The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient

 , ,
, ,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Tissue Samples and Animal Experiments
2.2. Study Design
2.3. Right Ventricle Pressure Measurements
2.4. Histology and Morphometry
2.5. Western Blot
2.6. Immunofluorescence Staining and Quantitative Analysis
2.7. Statistical Analysis
3. Results
3.1. BMPR2 Levels Are not Decreased in Whole Lung Lysates of iPAH and hPAH Patients
3.2. Decreased Levels of pSmad 1/5/8 in the Lung Vasculature of iPAH and hPAH Patients
3.3. Development of PAH in Animal Models
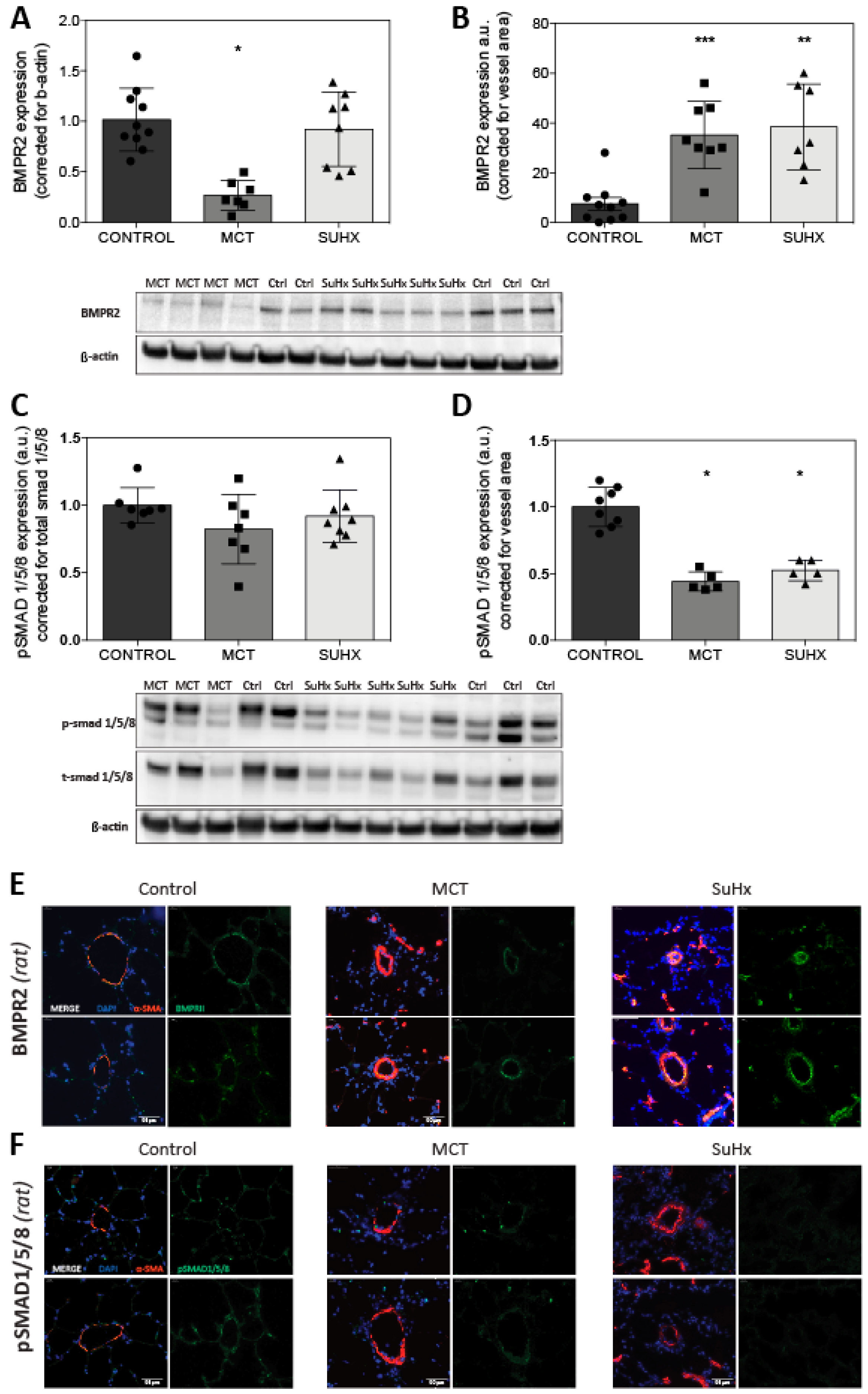
3.4. BMPR2 Expression Is Increased in MCT and SuHx Lung Vasculature
4. Discussion
4.1. Disturbed BMP Signaling in iPAH and hPAH
4.2. Reduced BMPR2 and pSmad 1/5/8 Expression in MCT and SuHx Animal Models of PAH
4.3. Interventions Aimed to Restore the TGF-β/BMP Balance in Experimental PAH
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated Clinical Classification of Pulmonary Hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.R.; Machado, R.D.; Pauciulo, M.W.; Morgan, N.V.; Humbert, M.; Elliott, G.C.; Ward, K.; Yacoub, M.; Mikhail, G.; Rogers, P.; et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-β family. J. Med. Genet. 2000, 37, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Austin, E.D.; Ma, L.; LeDuc, C.; Rosenzweig, E.B.; Borczuk, A.; Phillips, J.A.; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole Exome Sequencing to Identify a Novel Gene (Caveolin-1) Associated with Human Pulmonary Arterial HypertensionClinical Perspective. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef]
- Wang, G.; Fan, R.; Ji, R.; Zou, W.; Penny, D.J.; Varghese, N.P.; Fan, Y. Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulm. Med. 2016, 16, 17. [Google Scholar] [CrossRef]
- Girerd, B.; Montani, D.; Coulet, F.; Sztrymf, B.; Yaici, A.; Jaïs, X.; Tregouet, D.; Reis, A.; Drouin-Garraud, V.; Fraisse, A.; et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am. J. Respir. Crit. Care Med. 2010, 181, 851–861. [Google Scholar] [CrossRef]
- Shintani, M.; Yagi, H.; Nakayama, T.; Saji, T.; Matsuoka, R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J. Med. Genet. 2009, 46, 331–337. [Google Scholar] [CrossRef]
- Drake, K.M.; Dunmore, B.J.; McNelly, L.N.; Morrell, N.W.; Aldred, M.A. Correction of Nonsense BMPR2 and SMAD9 Mutations by Ataluren in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 403–409. [Google Scholar] [CrossRef]
- Pousada, G.; Baloira, A.; Fontán, D.; Núñez, M.; Valverde, D. Mutational and clinical analysis of the ENG gene in patients with pulmonary arterial hypertension. BMC Genet. 2016, 17, 72. [Google Scholar] [CrossRef]
