Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT) beyond SMARCA4 Mutations: A Comprehensive Genomic Analysis

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. DNA Extraction
2.3. Whole Exome Sequencing (WES)
2.4. Oligonucleotide CGH Microarrays
2.5. SMARCA2 Promoter Sequencing
2.6. RNA Sequencing (RNA-Seq), Real-Time RT-PCR and Differential Expression Analysis
2.7. Immunohistochemistry
2.8. Cell Culture and Viability Assays
3. Results
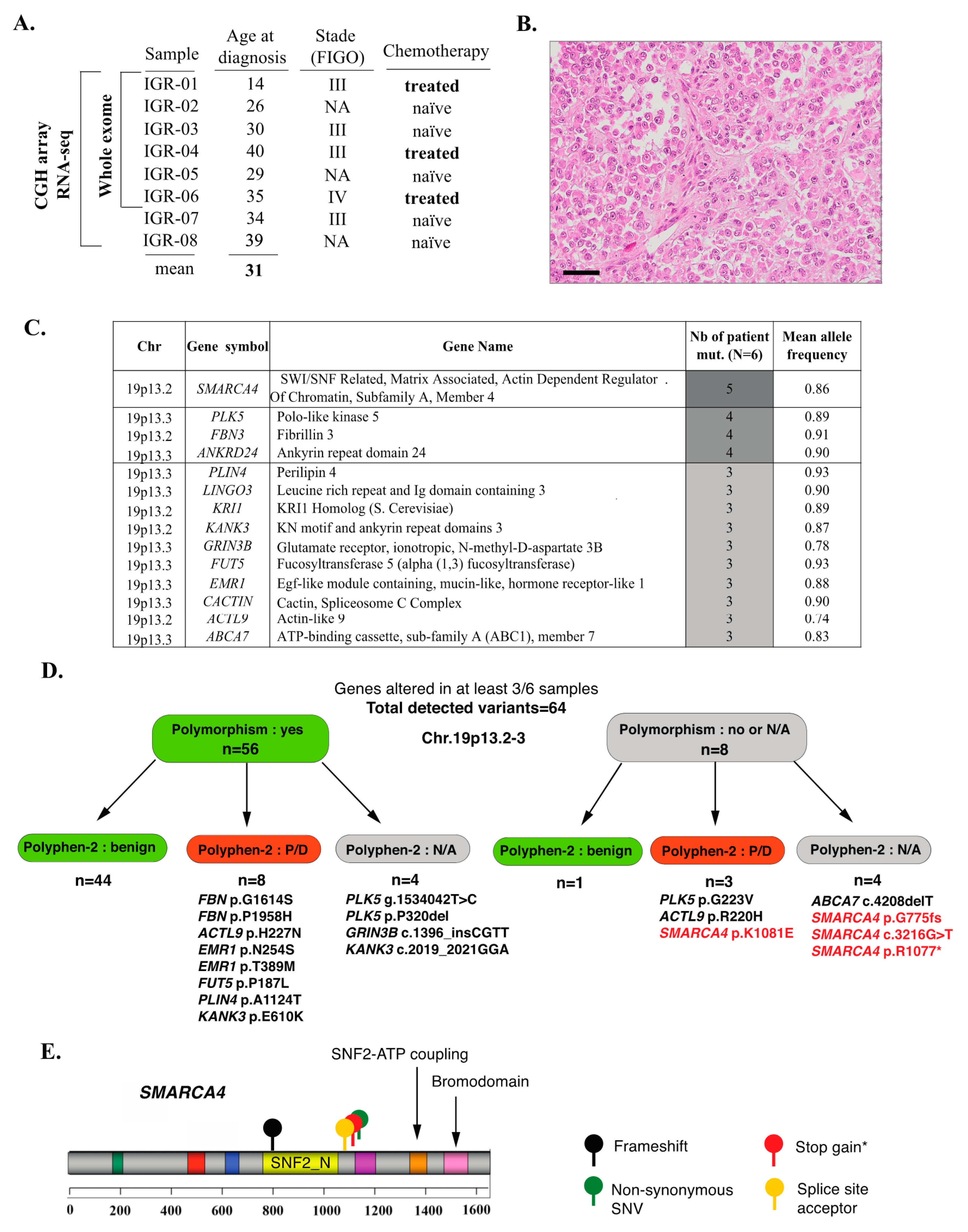
3.1. Clinical Data and Mutational Profiles of SCCOHT: A General Overview
3.2. Inactivating SMARCA4 Mutations and Related Findings
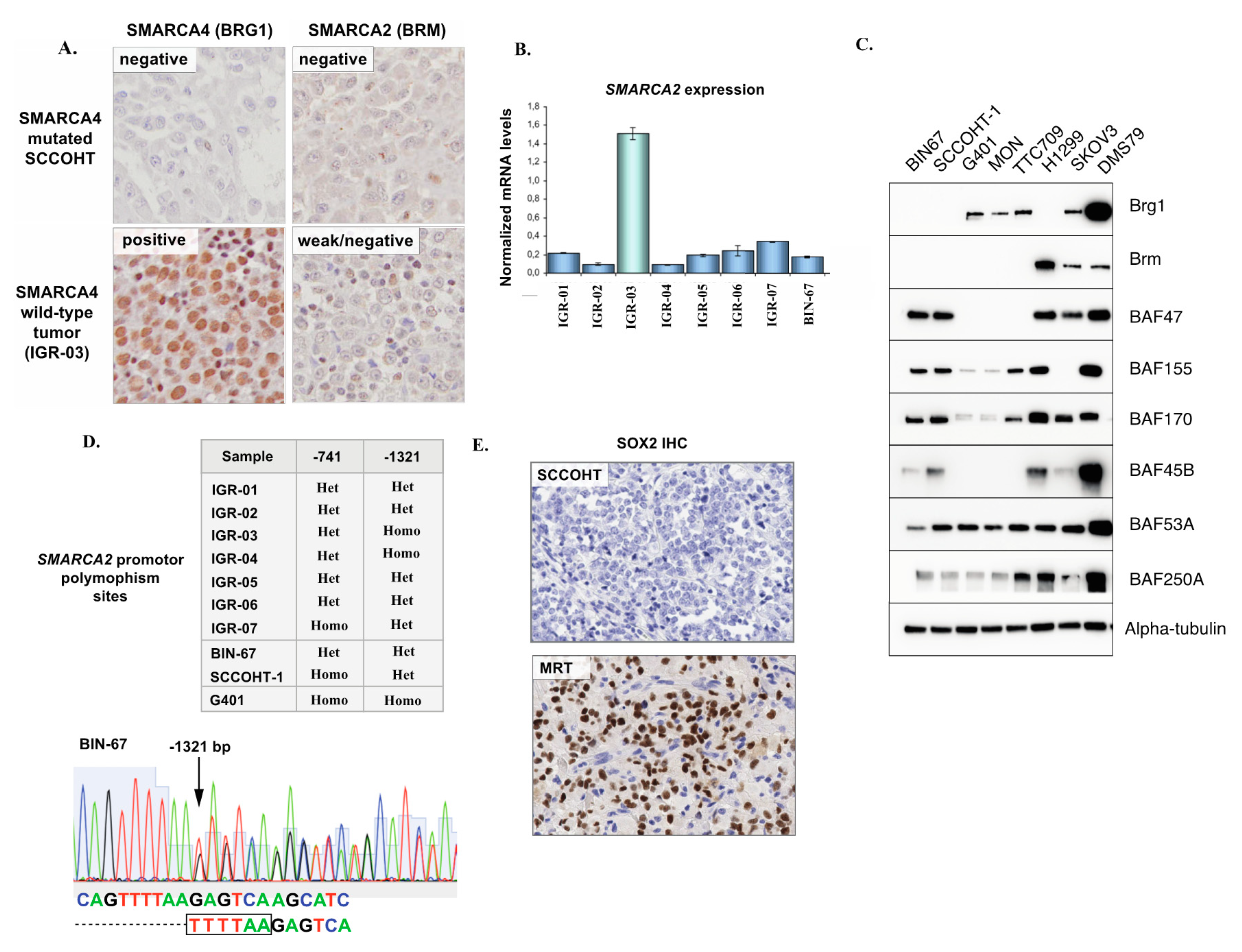
3.3. SMARCA2 Loss of Expression in SCCOHT
3.4. Validation of the p.G223V PLK5 Variant in a Larger Series of SCCOHT Samples
3.5. Somatic Copy Number Alterations (SCNAs) in SCCOHT
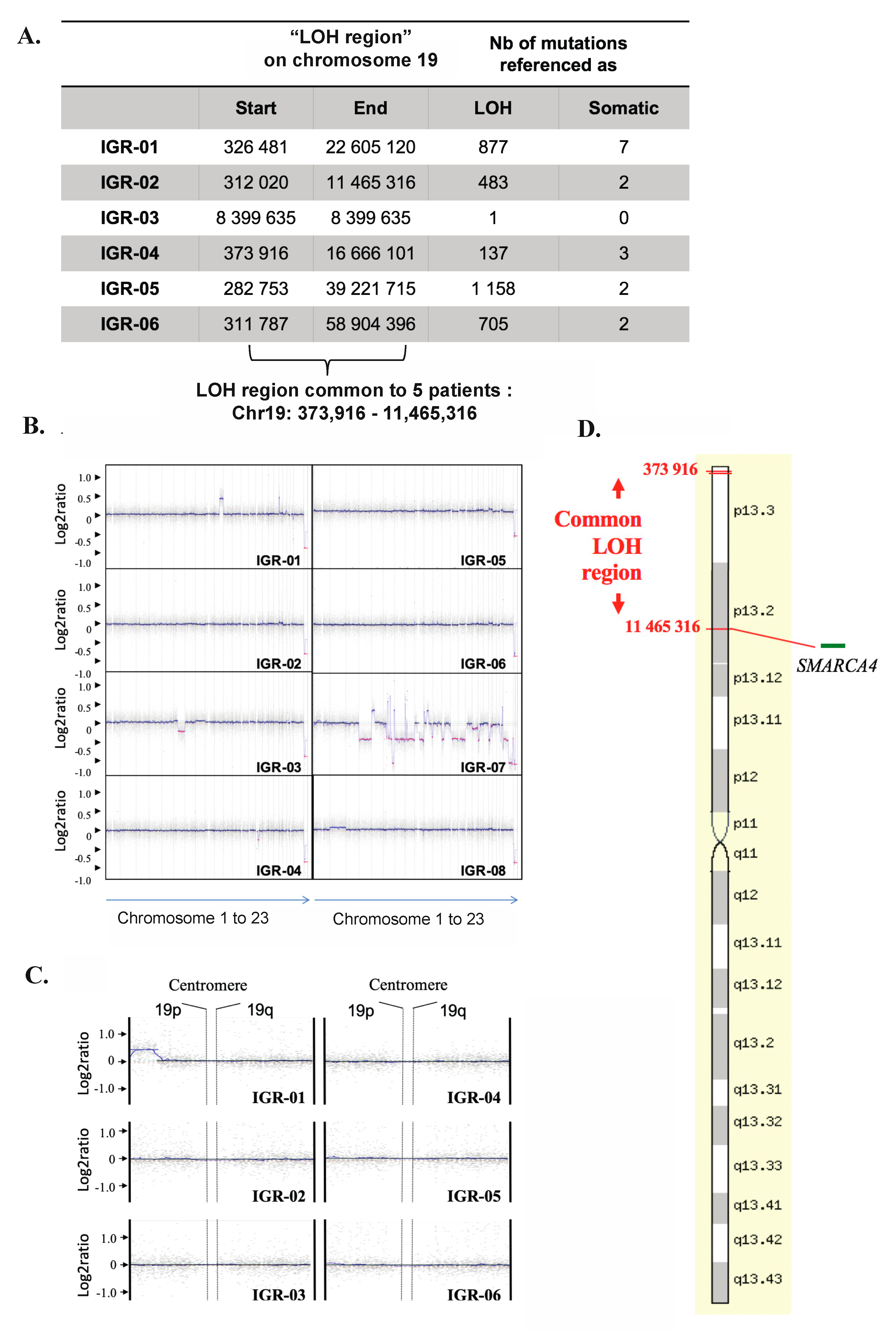
3.6. Copy-Neutral Loss-of-Heterozygosity (CN-LOH) at the 19p13.2-3 Locus
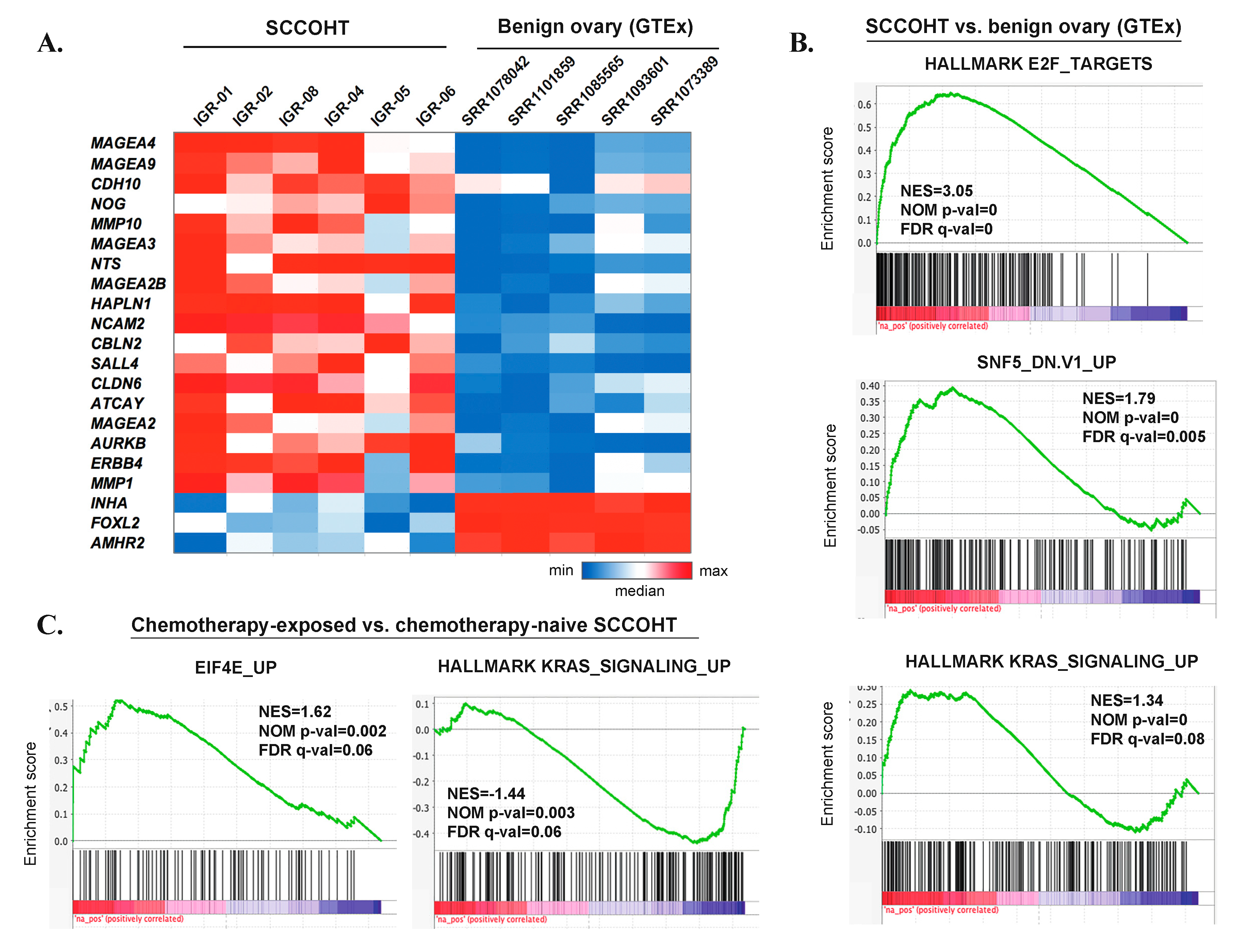
3.7. Gene Expression Profiles of SCCOHT
3.8. Genomic and Transcriptomic Profiles of Chemotherapy-Naïve Versus Chemotherapy-Exposed SCCOHT
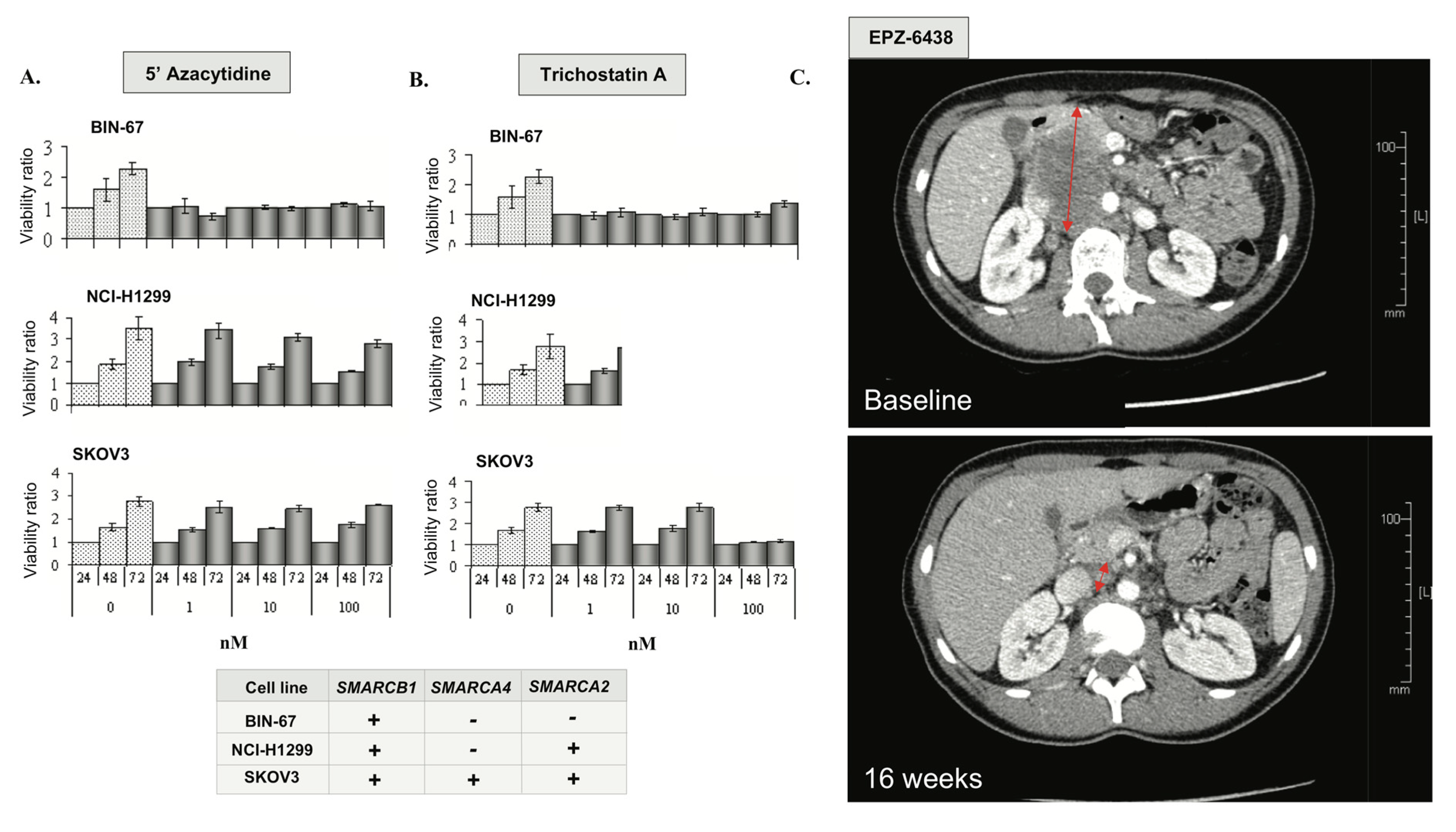
3.9. Epigenetic Vulnerabilities of SCCOHT Associated with SWI/SNF Deregulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Young, R.H.; Oliva, E.; Scully, R.E. Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases. Am. J. Surg. Pathol. 1994, 18, 1102–1116. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G. Ovarian neoplasms composed of small round cells: A review. Adv. Anat. Pathol. 2004, 11, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Kupryjańczyk, J.; Dansonka-Mieszkowska, A.; Moes-Sosnowska, J.; Plisiecka-Hałasa, J.; Szafron, Ł.; Podgórska, A.; Rzepecka, I.K.; Konopka, B.; Budziłowska, A.; Rembiszewska, A.; et al. Ovarian small cell carcinoma of hypercalcemic type—Evidence of germline origin and smarca4 gene inactivation. A pilot study. Pol. J. Pathol. 2013, 4, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Barondeau, J.; Rodgers, M.; Braun, L.; Azarow, K.; Forouhar, M.; Faucette, K. Small cell ovarian carcinoma: A rare, aggressive tumor masquerading as constipation in a teenager with a fatal outcome. J. Pediatr. Hematol. Oncol. 2010, 32, e139–e141. [Google Scholar] [CrossRef]
- Pautier, P.; Ribrag, V.; Duvillard, P. Results of a prospective dose-intensive regimen in 27 patients with small cell carcinoma of the ovary of the hypercalcemic type. Ann. Oncol. 2007, 18, 1985–1989. [Google Scholar] [CrossRef]
- Jelinic, P.; Mueller, J.J.; Olvera, N. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat. Genet. 2014, 46, 424–426. [Google Scholar] [CrossRef]
- Ramos, P.; Karnezis, A.N.; Craig, D.W.; Sekulic, A.; Russell, M.L.; Hendricks, W.P.D.; Corneveaux, J.J.; Barrett, M.T.; Shumansky, K.; Yang, Y.; et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat. Genet. 2014, 46, 427–429. [Google Scholar] [CrossRef]
