Immunotherapy in Myeloproliferative Diseases


Abstract
1. Introduction
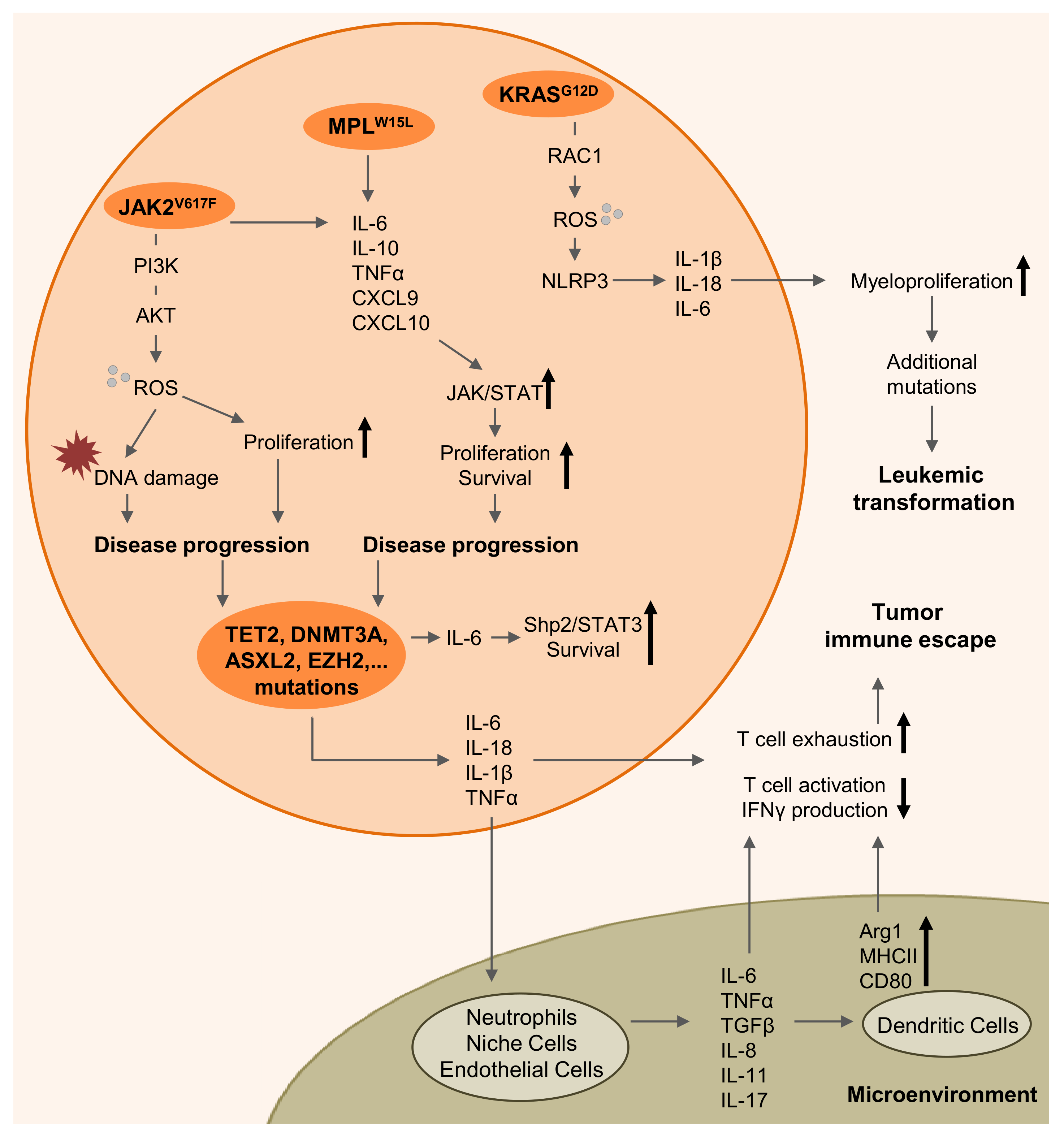
2. Myeloproliferative Diseases are Driven by Inflammation
3. Allogeneic Hematopoietic Stem-Cell Transplantation
4. MDSCs Mediate Leukemia Immune Escape
5. Interferon Alpha
6. JAK2 Inhibition
7. Targeting CD123
8. Tumor Vaccination
9. Immune Checkpoint Blockade
10. WT1-Specific T-Cells
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Campbell, P.J.; Green, A.R. The myeloproliferative disorders. N. Engl. J. Med. 2006, 355, 2452–2466. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 who classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L. Myeloproliferative neoplasms. N. Engl. J. Med. 2017, 377, 895–896. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef]
- Klausen, U.; Holmberg, S.; Holmstrom, M.O.; Jorgensen, N.G.D.; Grauslund, J.H.; Svane, I.M.; Andersen, M.H. Novel strategies for peptide-based vaccines in hematological malignancies. Front. Immunol. 2018, 9, 2264. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal jak2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of jak2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase jak2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase jak2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. Jak2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. Mpl515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. Mplw515l is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [PubMed]
- Chaligne, R.; James, C.; Tonetti, C.; Besancenot, R.; Le Couedic, J.P.; Fava, F.; Mazurier, F.; Godin, I.; Maloum, K.; Larbret, F.; et al. Evidence for mpl w515l/k mutations in hematopoietic stem cells in primitive myelofibrosis. Blood 2007, 110, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.A.; Campbell, P.J.; Scott, L.M.; Bench, A.J.; Erber, W.N.; Bareford, D.; Wilkins, B.S.; Reilly, J.T.; Hasselbalch, H.C.; Bowman, R.; et al. Mpl mutations in myeloproliferative disorders: Analysis of the pt-1 cohort. Blood 2008, 112, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in calr-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic calr mutations in myeloproliferative neoplasms with nonmutated jak2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Li, B.; Mascarenhas, J.O.; Rampal, R.K. Leukemic transformation of myeloproliferative neoplasms: Therapeutic and genomic considerations. Curr. Hematol. Malig. Rep. 2018, 13, 588–595. [Google Scholar] [CrossRef]
- Spivak, J.L. The chronic myeloproliferative disorders: Clonality and clinical heterogeneity. Semin. Hematol. 2004, 41, 1–5. [Google Scholar] [CrossRef]
- Cervantes, F.; Tassies, D.; Salgado, C.; Rovira, M.; Pereira, A.; Rozman, C. Acute transformation in nonleukemic chronic myeloproliferative disorders: Actuarial probability and main characteristics in a series of 218 patients. Acta Haematol. 1991, 85, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.A.; Atenafu, E.G.; Messner, H.A.; Craddock, K.J.; Brandwein, J.M.; Lipton, J.H.; Minden, M.D.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Treatment outcomes following leukemic transformation in philadelphia-negative myeloproliferative neoplasms. Blood 2013, 121, 2725–2733. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Li, C.Y.; Ketterling, R.P.; Schroeder, G.S.; Knudson, R.A.; Tefferi, A. Leukemic transformation in myelofibrosis with myeloid metaplasia: A single-institution experience with 91 cases. Blood 2005, 105, 973–977. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Thoennissen, N.H.; Krug, U.O.; Lee, D.H.; Kawamata, N.; Iwanski, G.B.; Lasho, T.; Weiss, T.; Nowak, D.; Koren-Michowitz, M.; Kato, M.; et al. Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of philadelphia chromosome-negative myeloproliferative neoplasms. Blood 2010, 115, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gonen, M.; et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent srsf2 mutations that are associated with adverse outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.A.; Delhommeau, F.; LeCouedic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kusec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a jak2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900. [Google Scholar] [CrossRef]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. P53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Swierczek, S.I.; Drummond, J.; Hickman, K.; Kim, S.J.; Walker, K.; Doddapaneni, H.; Muzny, D.M.; Gibbs, R.A.; Wheeler, D.A.; et al. Whole-exome sequencing of polycythemia vera revealed novel driver genes and somatic mutation shared by t cells and granulocytes. Leukemia 2014, 28, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Delic, S.; Rose, D.; Kern, W.; Nadarajah, N.; Haferlach, C.; Haferlach, T.; Meggendorfer, M. Application of an ngs-based 28-gene panel in myeloproliferative neoplasms reveals distinct mutation patterns in essential thrombocythaemia, primary myelofibrosis and polycythaemia vera. Br. J. Haematol. 2016, 175, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Srsf2 mutations in primary myelofibrosis: Significant clustering with idh mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef] [PubMed]
- Courtier, F.; Carbuccia, N.; Garnier, S.; Guille, A.; Adelaide, J.; Cervera, N.; Gelsi-Boyer, V.; Mozziconacci, M.J.; Rey, J.; Vey, N.; et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica 2017, 102, e11–e14. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couedic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in jak2 v617f myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Thomassen, M.; Riley, C.H.; Kjaer, L.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Skov, V. Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of nrf2 for genomic instability and disease progression. PLoS ONE 2014, 9, e112786. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The nlrp3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Osswald, L.; Saller, B.S.; Unger, S.; De Feo, D.; Vinnakota, J.M.; Konantz, M.; Uhl, F.M.; Becker, H.; Lubbert, M.; et al. Oncogenic kras(g12d) causes myeloproliferation via nlrp3 inflammasome activation. Nat. Commun. 2020, 11, 1659. [Google Scholar] [CrossRef]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. Jak-stat pathway activation in malignant and nonmalignant cells contributes to mpn pathogenesis and therapeutic response. Cancer Discov 2015, 5, 316–331. [Google Scholar] [CrossRef]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018, 33, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in polycythemia vera and contribute to the growth of clonal erythroblasts independently of jak2v617f. Oncogene 2011, 30, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of inflammatory signaling in tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell 2018, 23, 833–849 e835. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with tet2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, S.; Haque, T.; Gritsman, K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front. Oncol. 2017, 7, 265. [Google Scholar] [CrossRef]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. USA 2017, 114, E1490–E1499. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Yang, Y.; Akada, H.; Nath, D.; Hutchison, R.E.; Mohi, G. Loss of ezh2 cooperates with jak2v617f in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood 2016, 127, 3410–3423. [Google Scholar] [CrossRef]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2v617f and dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef]
