Chromatin Trapping of Factors Involved in DNA Replication and Repair Underlies Heat-Induced Radio- and Chemosensitization

 , , and
, , and 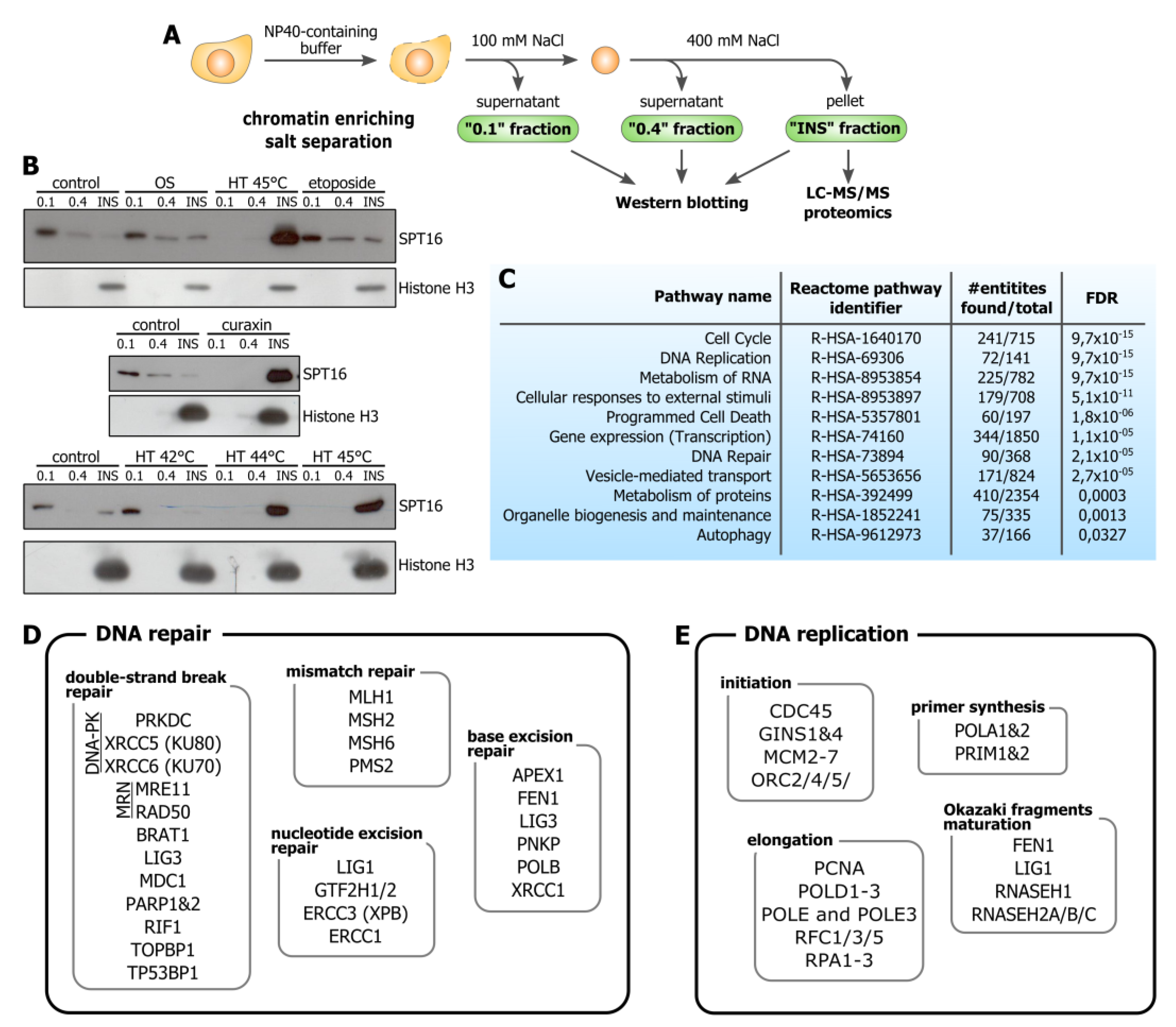{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Cell Culture, Drug Treatments, and Hyperthermia
2.3. Chromatin Enriching Salt Separation and Immunoblotting
2.4. Preparative SDS-PAGE and In-Gel Trypsin Digestion
2.5. LC-MS/MS Analysis
2.6. LC-MS/MS Data Analysis
2.7. Immunofluorescence Microscopy (Including Super-Resolution Microscopy)
2.8. Live-Cell Imaging and FRAP Analysis
2.9. In Situ Nick Translation
3. Results
3.1. Hyperthermia Induces C-Trapping
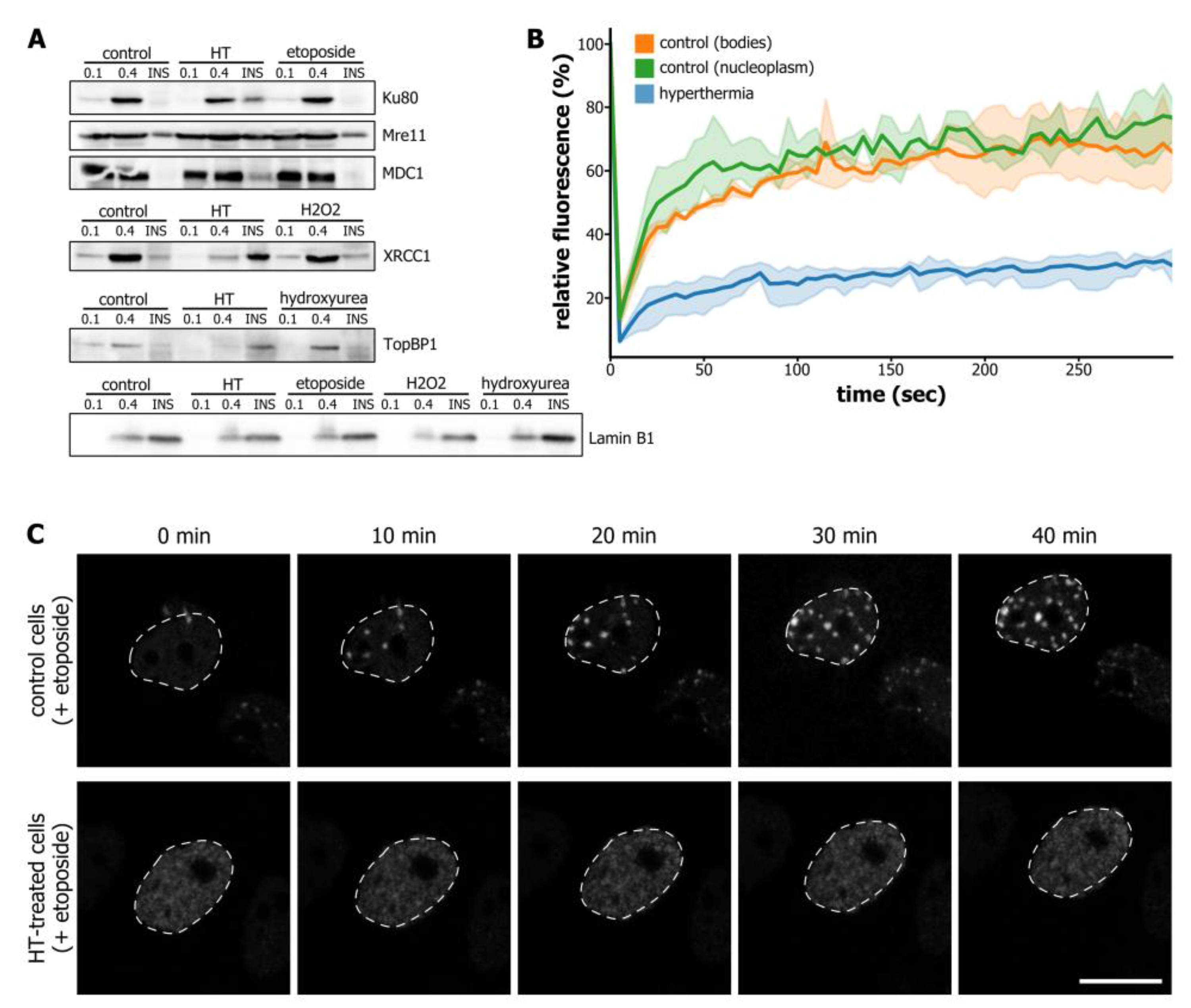
3.2. Hyperthermia-Induced C-Trapping Causes DNA Repair Deficiency
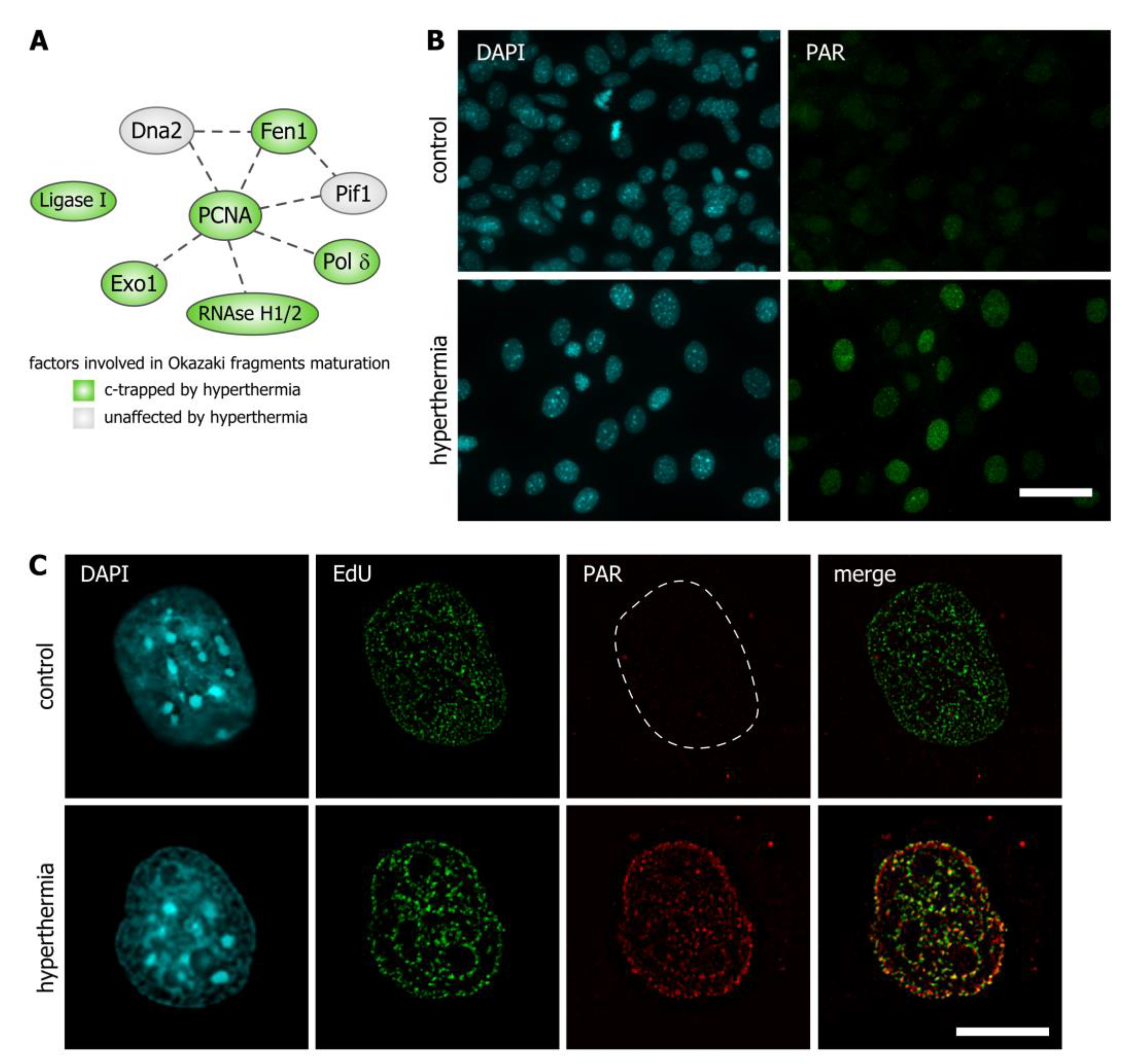
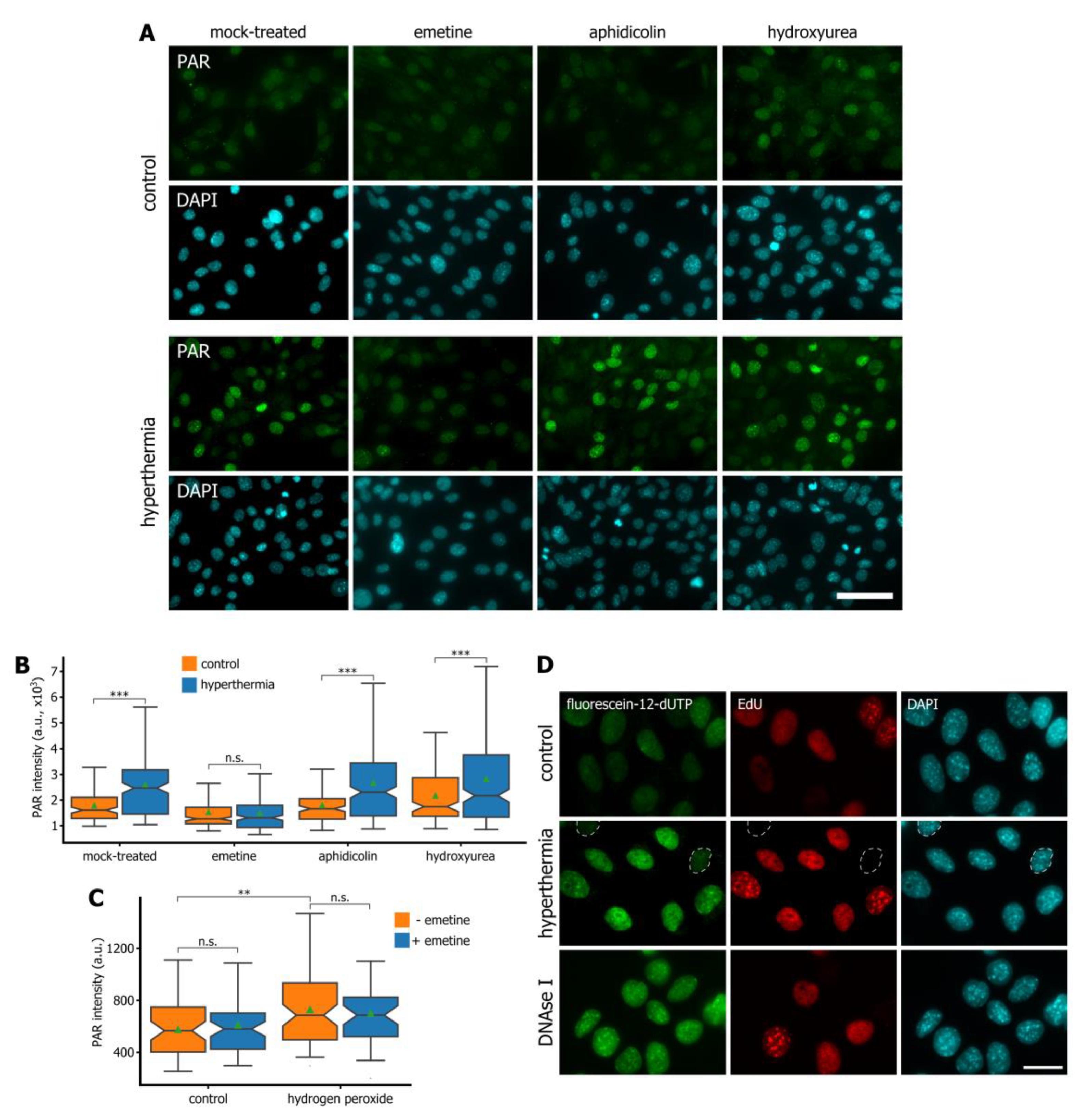
3.3. Hyperthermia Inhibits Maturation of Okazaki Fragments and Provokes a Corresponding PARP-Dependent DNA Damage Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, L.; Altman, M.B.; Laszlo, A.; Straube, W.; Zoberi, I.; Hallahan, D.E.; Chen, H. Ultrasound Hyperthermia Technology for Radiosensitization. Ultrasound Med. Biol. 2019, 45, 1025–1043. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.K.; Kim, J.C.; Shin, Y.; Han, S.M.; Won, W.R.; Her, J.; Park, J.Y.; Oh, K.T. Principles and applications of nanomaterial-based hyperthermia in cancer therapy. Arch. Pharm. Res. 2020, 43, 46–57. [Google Scholar] [CrossRef]