- Lin, D.I.; Chudnovsky, Y.; Duggan, B.; Zajchowski, D.; Greenbowe, J.; Ross, J.S.; Gay, L.M.; Ali, S.M.; Elvin, J.A. Comprehensive genomic profiling reveals inactivating SMARCA4 mutations and low tumor mutational burden in small cell carcinoma of the ovary, hypercalcemic-type. Gynecol. Oncol. 2017, 147, 626–633. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Patel, V.M.; Coon, M. Using drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Dai, J.; Wang, D.; Lu, H.; Dai, H.; Ye, H.; Gu, J.; Chen, S.; Huang, B. Assessment of tumor mutation burden calculation from gene panel sequencing data. Onco Targets 2019, 12, 3401–3409. [Google Scholar] [CrossRef]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Karolchik, D.M.; Baertsch, R. The UCSC genome browser database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef]
- Engström, P.G.; Steijger, T.; Sipos, B.; Grant, G.R.; Kahles, A.; Rätsch, G.; Rätsch, G.; Goldman, N.; Hubbard, T.J.; Harrow, J.; et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013, 10, 1185–1191. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.; del Angel, G.; Rivas, M.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Le Loarer, F.; Watson, S.; Pierron, G.; de Montpreville, V.T.; Ballet, S.; Firmin, N.; Auguste, A.; Pissaloux, D.; Boyault, S.; Paindavoine, S.; et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat. Genet. 2015, 47, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, A.N.; Wang, Y. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 2016, 238, 389–400. [Google Scholar] [CrossRef]