- Masarova, L.; Bose, P.; Verstovsek, S. The rationale for immunotherapy in myeloproliferative neoplasms. Curr. Hematol. Malig. Rep. 2019, 14, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Le Bousse-Kerdiles, M.C.; Chevillard, S.; Charpentier, A.; Romquin, N.; Clay, D.; Smadja-Joffe, F.; Praloran, V.; Dupriez, B.; Demory, J.L.; Jasmin, C.; et al. Differential expression of transforming growth factor-beta, basic fibroblast growth factor, and their receptors in cd34+ hematopoietic progenitor cells from patients with myelofibrosis and myeloid metaplasia. Blood 1996, 88, 4534–4546. [Google Scholar] [CrossRef] [PubMed]
- Bock, O.; Hoftmann, J.; Theophile, K.; Hussein, K.; Wiese, B.; Schlue, J.; Kreipe, H. Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. Am. J. Pathol. 2008, 172, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, J.J.; Pierre-Louis, O.; Hasselbalch, H.C.; Uzan, G.; Jasmin, C.; Martyre, M.C.; Le Bousse-Kerdiles, M.C.; French INSERM; the European EUMNET Networks on Myelofibrosis. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood 2008, 112, 3026–3035. [Google Scholar] [CrossRef]
- Flamant, L.; Toffoli, S.; Raes, M.; Michiels, C. Hypoxia regulates inflammatory gene expression in endothelial cells. Exp. Cell Res. 2009, 315, 733–747. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Zhan, H.; Cardozo, C.; Yu, W.; Wang, A.; Moliterno, A.R.; Dang, C.V.; Spivak, J.L. Microrna deregulation in polycythemia vera and essential thrombocythemia patients. Blood Cells Mol. Dis. 2013, 50, 190–195. [Google Scholar] [CrossRef][Green Version]
- Martyre, M.C.; Magdelenat, H.; Bryckaert, M.C.; Laine-Bidron, C.; Calvo, F. Increased intraplatelet levels of platelet-derived growth factor and transforming growth factor-beta in patients with myelofibrosis with myeloid metaplasia. Br. J. Haematol. 1991, 77, 80–86. [Google Scholar] [CrossRef]
- Hermouet, S.; Godard, A.; Pineau, D.; Corre, I.; Raher, S.; Lippert, E.; Jacques, Y. Abnormal production of interleukin (il)-11 and il-8 in polycythaemia vera. Cytokine 2002, 20, 178–183. [Google Scholar] [CrossRef]
- Panteli, K.E.; Hatzimichael, E.C.; Bouranta, P.K.; Katsaraki, A.; Seferiadis, K.; Stebbing, J.; Bourantas, K.L. Serum interleukin (il)-1, il-2, sil-2ra, il-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br. J. Haematol. 2005, 130, 709–715. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Bellomo, G.; D’Angelo, A.; Granata, A.; Russo, S.; Quartarone, E.; Musolino, C. Evaluation of interleukin-17 serum levels in patients with chronic myeloproliferative diseases. Tumori 2009, 95, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Slezak, S.; Jin, P.; Caruccio, L.; Ren, J.; Bennett, M.; Zia, N.; Adams, S.; Wang, E.; Ascensao, J.; Schechter, G.; et al. Gene and microrna analysis of neutrophils from patients with polycythemia vera and essential thrombocytosis: Down-regulation of micro rna-1 and -133a. J. Transl. Med. 2009, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of incb018424, a jak1 and jak2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Pahl, H.L. The hen or the egg: Inflammatory aspects of murine mpn models. Mediat. Inflamm. 2015, 2015, 101987. [Google Scholar] [CrossRef] [PubMed]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking il-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-pd-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Fujieda, K.; Senju, S.; Ikeda, T.; Oshiumi, H.; Nishimura, Y. Immune-suppressive effects of interleukin-6 on t-cell-mediated anti-tumor immunity. Cancer Sci. 2018, 109, 523–530. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/socs3 activation by il-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Ara, T.; Nakata, R.; Sheard, M.A.; Shimada, H.; Buettner, R.; Groshen, S.G.; Ji, L.; Yu, H.; Jove, R.; Seeger, R.C.; et al. Critical role of stat3 in il-6-mediated drug resistance in human neuroblastoma. Cancer Res. 2013, 73, 3852–3864. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing il-6 and tnf expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Fujieda, K.; Hirayama, M.; Ikeda, T.; Yuno, A.; Matsumura, K.; Fukuma, D.; Araki, K.; Mizuta, H.; Nakayama, H.; et al. Soluble il6r expressed by myeloid cells reduces tumor-specific th1 differentiation and drives tumor progression. Cancer Res. 2017, 77, 2279–2291. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Senju, S.; Matsumura, K.; Swain, S.L.; Nishimura, Y. Il-6-mediated environmental conditioning of defective th1 differentiation dampens antitumour immune responses in old age. Nat. Commun. 2015, 6, 6702. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Qu, Z.; Sun, F.; Han, L.; Li, L.; Yan, S.; Stabile, L.P.; Chen, L.F.; Siegfried, J.M.; Xiao, G. Myeloid stat3 promotes lung tumorigenesis by transforming tumor immunosurveillance into tumor-promoting inflammation. Cancer Immunol. Res. 2017, 5, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Narita, Y.; Kitamura, H.; Wakita, D.; Sumida, K.; Masuko, K.; Terada, S.; Nakano, K.; Nishimura, T. The key role of il-6-arginase cascade for inducing dendritic cell-dependent cd4(+) t cell dysfunction in tumor-bearing mice. J. Immunol. 2013, 190, 812–820. [Google Scholar] [CrossRef]
- Kitamura, H.; Kamon, H.; Sawa, S.; Park, S.J.; Katunuma, N.; Ishihara, K.; Murakami, M.; Hirano, T. Il-6-stat3 controls intracellular mhc class ii alphabeta dimer level through cathepsin s activity in dendritic cells. Immunity 2005, 23, 491–502. [Google Scholar] [CrossRef]
- Deeg, H.J.; Gooley, T.A.; Flowers, M.E.; Sale, G.E.; Slattery, J.T.; Anasetti, C.; Chauncey, T.R.; Doney, K.; Georges, G.E.; Kiem, H.P.; et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood 2003, 102, 3912–3918. [Google Scholar] [CrossRef]