- Van den Tempel, N.; Horsman, M.R.; Kanaar, R. Improving efficacy of hyperthermia in oncology by exploiting biological mechanisms. Int. J. Hyperth. 2016, 32, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; Kok, H.P.; Oei, S.B.; Horsman, M.R.; Stalpers, L.J.A.; Franken, N.A.P.; Crezee, J. Molecular and biological rationale of hyperthermia as radio- and chemosensitizer. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; Vriend, L.E.; Crezee, J.; Franken, N.A.; Krawczyk, P.M. Effects of hyperthermia on DNA repair pathways: One treatment to inhibit them all. Radiat. Oncol. 2015, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Razin, S.V. Heat Stress-Induced DNA Damage. Acta Nat. 2016, 8, 75–78. [Google Scholar] [CrossRef]
- Nesher, E.; Safina, A.; Aljahdali, I.; Portwood, S.; Wang, E.S.; Koman, I.; Wang, J.; Gurova, K.V. Role of Chromatin Damage and Chromatin Trapping of FACT in Mediating the Anticancer Cytotoxicity of DNA-Binding Small-Molecule Drugs. Cancer Res. 2018, 78, 1431–1443. [Google Scholar] [CrossRef]
- Gurova, K.V. Chromatin Stability as a Target for Cancer Treatment. BioEssays 2019, 41, e1800141. [Google Scholar] [CrossRef]
- Safina, A.; Cheney, P.; Pal, M.; Brodsky, L.; Ivanov, A.; Kirsanov, K.; Lesovaya, E.; Naberezhnov, D.; Nesher, E.; Koman, I.; et al. FACT is a sensor of DNA torsional stress in eukaryotic cells. Nucleic Acids Res. 2017, 45, 1925–1945. [Google Scholar] [CrossRef]