- Genestie, C.; Blanc-Durand, F.; Auguste, A.; Pautier, P.; Dunant, A.; Scoazec, J.-Y.; Gouy, S.; Morice, P.; Bentivegna, E.; Maulard, A.; et al. Clinical utility of SMARCA4 testing by immunohistochemistry in rare ovarian tumours. Br. J. Cancer 2019, 122, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; McKenzie, Z.M.; D’Avino, A.R.; Mashtalir, N.; Lareau, C.A.; St Pierre, R.; Wang, L.; Shilatifard, A.; Kadoch, C. The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity-independent genomic targeting. Nat. Genet. 2019, 51, 618–626. [Google Scholar] [CrossRef]
- Liu, G.; Gramling, S.; Munoz, D.; Cheng, D.; Azad, A.K.; Mirshams, M.; Chen, Z.; Xu, W.; Roberts, H.; Shepherd, F.A.; et al. Two novel BRM insertion promoter sequence variants are associated with loss of BRM expression and lung cancer risk. Oncogene 2011, 30, 3295–3304. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Nagel, I.; Oyen, F.; Bartelheim, K.; Russell, R.B.; Schüller, U.; Junckerstorff, R.; Rosenblum, M.; Alassiri, A.H.; Rossi, S.; et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014, 128, 453–456. [Google Scholar] [CrossRef]
- Venneti, S.; Le, P.; Martinez, D.; Xie, S.X.; Sullivan, L.M.; Rorke-Adams, L.B.; Bruce, P.; Alexander, J. Malignant rhabdoid tumors express stem cell factors, which relate to the expression of EZH2 and Id proteins. Am. J. Surg. Pathol. 2011, 35, 1463–1472. [Google Scholar] [CrossRef]