- Salit, R.B.; Deeg, H.J. Role of hematopoietic stem cell transplantation in patients with myeloproliferative disease. Hematol. Oncol. Clin. North. Am. 2014, 28, 1023–1035. [Google Scholar] [CrossRef][Green Version]
- Keyzner, A.; Han, S.; Shapiro, S.; Moshier, E.; Schorr, E.; Petersen, B.; Najfeld, V.; Kremyanskaya, M.; Isola, L.; Hoffman, R.; et al. Outcome of allogeneic hematopoietic stem cell transplantation for patients with chronic and advanced phase myelofibrosis. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2016, 22, 2180–2186. [Google Scholar] [CrossRef]
- Cervantes, F.; Rovira, M.; Urbano-Ispizua, A.; Rozman, M.; Carreras, E.; Montserrat, E. Complete remission of idiopathic myelofibrosis following donor lymphocyte infusion after failure of allogeneic transplantation: Demonstration of a graft-versus-myelofibrosis effect. Bone Marrow Transpl. 2000, 26, 697–699. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ditschkowski, M.; Beelen, D.W.; Trenschel, R.; Koldehoff, M.; Elmaagacli, A.H. Outcome of allogeneic stem cell transplantation in patients with myelofibrosis. Bone Marrow Transpl. 2004, 34, 807–813. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anderson, J.E.; Sale, G.; Appelbaum, F.R.; Chauncey, T.R.; Storb, R. Allogeneic marrow transplantation for primary myelofibrosis and myelofibrosis secondary to polycythaemia vera or essential thrombocytosis. Br. J. Haematol. 1997, 98, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C.L. Chronic myeloid leukemia. N. Engl. J. Med. 1999, 340, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Kroger, N.; Giorgino, T.; Scott, B.L.; Ditschkowski, M.; Alchalby, H.; Cervantes, F.; Vannucchi, A.; Cazzola, M.; Morra, E.; Zabelina, T.; et al. Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood 2015, 125, 3347–3350. [Google Scholar] [CrossRef] [PubMed]
- Rajantie, J.; Sale, G.E.; Deeg, H.J.; Amos, D.; Appelbaum, F.; Storb, R.; Clift, R.A.; Buckner, C.D. Adverse effect of severe marrow fibrosis on hematologic recovery after chemoradiotherapy and allogeneic bone marrow transplantation. Blood 1986, 67, 1693–1697. [Google Scholar] [CrossRef]
- Bartenstein, M.; Deeg, H.J. Hematopoietic stem cell transplantation for mds. Hematol. Oncol. Clin. North. Am. 2010, 24, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Nachtkamp, K.; Kundgen, A.; Strupp, C.; Giagounidis, A.; Kobbe, G.; Gattermann, N.; Haas, R.; Germing, U. Impact on survival of different treatments for myelodysplastic syndromes (mds). Leuk. Res. 2009, 33, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- de Lima, M.; Anagnostopoulos, A.; Munsell, M.; Shahjahan, M.; Ueno, N.; Ippoliti, C.; Andersson, B.S.; Gajewski, J.; Couriel, D.; Cortes, J.; et al. Nonablative versus reduced-intensity conditioning regimens in the treatment of acute myeloid leukemia and high-risk myelodysplastic syndrome: Dose is relevant for long-term disease control after allogeneic hematopoietic stem cell transplantation. Blood 2004, 104, 865–872. [Google Scholar] [CrossRef]
- Sierra, J.; Perez, W.S.; Rozman, C.; Carreras, E.; Klein, J.P.; Rizzo, J.D.; Davies, S.M.; Lazarus, H.M.; Bredeson, C.N.; Marks, D.I.; et al. Bone marrow transplantation from hla-identical siblings as treatment for myelodysplasia. Blood 2002, 100, 1997–2004. [Google Scholar]
- Scott, B.L.; Gooley, T.A.; Sorror, M.L.; Rezvani, A.R.; Linenberger, M.L.; Grim, J.; Sandmaier, B.M.; Myerson, D.; Chauncey, T.R.; Storb, R.; et al. The dynamic international prognostic scoring system for myelofibrosis predicts outcomes after hematopoietic cell transplantation. Blood 2012, 119, 2657–2664. [Google Scholar] [CrossRef]
- Alchalby, H.; Yunus, D.R.; Zabelina, T.; Kobbe, G.; Holler, E.; Bornhauser, M.; Schwerdtfeger, R.; Bethge, W.; Kvasnicka, H.M.; Busche, G.; et al. Risk models predicting survival after reduced-intensity transplantation for myelofibrosis. Br. J. Haematol. 2012, 157, 75–85. [Google Scholar] [CrossRef]
- Magenau, J.; Couriel, D.R. Hematopoietic stem cell transplantation for acute myeloid leukemia: To whom, when, and how. Curr. Oncol. Rep. 2013, 15, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; Labopin, M.; Nagler, A.; Niederwieser, D.; Castagna, L.; Tabrizi, R.; Stadler, M.; Kuball, J.; Cornelissen, J.; Vorlicek, J.; et al. Treatment, risk factors, and outcome of adults with relapsed aml after reduced intensity conditioning for allogeneic stem cell transplantation. Blood 2012, 119, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ritchey, J.; Prior, J.L.; Holt, M.; Shannon, W.D.; Deych, E.; Piwnica-Worms, D.R.; DiPersio, J.F. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood 2010, 116, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, O.C.; Dennis, M.; Jilani, N.Y.; Loke, J.; Siddique, S.; Ryan, G.; Nunnick, J.; Khanum, R.; Raghavan, M.; Cook, M.; et al. Azacitidine augments expansion of regulatory t cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (aml). Blood 2012, 119, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.M.; Estrov, Z.; Cortes, J.E.; Thomas, D.A.; Kadia, T.; Pierce, S.; Jabbour, E.; Borthakur, G.; Rumi, E.; et al. Long-term outcomes of 107 patients with myelofibrosis receiving jak1/jak2 inhibitor ruxolitinib: Survival advantage in comparison to matched historical controls. Blood 2012, 120, 1202–1209. [Google Scholar] [CrossRef]
- Jaekel, N.; Behre, G.; Behning, A.; Wickenhauser, C.; Lange, T.; Niederwieser, D.; Al-Ali, H.K. Allogeneic hematopoietic cell transplantation for myelofibrosis in patients pretreated with the jak1 and jak2 inhibitor ruxolitinib. Bone Marrow Transpl. 2014, 49, 179–184. [Google Scholar] [CrossRef] [PubMed]
- McLornan, D.P.; Yakoub-Agha, I.; Robin, M.; Chalandon, Y.; Harrison, C.N.; Kroger, N. State-of-the-art review: Allogeneic stem cell transplantation for myelofibrosis in 2019. Haematologica 2019, 104, 659–668. [Google Scholar] [CrossRef]
- Wang, J.C.; Kundra, A.; Andrei, M.; Baptiste, S.; Chen, C.; Wong, C.; Sindhu, H. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk. Res. 2016, 43, 39–43. [Google Scholar] [CrossRef]