- Chang, H.W.; Valieva, M.E.; Safina, A.; Chereji, R.V.; Wang, J.; Kulaeva, O.I.; Morozov, A.V.; Kirpichnikov, M.P.; Feofanov, A.V.; Gurova, K.V.; et al. Mechanism of FACT removal from transcribed genes by anticancer drugs curaxins. Sci. Adv. 2018, 4, eaav2131. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef]
- Federation, A.J.; Nandakumar, V.; Searle, B.C.; Stergachis, A.; Wang, H.; Pino, L.K.; Merrihew, G.; Ting, Y.S.; Howard, N.; Kutyavin, T.; et al. Highly Parallel Quantification and Compartment Localization of Transcription Factors and Nuclear Proteins. Cell Rep. 2020, 30, 2463–2471.e2465. [Google Scholar] [CrossRef]
- Zhao, Z.; Dammert, M.A.; Hoppe, S.; Bierhoff, H.; Grummt, I. Heat shock represses rRNA synthesis by inactivation of TIF-IA and lncRNA-dependent changes in nucleosome positioning. Nucleic Acids Res. 2016, 44, 8144–8152. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Takeshita, S.; Okaichi, K.; Okumura, Y.; Ohnishi, T. Heat exposure enhances radiosensitivity by depressing DNA-PK kinase activity during double strand break repair. Int. J. Hyperth. 2014, 30, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Burgman, P.; Ouyang, H.; Peterson, S.; Chen, D.J.; Li, G.C. Heat inactivation of Ku autoantigen: Possible role in hyperthermic radiosensitization. Cancer Res. 1997, 57, 2847–2850. [Google Scholar]