- Andrysik, Z.; Bernstein, W.Z.; Deng, L.; Myer, D.L.; Li, Y.-Q.; Tischfield, J.A.; Stambrook, P.J. The novel mouse Polo-like kinase 5 responds to DNA damage and localizes in the nucleolus. Nucleic Acids Res. 2010, 38, 2931–2943. [Google Scholar] [CrossRef]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.-J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S.Y.; Karnezis, A.N.; Colborne, S.; Santos, N.D.; Lang, J.D. The histone methyltransferase EZH2 is a therapeutic target in small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 2017, 242, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.-C.; Toulmonde, M.; Michot, J.-M.; Lucchesi, C.; Varga, A. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Eichhorn, J.H.; Young, R.H.; Scully, R.E. Primary ovarian small cell carcinoma of pulmonary type. A clinicopathologic, immunohistologic, and flow cytometric analysis of 11 cases. Am. J. Surg. Pathol. 1992, 16, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Förster, C.; Ostertag, H.; Schmitt, J.; Roessner, A. Small cell carcinoma of the ovary, hypercalcemic type. A case report with immunohistochemical, ultrastructural and cytophotometric analysis and review of the literature. Gen. Diagn. Pathol. 1997, 142, 365–370. [Google Scholar]
- O’Keefe, C.; McDevitt, M.A.; Maciejewski, J.P. Copy neutral loss of heterozygosity: A novel chromosomal lesion in myeloid malignancies. Blood 2010, 115, 2731–2739. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- David, M.P.; Venkatramani, R.; Lopez-Terrada, D.H.; Roy, A.; Patil, N.; Fisher, K.E. Multimodal molecular analysis of an atypical small cell carcinoma of the ovary, hypercalcemic type. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002956. [Google Scholar] [CrossRef]