- Draghiciu, O.; Lubbers, J.; Nijman, H.W.; Daemen, T. Myeloid derived suppressor cells-an overview of combat strategies to increase immunotherapy efficacy. OncoImmunology 2015, 4, e954829. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of cd8+ t cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Nagaraj, S.; Gabrilovich, D.I. Tumor-associated cd8+ t cell tolerance induced by bone marrow-derived immature myeloid cells. J. Immunol. 2005, 175, 4583–4592. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+cd115+ immature myeloid suppressor cells mediate the development of tumor-induced t regulatory cells and t-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Harari, O.; Liao, J.K. Inhibition of mhc ii gene transcription by nitric oxide and antioxidants. Curr. Pharm. Des. 2004, 10, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Srivastava, S.; Kandpal, U.; Sade, H.; Lewis, V.; Sarin, A.; George, A.; Bal, V.; Durdik, J.M.; Rath, S. Inducible nitric oxide synthase in t cells regulates t cell death and immune memory. J. Clin. Investig. 2004, 113, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Hernandez, C.P.; Quiceno, D.; Dubinett, S.M.; Zabaleta, J.; Ochoa, J.B.; Gilbert, J.; Ochoa, A.C. Arginase i in myeloid suppressor cells is induced by cox-2 in lung carcinoma. J. Exp. Med. 2005, 202, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates t-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef]
- Modolell, M.; Choi, B.S.; Ryan, R.O.; Hancock, M.; Titus, R.G.; Abebe, T.; Hailu, A.; Muller, I.; Rogers, M.E.; Bangham, C.R.; et al. Local suppression of t cell responses by arginase-induced l-arginine depletion in nonhealing leishmaniasis. PLoS Negl. Trop. Dis. 2009, 3, e480. [Google Scholar] [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by cb-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Barosi, G. An immune dysregulation in mpn. Curr. Hematol. Malig. Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yao, Y.; Shen, Q.; Li, G.; Hu, L.; Zhang, X. Demethylating agent decitabine disrupts tumor-induced immune tolerance by depleting myeloid-derived suppressor cells. J. Cancer Res. Clin. Oncol. 2017, 143, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Verstovsek, S.; Cortes, J.E.; Tse, S.; Gasior, Y.; Jain, N.; Jabbour, E.J.; Estrov, Z.; Alvarado, Y.; DiNardo, C.D.; et al. A phase 1/2 study of ruxolitinib and decitabine in patients with post-myeloproliferative neoplasm acute myeloid leukemia. Leukemia 2020. [Google Scholar] [CrossRef]
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Rajapakshe, K.; Krieger, S.; Sun, B.; Mill, C.P.; DiNardo, C.; Pemmaraju, N.; Kadia, T.; et al. Bet protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary aml cells. Leukemia 2017, 31, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Tichelli, A.; Gratwohl, A.; Berger, C.; Lori, A.; Würsch, A.; Dieterle, A.; Thomssen, C.; Nissen, C.; Holdener, E.; Speck, B. Treatment of thrombocytosis in myeloproliferative disorders with interferon alpha-2a. Blut 1989, 58, 15–19. [Google Scholar] [CrossRef]
- Mullally, A.; Bruedigam, C.; Poveromo, L.; Heidel, F.H.; Purdon, A.; Vu, T.; Austin, R.; Heckl, D.; Breyfogle, L.J.; Kuhn, C.P.; et al. Depletion of jak2v617f myeloproliferative neoplasm-propagating stem cells by interferon-alpha in a murine model of polycythemia vera. Blood 2013, 121, 3692–3702. [Google Scholar] [CrossRef]
- Gowin, K.; Thapaliya, P.; Samuelson, J.; Harrison, C.; Radia, D.; Andreasson, B.; Mascarenhas, J.; Rambaldi, A.; Barbui, T.; Rea, C.J.; et al. Experience with pegylated interferon alpha-2a in advanced myeloproliferative neoplasms in an international cohort of 118 patients. Haematologica 2012, 97, 1570–1573. [Google Scholar] [CrossRef]
- Riley, C.H.; Brimnes, M.K.; Hansen, M.; Jensen, M.K.; Hasselbalch, H.C.; Kjaer, L.; Straten, P.T.; Svane, I.M. Interferon-alpha induces marked alterations in circulating regulatory t cells, nk cell subsets, and dendritic cells in patients with jak2v617f-positive essential thrombocythemia and polycythemia vera. Eur J. Haematol. 2016, 97, 83–92. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Cassinat, B.; Chevret, S.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Grandchamp, B.; Chomienne, C.; Fenaux, P. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008, 112, 3065–3072. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Cassinat, B.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Menot, M.L.; Massonnet, G.; Dutel, J.L.; Ghomari, K.; et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon alpha-2a. Blood 2006, 108, 2037–2040. [Google Scholar] [CrossRef]
- Ianotto, J.C.; Chauveau, A.; Boyer-Perrard, F.; Gyan, E.; Laribi, K.; Cony-Makhoul, P.; Demory, J.L.; de Renzis, B.; Dosquet, C.; Rey, J.; et al. Benefits and pitfalls of pegylated interferon-alpha2a therapy in patients with myeloproliferative neoplasm-associated myelofibrosis: A french intergroup of myeloproliferative neoplasms (fim) study. Haematologica 2018, 103, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Proietti, E.; Belardelli, F. Ifn-alpha and novel strategies of combination therapy for cancer. Ann. N. Y. Acad. Sci. 2007, 1112, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A. Interferon: The pathways of discovery i. Molecular and cellular aspects. Cytokine Growth Factor Rev. 2006, 17, 381–409. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Erickson, S.; Szeps, M.; Gruber, A.; Sangfelt, O.; Einhorn, S.; Pisa, P.; Grander, D. Interferon alpha down-regulates telomerase reverse transcriptase and telomerase activity in human malignant and nonmalignant hematopoietic cells. Blood 2000, 96, 4313–4318. [Google Scholar] [CrossRef]
- Riley, C.H.; Hansen, M.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M.; Jensen, M.K. Expansion of circulating cd56bright natural killer cells in patients with jak2-positive chronic myeloproliferative neoplasms during treatment with interferon-alpha. Eur J. Haematol. 2015, 94, 227–234. [Google Scholar] [CrossRef]
- Huangfu, W.C.; Qian, J.; Liu, C.; Liu, J.; Lokshin, A.E.; Baker, D.P.; Rui, H.; Fuchs, S.Y. Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 2012, 31, 161–172. [Google Scholar] [CrossRef]
- Di Bona, D.; Cippitelli, M.; Fionda, C.; Camma, C.; Licata, A.; Santoni, A.; Craxi, A. Oxidative stress inhibits ifn-alpha-induced antiviral gene expression by blocking the jak-stat pathway. J. Hepatol. 2006, 45, 271–279. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Holmstrom, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: Minimal residual disease and cure? Semin. Immunopathol. 2019, 41, 5–19. [Google Scholar] [CrossRef]
- Zeiser, R.; von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef]
- Bjorn, M.E.; de Stricker, K.; Kjaer, L.; Ellemann, K.; Hasselbalch, H.C. Combination therapy with interferon and jak1-2 inhibitor is feasible: Proof of concept with rapid reduction in jak2v617f-allele burden in polycythemia vera. Leuk Res. Rep. 2014, 3, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, S.U.; Kjaer, L.; Bjorn, M.E.; Knudsen, T.A.; Sorensen, A.L.; Andersen, C.B.L.; Bjerrum, O.W.; Brochmann, N.; Fassi, D.E.; Kruse, T.A.; et al. Safety and efficacy of combination therapy of interferon-alpha2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018, 7, 3571–3581. [Google Scholar] [CrossRef]