- Qi, D.; Hu, Y.; Li, J.; Peng, T.; Su, J.; He, Y.; Ji, W. Hyperthermia Induces Apoptosis of 786-O Cells through Suppressing Ku80 Expression. PLoS ONE 2015, 10, e0122977. [Google Scholar] [CrossRef]
- Seno, J.D.; Dynlacht, J.R. Intracellular redistribution and modification of proteins of the Mre11/Rad50/Nbs1 DNA repair complex following irradiation and heat-shock. J. Cell. Physiol. 2004, 199, 157–170. [Google Scholar] [CrossRef]
- Bhatti, S.; Kozlov, S.; Farooqi, A.A.; Naqi, A.; Lavin, M.; Khanna, K.K. ATM protein kinase: The linchpin of cellular defenses to stress. Cell. Mol. Life Sci. CMLS 2011, 68, 2977–3006. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Dynlacht, J.R.; Dikomey, E. Mechanism of radiosensitization by hyperthermia (> or = 43 degrees C) as derived from studies with DNA repair defective mutant cell lines. Int. J. Hyperth. 2004, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grofte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. Embo J. 2019, 38, e101379. [Google Scholar] [CrossRef] [PubMed]
- Velichko, A.K.; Petrova, N.V.; Kantidze, O.L.; Razin, S.V. Dual effect of heat shock on DNA replication and genome integrity. Mol. Boil. Cell 2012, 23, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Velichko, A.K.; Markova, E.N.; Petrova, N.V.; Razin, S.V.; Kantidze, O.L. Mechanisms of heat shock response in mammals. Cell. Mol. Life Sci. CMLS 2013, 70, 4229–4241. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331.e3. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Vassilev, L.T.; Wu, J.; Sogo, J.M.; Nallaseth, F.S.; DePamphilis, M.L. Emetine allows identification of origins of mammalian DNA replication by imbalanced DNA synthesis, not through conservative nucleosome segregation. Embo J. 1991, 10, 4351–4360. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, P.M.; Eppink, B.; Essers, J.; Stap, J.; Rodermond, H.; Odijk, H.; Zelensky, A.; van Bree, C.; Stalpers, L.J.; Buist, M.R.; et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc. Natl. Acad. Sci. USA 2011, 108, 9851–9856. [Google Scholar] [CrossRef]
- Van den Tempel, N.; Zelensky, A.N.; Odijk, H.; Laffeber, C.; Schmidt, C.K.; Brandsma, I.; Demmers, J.; Krawczyk, P.M.; Kanaar, R. On the Mechanism of Hyperthermia-Induced BRCA2 Protein Degradation. Cancers 2019, 11, 97. [Google Scholar] [CrossRef]
- Lepock, J.R. Role of nuclear protein denaturation and aggregation in thermal radiosensitization. Int. J. Hyperth. 2004, 20, 115–130. [Google Scholar] [CrossRef]
- Roti Roti, J.L. Heat-induced alterations of nuclear protein associations and their effects on DNA repair and replication. Int. J. Hyperth. 2007, 23, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, A.; Davidson, T.; Harvey, A.; Sim, J.E.; Malyapa, R.S.; Spitz, D.R.; Roti Roti, J.L. Alterations in heat-induced radiosensitization accompanied by nuclear structure alterations in Chinese hamster cells. Int. J. Hyperth. 2006, 22, 43–60. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luzhin, A.V.; Avanesyan, B.; Velichko, A.K.; Shender, V.O.; Ovsyannikova, N.; Arapidi, G.P.; Shnaider, P.V.; Petrova, N.V.; Kireev, I.I.; Razin, S.V.; et al. Chromatin Trapping of Factors Involved in DNA Replication and Repair Underlies Heat-Induced Radio- and Chemosensitization. Cells 2020, 9, 1423. https://doi.org/10.3390/cells9061423
Luzhin AV, Avanesyan B, Velichko AK, Shender VO, Ovsyannikova N, Arapidi GP, Shnaider PV, Petrova NV, Kireev II, Razin SV, et al. Chromatin Trapping of Factors Involved in DNA Replication and Repair Underlies Heat-Induced Radio- and Chemosensitization. Cells. 2020; 9(6):1423. https://doi.org/10.3390/cells9061423
Chicago/Turabian StyleLuzhin, Artem V., Bogdan Avanesyan, Artem K. Velichko, Victoria O. Shender, Natalia Ovsyannikova, Georgij P. Arapidi, Polina V. Shnaider, Nadezhda V. Petrova, Igor I. Kireev, Sergey V. Razin, and et al. 2020. "Chromatin Trapping of Factors Involved in DNA Replication and Repair Underlies Heat-Induced Radio- and Chemosensitization" Cells 9, no. 6: 1423. https://doi.org/10.3390/cells9061423
APA StyleLuzhin, A. V., Avanesyan, B., Velichko, A. K., Shender, V. O., Ovsyannikova, N., Arapidi, G. P., Shnaider, P. V., Petrova, N. V., Kireev, I. I., Razin, S. V., & Kantidze, O. L. (2020). Chromatin Trapping of Factors Involved in DNA Replication and Repair Underlies Heat-Induced Radio- and Chemosensitization. Cells, 9(6), 1423. https://doi.org/10.3390/cells9061423