- Medina, P.P.; Carretero, J.; Fraga, M.F.; Esteller, M.; Sidransky, D.; Sanchez-Cespedes, M. Genetic and epigenetic screening for gene alterations of the chromatin-remodeling factor, SMARCA4/BRG1, in lung tumors. Genes Chromosomes Cancer 2004, 41, 170–177. [Google Scholar] [CrossRef]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 3128–3133. [Google Scholar] [CrossRef]
- Kahali, B.; Yu, J.; Marquez, S.B.; Thompson, K.e.n.n.e.t.h.W.; Liang, S.Y.; Lu, L. The silencing of the SWI/SNF subunit and anticancer gene BRM in Rhabdoid tumors. Oncotarget 2014, 5, 3316–3332. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coatham, M.; Li, X.; Karnezis, A.N.; Hoang, L.N.; Tessier-Cloutier, B.; Meng, B. Concurrent ARID1A and ARID1B inactivation in endometrial and ovarian dedifferentiated carcinomas. Mod. Pathol. 2016, 29, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.D.; Hendricks, W.P.D.; Orlando, K.A.; Yin, H.; Kiefer, J.; Ramos, P. Ponatinib shows potent antitumor activity in small cell carcinoma of the ovary hypercalcemic type (SCCOHT) through multikinase inhibition. Clin. Cancer Res. 2018, 24, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Muscat, A.; Popovski, D.; Jayasekara, W.S.N.; Rossello, F.J.; Ferguson, M.; Marini, K.D. Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of malignant rhabdoid tumors. Clin. Cancer Res. 2016, 22, 3560–3570. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Localization | Gene Symbol | Description | Mean Log2Ratio |
|---|---|---|---|
| 12q12-q14 | SHMT2 | Serine hydroxymethyltransferase 2 (mitochondrial) | 2.33 |
| 12q13.3 | NDUFA4L2 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4-like 2 | 2.33 |
| 12q13.3 | NXPH4 | Neurexophilin 4 | 2.33 |
| 12q13.3 | LRP1 | Low density lipoprotein receptor-related protein 1 | 2.33 |
| 14q32.2 | BEGAIN | Brain-enriched guanylate kinase-associated | 2.23 |
| 14q32.2 | LINC00523 | Long intergenic non-protein coding RNA 523 | 2.23 |
| 16q24 | CBFA2T3 | Core-binding factor, runt domain, alpha subunit 2; translocated to, 3 | 1.40 |
| 16q24 | APRT | Adenine phosphoribosyltransferase | 1.40 |
| 16q24.3 | ACSF3 | Acyl-CoA Synthetase Family Member 3 | 1.40 |
| 16q24.3 | CTU2 | Cytosolic thiouridylase subunit 2 homolog (S. pombe) | 1.40 |
| 16q24.3 | GALNS | Galactosamine (N-acetyl)-6-sulfate sulfatase | 1.40 |
| 16q24.3 | MIR4722 | MicroRNA 4722 | 1.40 |
| 16q24.3 | PABPN1L | Poly(A) binding protein, nuclear 1-like (cytoplasmic) | 1.40 |
| 16q24.3 | CDT1 | Chromatin licensing and DNA replication factor 1 | 1.40 |
| 16q24.3 | PIEZO1 | Piezo-type mechanosensitive ion channel component 1 | 1.40 |