- Yacoub, A.; Mascarenhas, J.; Kosiorek, H.; Prchal, J.T.; Berenzon, D.; Baer, M.R.; Ritchie, E.; Silver, R.T.; Kessler, C.; Winton, E.; et al. Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood 2019, 134, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Pettit, K.; Odenike, O. Novel therapies for myelofibrosis. Curr. Hematol. Malig. Rep. 2017, 12, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Kvasnicka, H.M.; Thiele, J.; Bueso-Ramos, C.E.; Sun, W.; Cortes, J.; Kantarjian, H.M.; Verstovsek, S. Long-term effects of ruxolitinib versus best available therapy on bone marrow fibrosis in patients with myelofibrosis. J. Hematol. Oncol. 2018, 11, 42. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Verstovsek, S.; Guglielmelli, P.; Griesshammer, M.; Burn, T.C.; Naim, A.; Paranagama, D.; Marker, M.; Gadbaw, B.; Kiladjian, J.J. Ruxolitinib reduces jak2 p.V617f allele burden in patients with polycythemia vera enrolled in the response study. Ann. Hematol. 2017, 96, 1113–1120. [Google Scholar] [CrossRef]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on jak2p.V617f allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef]
- Greenfield, G.; McPherson, S.; Mills, K.; McMullin, M.F. The ruxolitinib effect: Understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef]
- Bjorn, M.E.; Hasselbalch, H.C. The impact of ruxolitinib treatment on inflammation-mediated comorbidities in myelofibrosis and related neoplasms. Clin. Case Rep. 2015, 3, 499–503. [Google Scholar] [CrossRef]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: A randomized clinical trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Heine, A.; Held, S.A.; Daecke, S.N.; Wallner, S.; Yajnanarayana, S.P.; Kurts, C.; Wolf, D.; Brossart, P. The jak-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013, 122, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.; Heine, A.; Quast, T.; Kolanus, W.; Trebicka, J.; Brossart, P.; Wolf, D. The jak inhibitor ruxolitinib impairs dendritic cell migration via off-target inhibition of rock. Leukemia 2016, 30, 2119–2123. [Google Scholar] [CrossRef]
- Schonberg, K.; Rudolph, J.; Vonnahme, M.; Parampalli Yajnanarayana, S.; Cornez, I.; Hejazi, M.; Manser, A.R.; Uhrberg, M.; Verbeek, W.; Koschmieder, S.; et al. Jak inhibition impairs nk cell function in myeloproliferative neoplasms. Cancer Res. 2015, 75, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Parampalli Yajnanarayana, S.; Stubig, T.; Cornez, I.; Alchalby, H.; Schonberg, K.; Rudolph, J.; Triviai, I.; Wolschke, C.; Heine, A.; Brossart, P.; et al. Jak1/2 inhibition impairs t cell function in vitro and in patients with myeloproliferative neoplasms. Br. J. Haematol. 2015, 169, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Massa, M.; Rosti, V.; Campanelli, R.; Fois, G.; Barosi, G. Rapid and long-lasting decrease of t-regulatory cells in patients with myelofibrosis treated with ruxolitinib. Leukemia 2014, 28, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Wysham, N.G.; Sullivan, D.R.; Allada, G. An opportunistic infection associated with ruxolitinib, a novel janus kinase 1,2 inhibitor. Chest 2013, 143, 1478–1479. [Google Scholar] [CrossRef]
- Caocci, G.; Murgia, F.; Podda, L.; Solinas, A.; Atzeni, S.; La Nasa, G. Reactivation of hepatitis b virus infection following ruxolitinib treatment in a patient with myelofibrosis. Leukemia 2014, 28, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Wathes, R.; Moule, S.; Milojkovic, D. Progressive multifocal leukoencephalopathy associated with ruxolitinib. N. Engl. J. Med. 2013, 369, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. Cd123 as a therapeutic target in the treatment of hematological malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef] [PubMed]
- Lasho, T.; Finke, C.; Kimlinger, T.K.; Zblewski, D.; Chen, D.; Patnaik, M.M.; Hanson, C.A.; Brooks, C.; Tefferi, A.; Pardanani, A. Expression of cd123 (il-3r-alpha), a therapeutic target of sl-401, on myeloproliferative neoplasms. Blood 2014, 124. [Google Scholar] [CrossRef]
- Broughton, S.E.; Dhagat, U.; Hercus, T.R.; Nero, T.L.; Grimbaldeston, M.A.; Bonder, C.S.; Lopez, A.F.; Parker, M.W. The gm-csf/il-3/il-5 cytokine receptor family: From ligand recognition to initiation of signaling. Immunol. Rev. 2012, 250, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Frankel, A. Cd 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark. Res. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Gupta, V.; Ali, H.; Yacoub, A.; Wang, E.S.; Lee, S.; Schiller, G.J.; Sardone, M.; Wysowskyj, H.; Chen, J.; et al. Results from a phase 1/2 clinical trial of tagraxofusp (sl-401) in patients with intermediate, or high risk, relapsed/refractory myelofibrosis. Blood 2019, 134, 558. [Google Scholar] [CrossRef]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevated expression of il-3ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef]
- Alkharabsheh, O.; Frankel, A.E. Clinical activity and tolerability of sl-401 (tagraxofusp): Recombinant diphtheria toxin and interleukin-3 in hematologic malignancies. Biomedicines 2019, 7, 6. [Google Scholar] [CrossRef]
- Hall, P.D.; Willingham, M.C.; Kreitman, R.J.; Frankel, A.E. Dt388-gm-csf, a novel fusion toxin consisting of a truncated diphtheria toxin fused to human granulocyte-macrophage colony-stimulating factor, prolongs host survival in a Sci.d mouse model of acute myeloid leukemia. Leukemia 1999, 13, 629–633. [Google Scholar] [CrossRef]
- Frankel, A.; Liu, J.S.; Rizzieri, D.; Hogge, D. Phase i clinical study of diphtheria toxin-interleukin 3 fusion protein in patients with acute myeloid leukemia and myelodysplasia. Leuk. Lymphoma 2008, 49, 543–553. [Google Scholar] [CrossRef]
- Black, J.H.; McCubrey, J.A.; Willingham, M.C.; Ramage, J.; Hogge, D.E.; Frankel, A.E. Diphtheria toxin-interleukin-3 fusion protein (dt(388)il3) prolongs disease-free survival of leukemic immunocompromised mice. Leukemia 2003, 17, 155–159. [Google Scholar] [CrossRef]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A.; et al. Monoclonal antibody-mediated targeting of cd123, il-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 2009, 5, 31–42. [Google Scholar] [CrossRef]
- Nievergall, E.; Ramshaw, H.S.; Yong, A.S.; Biondo, M.; Busfield, S.J.; Vairo, G.; Lopez, A.F.; Hughes, T.P.; White, D.L.; Hiwase, D.K. Monoclonal antibody targeting of il-3 receptor alpha with csl362 effectively depletes cml progenitor and stem cells. Blood 2014, 123, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Busfield, S.J.; Biondo, M.; Wong, M.; Ramshaw, H.S.; Lee, E.M.; Ghosh, S.; Braley, H.; Panousis, C.; Roberts, A.W.; He, S.Z.; et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-cd123 mab engineered for optimal adcc. Leukemia 2014, 28, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Roboz, G.J.; Walter, R.B.; Altman, J.K.; Ferguson, A.; Curcio, T.J.; Orlowski, K.F.; Garrett, L.; Busfield, S.J.; Barnden, M.; et al. First-in man, phase 1 study of csl362 (anti-il3r alpha/anti-cd123 monoclonal antibody) in patients with cd123+acute myeloid leukemia (aml) in cr at high risk for early relapse. Blood 2014, 124. [Google Scholar] [CrossRef]
