyunsaturated fatty acids. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 122. [Google Scholar] [CrossRef]
- Dabkowski, E.R.; O’Connell, K.A.; Xu, W.; Ribeiro, R.F.; Hecker, P.A.; Shekar, K.C.; Daneault, C.; Des Rosiers, C.; Stanley, W.C. Docosahexaenoic acid supplementation alters key properties of cardiac mitochondria and modestly attenuates development of left ventricular dysfunction in pressure overload-induced heart failure. Cardiovasc. Drugs Ther. 2013, 27, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, L.; Hu, W.; Zheng, Q.; Xiang, W. Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by omega-3 fatty acid–induced up-regulation of mitofusin 2. Metabolism 2011, 60, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, P.; Zhang, J.H.; Li, Y.; Xu, S.; Wang, C.; Wang, L.; Zhang, G.; Dai, J.; Zhu, S. Docosahexaenoic acid alleviates oxidative stress-based apoptosis via improving mitochondrial dynamics in early brain injury after subarachnoid hemorrhage. Cell. Mol. Neurobiol. 2018, 38, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Liu, C.; Chen, Q.; Liu, N.; Yan, Y.; Liu, B. SIRT3: A new regulator of cardiovascular diseases. Oxidative Med. Cell. Longev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Parodi-Rullán, R.M.; Chapa-Dubocq, X.R.; Javadov, S. Acetylation of mitochondrial proteins in the heart: The role of SIRT3. Front. Physiol. 2018, 9, 1094. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.-Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, K.L.; Keshavarz-Bahaghighat, H.; Darwesh, A.M.; Sosnowski, D.K.; Seubert, J.M. Age and Sex Differences in Hearts of Soluble Epoxide Hydrolase Null Mice. Front. Physiol. 2020, 11, 48. [Google Scholar] [CrossRef]
- Liu, L.; Huang, X.; Gao, J.; Guo, Y.; Di, Y.; Sun, S.; Deng, X.; Cao, J. Improved endogenous epoxyeicosatrienoic acid production mends heart function via increased PGC 1α-mitochondrial functions in metabolic syndrome. J. Pharm. Sci. 2018, 138, 138–145. [Google Scholar] [CrossRef]
- Fajemiroye, J.O.; Cunha, L.C.d.; Saavedra-Rodríguez, R.; Rodrigues, K.L.; Naves, L.M.; Mourão, A.A.; Silva, E.F.d.; Williams, N.E.E.; Martins, J.L.R.; Sousa, R.B. Aging-induced biological changes and cardiovascular diseases. Biomed. Res. Int. 2018, 2018. [Google Scholar] [CrossRef]
- Merz, A.A.; Cheng, S. Sex differences in cardiovascular ageing. Heart 2016, 102, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.A.; Kalasky, M.J.; Proctor, D.N. Evidence for sex differences in cardiovascular aging and adaptive responses to physical activity. Eur. J. Appl. Physiol. 2010, 110, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V.; Kararigas, G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol. Rev. 2017, 97, 1–37. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V.; Oertelt-Prigione, S.; Seeland, U.; Hetzer, R. Sex and gender differences in myocardial hypertrophy and heart failure. Circ. J. 2010, 74, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.R.; Patel, H.N.; Mehta, L.S.; Sanghani, R.M.; Lundberg, G.P.; Lewis, S.J.; Mendelson, M.A.; Wood, M.J.; Volgman, A.S.; Mieres, J.H. Sex differences in ischemic heart disease: Advances, obstacles, and next steps. Circ. Cardiovasc. Qual. Outcomes 2018, 11, e004437. [Google Scholar] [CrossRef]
- Bhupathy, P.; Haines, C.D.; Leinwand, L.A. Influence of sex hormones and phytoestrogens on heart disease in men and women. Women’s Health 2010, 6, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Kaley, G. Gender-specific regulation of cardiovascular function: Estrogen as key player. Microcirculation 2004, 11, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Borrás, C.; Sastre, J.; García-Sala, D.; Lloret, A.; Pallardó, F.V.; Viña, J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic. Biol. Med. 2003, 34, 546–552. [Google Scholar] [CrossRef]
- Colom, B.; Oliver, J.; Roca, P.; Garcia-Palmer, F.J. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc. Res. 2007, 74, 456–465. [Google Scholar] [CrossRef]
- Justo, R.; Frontera, M.; Pujol, E.; Rodríguez-Cuenca, S.; Lladó, I.; García-Palmer, F.J.; Roca, P.; Gianotti, M. Gender-related differences in morphology and thermogenic capacity of brown adipose tissue mitochondrial subpopulations. Life Sci. 2005, 76, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Pinot, F.; Grant, D.F.; Spearow, J.L.; Parker, A.G.; Hammock, B.D. Differential regulation of soluble epoxide hydrolase by clofibrate and sexual hormones in the liver and kidneys of mice. Biochem. Pharm. 1995, 50, 501–508. [Google Scholar] [CrossRef]
- Qin, J.; Le, Y.; Froogh, G.; Kandhi, S.; Jiang, H.; Luo, M.; Sun, D.; Huang, A. Sexually dimorphic adaptation of cardiac function: Roles of epoxyeicosatrienoic acid and peroxisome proliferator-activated receptors. Physiol. Rep. 2016, 4, e12838. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Miyata, M.; Tohkin, M.; Nagata, K.; Bend, J.R.; Gonzalez, F.J. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J. Biol. Chem. 2000, 275, 40504–40510. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keshavarz-Bahaghighat, H.; Darwesh, A.M.; Sosnowski, D.K.; Seubert, J.M. Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids. Cells 2020, 9, 1565. https://doi.org/10.3390/cells9071565
Keshavarz-Bahaghighat H, Darwesh AM, Sosnowski DK, Seubert JM. Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids. Cells. 2020; 9(7):1565. https://doi.org/10.3390/cells9071565
Chicago/Turabian StyleKeshavarz-Bahaghighat, Hedieh, Ahmed M. Darwesh, Deanna K. Sosnowski, and John M. Seubert. 2020. "Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids" Cells 9, no. 7: 1565. https://doi.org/10.3390/cells9071565
APA StyleKeshavarz-Bahaghighat, H., Darwesh, A. M., Sosnowski, D. K., & Seubert, J. M. (2020). Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids. Cells, 9(7), 1565. https://doi.org/10.3390/cells9071565