| 16q24.3 | TRAPPC2L | Trafficking protein particle complex 2-like | 1.40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auguste, A.; Blanc-Durand, F.; Deloger, M.; Le Formal, A.; Bareja, R.; Wilkes, D.C.; Richon, C.; Brunn, B.; Caron, O.; Devouassoux-Shisheboran, M.; et al. Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT) beyond SMARCA4 Mutations: A Comprehensive Genomic Analysis. Cells 2020, 9, 1496. https://doi.org/10.3390/cells9061496
Auguste A, Blanc-Durand F, Deloger M, Le Formal A, Bareja R, Wilkes DC, Richon C, Brunn B, Caron O, Devouassoux-Shisheboran M, et al. Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT) beyond SMARCA4 Mutations: A Comprehensive Genomic Analysis. Cells. 2020; 9(6):1496. https://doi.org/10.3390/cells9061496
Chicago/Turabian StyleAuguste, Aurélie, Félix Blanc-Durand, Marc Deloger, Audrey Le Formal, Rohan Bareja, David C. Wilkes, Catherine Richon, Béatrice Brunn, Olivier Caron, Mojgan Devouassoux-Shisheboran, and et al. 2020. "Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT) beyond SMARCA4 Mutations: A Comprehensive Genomic Analysis" Cells 9, no. 6: 1496. https://doi.org/10.3390/cells9061496
APA StyleAuguste, A., Blanc-Durand, F., Deloger, M., Le Formal, A., Bareja, R., Wilkes, D. C., Richon, C., Brunn, B., Caron, O., Devouassoux-Shisheboran, M., Gouy, S., Morice, P., Bentivegna, E., Sboner, A., Elemento, O., Rubin, M. A., Pautier, P., Genestie, C., Cyrta, J., & Leary, A. (2020). Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT) beyond SMARCA4 Mutations: A Comprehensive Genomic Analysis. Cells, 9(6), 1496. https://doi.org/10.3390/cells9061496