- Smith, B.D.; Roberts, A.W.; Roboz, G.J.; DeWitte, M.; Ferguson, A.; Garrett, L.; Curcio, T.; Orlowski, K.F.; Dasen, S.; Bensen-Kennedy, D.M.; et al. Minimal residual disease (mrd) as exploratory endpoint in a phase 1 study of the anti-cd123 mab csl362 given as post-remission therapy in adult acute myeloid leukemia (aml). Blood 2015, 126. [Google Scholar] [CrossRef]
- Syed, K.; Pietsch, C.; Axel, A.; Forslund, A.; Sasser, K.; Salvati, M. Preclinical evaluation of csl362/jnj-56022473 in combination with decitabine or azacitidine in in vitro assays. Blood 2015, 126, 3819. [Google Scholar] [CrossRef]
- Li, F.; Sutherland, M.K.; Yu, C.; Walter, R.B.; Westendorf, L.; Valliere-Douglass, J.; Pan, L.; Cronkite, A.; Sussman, D.; Klussman, K.; et al. Characterization of sgn-cd123a, a potent cd123-directed antibody-drug conjugate for acute myeloid leukemia. Mol. Cancer Ther. 2018, 17, 554–564. [Google Scholar] [CrossRef]
- Akiyama, T.; Takayanagi, S.; Maekawa, Y.; Miyawaki, K.; Jinnouchi, F.; Jiromaru, T.; Sugio, T.; Daitoku, S.; Kusumoto, H.; Shimabe, M.; et al. First preclinical report of the efficacy and pd results of khk2823, a non-fucosylated fully human monoclonal antibody against il-3r alpha. Blood 2015, 126, 1349. [Google Scholar] [CrossRef]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A cd123-targeting antibody-drug conjugate, imgn632, designed to eradicate aml while sparing normal bone marrow cells. Blood Adv. 2018, 2, 848–858. [Google Scholar] [CrossRef]
- Holmstrom, M.O.; Riley, C.H.; Skov, V.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. Spontaneous t-cell responses against the immune check point programmed-death-ligand 1 (pd-l1) in patients with chronic myeloproliferative neoplasms correlate with disease stage and clinical response. OncoImmunology 2018, 7, e1433521. [Google Scholar] [CrossRef]
- Holmström, M.O.; Hjortsø, M.D.; Ahmad, S.M.; Met, Ö.; Martinenaite, E.; Riley, C.; Straten, P.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H.; et al. The jak2v617f mutation is a target for specific t-cells in the jak2v617f positive myeloproliferative neoplasms. Leukemia 2017, 31, 495–498. [Google Scholar]
- Holmström, M.O.; Martinenaite, E.; Ahmad, S.M.; Met, Ö.; Friese, C.; Kjær, L.; Riley, C.H.; Thor Straten, P.; Svane, I.M.; Hasselbalch, H.C.; et al. The calreticulin (calr) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 2018, 32, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Schischlik, F.; Jager, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Milosevic Feenstra, J.D.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, M.O.; Hasselbalch, H.C.; Andersen, M.H. The jak2v617f and calr exon 9 mutations are shared immunogenic neoantigens in hematological malignancy. OncoImmunology 2017, 6, e1358334. [Google Scholar] [CrossRef] [PubMed]
- Pecquet, C.; Balligand, T.; Chachoua, I.; Roy, A.; Vertenoeil, G.; Colau, D.; Fertig, E.; Marty, C.; Nivarthi, H.; Defour, J.P.; et al. Secreted mutant calreticulins as rogue cytokines trigger thrombopoietin receptor activation specifically in calr mutated cells: Perspectives for mpn therapy. Blood 2018, 132. [Google Scholar] [CrossRef]
- Jorgensen, M.A.; Holmstrom, M.O.; Martinenaite, E.; Riley, C.H.; Hasselbalch, H.C.; Andersen, M.H. Spontaneous t-cell responses against arginase-1 in the chronic myeloproliferative neoplasms relative to disease stage and type of driver mutation. OncoImmunology 2018, 7, e1468957. [Google Scholar] [CrossRef] [PubMed]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharm. 2009, 158, 638–651. [Google Scholar] [CrossRef]
- Munder, M.; Mollinedo, F.; Calafat, J.; Canchado, J.; Gil-Lamaignere, C.; Fuentes, J.M.; Luckner, C.; Doschko, G.; Soler, G.; Eichmann, K.; et al. Arginase i is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood 2005, 105, 2549–2556. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant pd-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Zamani, M.R.; Pourvahedi, M.; Golchehre, Z.; Hosseini Bereshneh, A.; Rezaei, N. Cancer/testis antigens: Expression, regulation, tumor invasion, and use in immunotherapy of cancers. Immunol. Invest. 2016, 45, 619–640. [Google Scholar] [CrossRef]
- Almstedt, M.; Blagitko-Dorfs, N.; Duque-Afonso, J.; Karbach, J.; Pfeifer, D.; Jager, E.; Lubbert, M. The DNA demethylating agent 5-aza-2’-deoxycytidine induces expression of ny-eso-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk. Res. 2010, 34, 899–905. [Google Scholar] [CrossRef]
- Liang, G.; Gonzales, F.A.; Jones, P.A.; Orntoft, T.F.; Thykjaer, T. Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2’-deoxycytidine. Cancer Res. 2002, 62, 961–966. [Google Scholar] [PubMed]
- Karpf, A.R.; Lasek, A.W.; Ririe, T.O.; Hanks, A.N.; Grossman, D.; Jones, D.A. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine. Mol. Pharm. 2004, 65, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Siebenkas, C.; Chiappinelli, K.B.; Guzzetta, A.A.; Sharma, A.; Jeschke, J.; Vatapalli, R.; Baylin, S.B.; Ahuja, N. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE 2017, 12, e0179501. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Paluch, B.E.; Matsuzaki, J.; James, S.R.; Collamat-Lai, G.; Blagitko-Dorfs, N.; Ford, L.A.; Naqash, R.; Lubbert, M.; Karpf, A.R.; et al. Induction of cancer testis antigen expression in circulating acute myeloid leukemia blasts following hypomethylating agent monotherapy. Oncotarget 2016, 7, 12840–12856. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, O.; Agathanggelou, A.; Novitzky-Basso, I.; Siddique, S.; McSkeane, T.; Ryan, G.; Vyas, P.; Cavenagh, J.; Stankovic, T.; Moss, P.; et al. Induction of a cd8+ t-cell response to the mage cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood 2010, 116, 1908–1918. [Google Scholar] [CrossRef]
- Krishnadas, D.K.; Shusterman, S.; Bai, F.; Diller, L.; Sullivan, J.E.; Cheerva, A.C.; George, R.E.; Lucas, K.G. A phase i trial combining decitabine/dendritic cell vaccine targeting mage-a1, mage-a3 and ny-eso-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol. Immunother. 2015, 64, 1251–1260. [Google Scholar] [CrossRef]
- Maslak, P.G.; Dao, T.; Bernal, Y.; Chanel, S.M.; Zhang, R.; Frattini, M.; Rosenblat, T.; Jurcic, J.G.; Brentjens, R.J.; Arcila, M.E.; et al. Phase 2 trial of a multivalent wt1 peptide vaccine (galinpepimut-s) in acute myeloid leukemia. Blood Adv. 2018, 2, 224–234. [Google Scholar] [CrossRef]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of pd-l1, pd-l2, pd-1 and ctla4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Orskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Gronbaek, K. Hypomethylation and up-regulation of pd-1 in t cells by azacytidine in mds/aml patients: A rationale for combined targeting of pd-1 and DNA methylation. Oncotarget 2015, 6, 9612–9626. [Google Scholar] [CrossRef]
- Cimen Bozkus, C.; Roudko, V.; Finnigan, J.P.; Mascarenhas, J.; Hoffman, R.; Iancu-Rubin, C.; Bhardwaj, N. Immune checkpoint blockade enhances shared neoantigen-induced t-cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov. 2019, 9, 1192–1207. [Google Scholar] [CrossRef]
- Bozkus, C.C.; Finnigan, J.P.; Mascarenhas, J.; Hoffman, R.; Bhardwaj, N.; Iancu-Rubin, C. Immune checkpoint blockade enhances mutated calreticulin-induced t cell immunity in myeloproliferative neoplasms. Blood 2017, 130. [Google Scholar]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic jak2(v617f) causes pd-l1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. Pd-l2 is a second ligand for pd-1 and inhibits t cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, F.; Yao, S.; Shin, T.; Flies, A.; Flies, S.; Xu, H.; Tamada, K.; Pardoll, D.M.; Chen, L. Interaction between b7-h1 and pd-1 determines initiation and reversal of t-cell anergy. Blood 2007, 110, 180–185. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit t cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Prestipino, A.; Zeiser, R. Clinical implications of tumor-intrinsic mechanisms regulating pd-l1. Sci. Transl. Med. 2019, 11, 478. [Google Scholar] [CrossRef]
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. Ctla4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef]
- Kadia, T.M.; Cortes, J.E.; Ghorab, A.; Ravandi, F.; Jabbour, E.; Daver, N.G. Nivolumab (nivo) maintenance (maint) in high-risk (hr) acute myeloid leukemia (aml) patients. J. Clin. Oncol. 2018, 36, 7014. [Google Scholar] [CrossRef]
- Daver, N.G.; Garcia-Manero, G.; Basu, S.; Cortes, J.E.; Ravandi, F.; Kadia, T.M.; Konopleva, M.Y.; Jabbour, E.J.; DiNardo, C.D.; Assi, R.; et al. Safety, efficacy, and biomarkers of response to azacitidine (aza) with nivolumab (nivo) and aza with nivo and ipilimumab (ipi) in relapsed/refractory acute myeloid leukemia: A non-randomized, phase 2 study. Blood 2018, 132, 906. [Google Scholar] [CrossRef]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson, P.A.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: A single-arm, phase 2 study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef]
- Cottin, L.; Riou, J.; Boyer, F.; Bouvier, A.; Zannetti, A.; Blouet, A.; Truchan-Graczyk, M.; Jouanneau-Courville, R.; Beucher, A.; Ribourtout, B.; et al. Wt1 gene is overexpressed in myeloproliferative neoplasms, especially in myelofibrosis. Blood Cells Mol. Dis. 2019, 75, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, S.; Sener, E.F.; Akalin, H.; Keklik, M.; Kaynar, L.; Ozkul, Y. Does the level of wt1 expression predict the outcome in philadelphia-negative myeloproliferative neoplasms? Genet. Test. Mol. Biomark. 2015, 19, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Sohn, H.J.; Hong, J.A.; Lee, H.J.; Sohn, D.H.; Shin, C.A.; Cho, H.I.; Min, W.S.; Kim, T.G. Post-transplant immunotherapy with wt1-specific ctls for high-risk acute myelogenous leukemia: A prospective clinical phase i/ii trial. Bone Marrow Transpl. 2019, 54, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Sugiyama, H.; Ogawa, H.; Nakagawa, M.; Yamagami, T.; Miwa, H.; Kita, K.; Hiraoka, A.; Masaoka, T.; Nasu, K.; et al. Wt1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood 1994, 84, 3071–3079. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Kim, H.J.; Kwak, D.H.; Park, S.S.; Jeon, Y.W.; Lee, S.E.; Cho, B.S.; Eom, K.S.; Kim, Y.J.; Lee, S.; et al. High wt1 expression is an early predictor for relapse in patients with acute promyelocytic leukemia in first remission with negative pml-rara after anthracycline-based chemotherapy: A single-center cohort study. J. Hematol. Oncol. 2017, 10, 30. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting wt1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Ragnarsson, G.B.; Nguyen, H.N.; Chaney, C.N.; Pufnock, J.S.; Schmitt, T.M.; Duerkopp, N.; Roberts, I.M.; Pogosov, G.L.; Ho, W.Y.; et al. Transferred wt1-reactive cd8+ t cells can mediate antileukemic activity and persist in post-transplant patients. Sci. Transl. Med. 2013, 5, 174ra127. [Google Scholar] [CrossRef]
- Rezvani, K.; Yong, A.S.; Savani, B.N.; Mielke, S.; Keyvanfar, K.; Gostick, E.; Price, D.A.; Douek, D.C.; Barrett, A.J. Graft-versus-leukemia effects associated with detectable wilms tumor-1 specific t lymphocytes after allogeneic stem-cell transplantation for acute lymphoblastic leukemia. Blood 2007, 110, 1924–1932. [Google Scholar] [CrossRef]
- Mailander, V.; Scheibenbogen, C.; Thiel, E.; Letsch, A.; Blau, I.W.; Keilholz, U. Complete remission in a patient with recurrent acute myeloid leukemia induced by vaccination with wt1 peptide in the absence of hematological or renal toxicity. Leukemia 2004, 18, 165–166. [Google Scholar] [CrossRef]


{kind=link}
| Trial | Treatment | Diagnosis | Outcome Measures | Status, Response, Comments |
|---|---|---|---|---|
| NCT02910258 | Pegylated interferon-α2a | PMF, SMF | Primary: ORR; Secondary: OS, AE | Completed; observational study; IFNα2a might improve OS, and leukemia free survival; IFNα2a increased risk of GvHD if given before allo-HSCT |
| NCT00241241 (Phase II) | Pegylated interferon-α2a | PV; previously untreated patients or treated with phlebotomy or HU | Primary: ORR; Secondary: safety, molecular response | Completed; Decrease of JAK2 allele frequency in 89% of patients; peg-IFN-α2a targets mutant clone; molecular CR in 7 patients; low toxicity |
| NCT02742324 (Phase I/II) | Ruxolitinib; Pegylated interferon-α2a | PMF, SMF | Primary: safety, DLT; Secondary: Molecular response | Recruiting; No DLT, well-tolerated combination therapy; decreased spleen size and JAK2 allele burden; improvement in blood counts; 63% with complete hematological response; decrease of other driver mutations |
| NCT01259856 (Phase III) | PEGASYS (peg-IFN-α2a); Hydroxyurea; Aspirin | High-risk PV and ET | Primary: CR, PR; Secondary: AE, change in TSS, JAK2 allele burden, disease progression, death | Completed; ORR at 12 months was 69.2% (ET) and 60% (PV); CR was higher in CALR-mutant ET compared to CALR non-mutant; significant rate of AE (manageable); PEG for patients being intolerant or resistant to HU |
| Trial | Treatment | Diagnosis | Outcome Measures | Status, Response, Comments |
|---|---|---|---|---|
| NCT02268253 (Phase II) | Tagraxofusp (SL-401; CD123-directed cytotoxin) | R/R MF, advanced MF, high-risk MF, CMML | Primary: AE, ORR | Recruiting; Tagraxofusp has single-agent activity; well tolerated |
| NCT04317781 (Phase II) | Tagraxofusp (CD123-directed cytotoxin) | BPDCN after autologous or allogeneic HSCT | Primary: AE; Secondary: PFS, OS | Recruiting |
| NCT04342962 (Phase II) | Tagraxofusp (CD123-directed cytotoxin) | R/R CD123+ AML, BPDCN-IF | Primary: ORR; Secondary: AE, OS, EFS, | Not yet recruiting (estimated July 2020) |
| NCT01632852 (Phase I) | CSL362 (Anti-IL3Rα/Anti-CD123 Monoclonal Antibody) | CD123+ AML in CR or CR with incomplete platelet recovery at high risk of early relapse | Primary: AE, DLT; Secondary: PK, immunogenicity | Completed; Study to generate dose and dosing schedule |
| NCT02472145 (Phase II/III) | Decitabine (HMA); Talacotuzumab (anti-CD123) | R/R AML, de novo AML, patients not eligible for curative therapy | Primary: CRR, OS; Secondary: EFS, ORR, DOR, CR | Completed; Lack of efficacy and high toxicity of combination therapy |
| NCT02848248 (Phase I) | SGN-CD123A (anti-CD123 ADC) | R/R AML | Primary: AE, DLT, LA; Secondary: PK, immunogenicity, OS, ORR | Terminated |
| NCT02181699 (Phase I) | KHK2823 (anti-CD123) | R/R AML, R/R MDS, patients not eligible for curative therapy | Primary: AE; Secondary: PK, ORR, OS, EFS, RFS, PFS, DFS, immunogenicity, PD | Terminated; Failed treatment response |
| NCT03386513 (Phase I/II) | IMGN632 (anti-CD123, DGN549 ADC) | R/R AML, R/R BPDCN, R/R ALL, high-risk MDS, MPN, CMML | Primary: MTD, RP2D; Secondary: AE, ORR, PK, Immunogenicity | Recruiting; Objective responses in one third of patients during initial dose escalation; no DLT |
| Trial | Treatment | Diagnosis | Outcome Measures | Status, Response, Comments |
|---|---|---|---|---|
| NCT03566446 (Phase I) | CALRLong36 peptide (Ex 9 mut) vaccine | CALR-mutant MPN (ET, PMF, MPN unclassifiable) | Primary: AE; Secondary: Immune response (T-cell cytokine release), mutation status, ORR | Active; No trial results posted yet; CALRLong36 peptide did show prompt responses in vitro |
| NCT01266083 (Phase II) | WT1 peptide vaccine | AML, ALL; patients being in CR; patients with WT1+ disease | Primary: AE, OS; Secondary: DFS, Immunol. ogic response, effects on MRD, OS | Completed; Vaccine was well tolerated; AEs: injection site reaction, fatigue, skin induration; vaccine-stimulated specific immune response |
| NCT02750995 (Phase I) | NPMW-peptide vaccine (against long peptide sequences from NY-ESO-1, PRAME, MAGE-A3, WT-1); Azacitidine | High-risk MDS, AML (<30% blasts) | Primary: AE; Secondary: specific T-cell reactivity, ORR | Recruiting |
| NCT03358719 (Phase I) | DEC-205/NY-ESO-1 Fusion Protein CDX-1401; Decitabine; Nivolumab | AML (<30% blasts), MDS, high-risk MDS, CMML, refractory anemia | Primary: AE; Secondary: immune profile, PB and BM response, CRR, PRR | Active; final data collection for primary outcome measure |
| Trial | Treatment | Diagnosis | Outcome Measures | Status, Response, Comments |
|---|---|---|---|---|
| NCT02421354 (Phase II) | Nivolumab (anti-PD-1) | Hepatomegaly, MF transformation in ET, PV, PMF, Splenomegaly | Primary: efficacy in MF; Secondary: AE; Tertiary: time to response, symptom burden, BM fibrosis, JAK2 allele burden | Terminated; 8 patients enrolled; terminated due to serious AE (75%) and other AE (87.5%) |
| NCT02871323 (Phase I) | Durvalumab (anti-PD-L1) | PMF, PV | Primary: AE; Secondary: MF symptom burden, response in PB and BM, cytokine profile | Withdrawn before enrollment of patients |
| NCT03065400 (Phase II) | Pembrolizumab (anti-PD-1) | Chronic phase MF, PMF, post-ET MF, PV, MPN-AP/BP | Primary: clinical improvement; Secondary: MPN-AP/BP patients that achieve complete morphologic remission of blasts | Completed; No results posted yet |
| NCT01822509 (Phase I/Ib) | Ipilimumab (anti-CTLA-4), Nivolumab (anti-PD-1) | Patients relapsed from hematologic malignancies after HSCT (MPN, ALL, AML, CLL, CML, MDS, Hodgkin lymphoma, Non-Hodgkin lymphoma) | Primary: MTD, DLT, AE; Secondary: clinical response, PFS, OS, immune cell numbers, cytokine production | Active; 21% AE; 14% Ipilimumab discontinuation (GvHD); 23% CR, 9% PR, 27% decreased tumor burden; infiltration of CD8 T-cells, decreased Treg activation; Nivolumab: 23% PFS, 56% OS; severe AE and GvHD |
| NCT00060372 (Phase I) | Ipilimumab (anti-CTLA-4); Donor lymphocytes | Patients with persistent or progressive cancer after allogeneic stem-cell transplant | Primary: incidence of aGvHD, graft rejection, immune reaction; Secondary: cGvHD, DFS, OS, ORR, T-cell activation | Completed; Induction of graft-versus-tumor effects after HSCT; Ipilimumab safe to use and causes anti-tumor response |
| NCT02768792 (Phase II) | Cytarabine (HiDAC); Pembrolizumab (anti-PD-1) | R/R AML | Primary: CR; Secondary: AE, PR, CR, RFS, PFS, OS | Active; 46% ORR, 38% CR/CRi, OS 8.9 months, DFS 5.7 months |
| NCT02532231 (Phase II) | Nivolumab (anti-PD-1) | AML in remission at high risk of relapse | Primary: recurrence-free survival; Secondary: Immunol. ogic response, OS, AE | Recruiting; OS 86% (12 months) and 67% (18 months); therapy well tolerated; detectable MRD while on therapy |
| NCT02397720 (Phase II) | Azacitidine, Ipilimumab (anti-CTLA-4), Nivolumab (anti-PD-1) | R/R AML, newly diagnosed AML | Primary: MTD, ORR, AE; Secondary: DFS, OS, PFS | Recruiting; 21% CR/CRi, 26% BM blast reduction (>50%), 23% disease progression; 12–14% AE; pts with CR: higher CD8 Tc infiltration into BM; well tolerated |
| NCT02464657 (Phase I/II) | Idarubicin, Cytarabine, Nivolumab (anti-PD-1), Solu-medrol, Dexamethasone | High-risk MDS, AML | Primary: MTD; Secondary: EFS | Active; Addition of Nivolumab to chemotherapy feasible in AML or high-risk MDS patients; GvHD needs to be improved |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, L.M.; Zeiser, R. Immunotherapy in Myeloproliferative Diseases. Cells 2020, 9, 1559. https://doi.org/10.3390/cells9061559
Braun LM, Zeiser R. Immunotherapy in Myeloproliferative Diseases. Cells. 2020; 9(6):1559. https://doi.org/10.3390/cells9061559
Chicago/Turabian StyleBraun, Lukas M., and Robert Zeiser. 2020. "Immunotherapy in Myeloproliferative Diseases" Cells 9, no. 6: 1559. https://doi.org/10.3390/cells9061559
APA StyleBraun, L. M., & Zeiser, R. (2020). Immunotherapy in Myeloproliferative Diseases. Cells, 9(6), 1559. https://doi.org/10.3390/cells9061559