- Dewachter, L.; Adnot, S.; Guignabert, C.; Tu, L.; Marcos, E.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; et al. Bone morphogenetic protein signalling in heritable versus idiopathic pulmonary hypertension. Eur. Respir. J. 2009, 34, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.R.; Ormiston, M.L.; Perez-Iratxeta, C.; Courtman, D.W.; Jiang, B.; Ferrer, E.; Caruso, P.; Southwood, M.; Foster, W.S.; Morrell, N.W.; et al. Proteomic Analysis Implicates Translationally Controlled Tumor Protein as a Novel Mediator of Occlusive Vascular Remodeling in Pulmonary Arterial Hypertension. Circulation 2014, 129, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C. Primary Pulmonary Hypertension Is Associated with Reduced Pulmonary Vascular Expression of Type II Bone Morphogenetic Protein Receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Yeager, M.E.; Zaiman, A.; Cool, C.D.; Voelkel, N.F.; Tuder, R.M. Impaired Transforming Growth Factor-β Signaling in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2004, 170, 1340–1348. [Google Scholar] [CrossRef]
- Morrell, N.W.; Bloch, D.B.; ten Dijke, P.; Goumans, M.-J.T.H.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signalling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef]
- Nasim, M.T.; Ogo, T.; Chowdhury, H.M.; Zhao, L.; Chen, C.; Rhodes, C.; Trembath, R.C. BMPR-II deficiency elicits pro-proliferative and anti-apoptotic responses through the activation of TGFβ-TAK1-MAPK pathways in PAH. Hum. Mol. Genet. 2012, 21, 2548–2558. [Google Scholar] [CrossRef]
- Soon, E.; Crosby, A.; Southwood, M.; Yang, P.; Tajsic, T.; Toshner, M.; Appleby, S.; Shanahan, C.M.; Bloch, K.D.; Pepke-Zaba, J.; et al. BMPR-II Deficiency Promotes Pulmonary Hypertension via Increased Inflammatory Cytokine Production. Am. J. Respir. Crit. Care Med. 2015, 192, 859–872. [Google Scholar] [CrossRef]
- Upton, P.D.; Morrell, N.W. The transforming growth factor-β–bone morphogenetic protein type signalling pathway in pulmonary vascular homeostasis and disease. Exp. Physiol. 2013, 98, 1262–1266. [Google Scholar] [CrossRef]
- Dorfmüller, P.; Perros, F.; Balabanian, K.; Humbert, M. Inflammation in pulmonary arterial hypertension. Eur. Respir. J. 2003, 22, 358–363. [Google Scholar] [CrossRef]
- Goumans, M.-J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Gore, B.; Izikki, M.; Mercier, O.; Dewachter, L.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; Lebrin, F.; et al. Key Role of the Endothelial TGF-β/ALK1/Endoglin Signaling Pathway in Humans and Rodents Pulmonary Hypertension. PLoS ONE 2014, 9, e100310. [Google Scholar] [CrossRef]
- Davies, R.J.; Holmes, A.M.; Deighton, J.; Long, L.; Yang, X.; Barker, L.; Walker, C.; Budd, D.C.; Upton, P.D.; Morrell, N.W. BMP type II receptor deficiency confers resistance to growth inhibition by TGF-β in pulmonary artery smooth muscle cells: Role of proinflammatory cytokines. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 302, L604–L615. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Vizán, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Müller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. eLife 2018, 7, e31756. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef] [PubMed]
- Nickel, N.P.; Spiekerkoetter, E.; Gu, M.; Li, C.G.; Li, H.; Kaschwich, M.; Diebold, I.; Hennigs, J.K.; Kim, K.-Y.; Miyagawa, K.; et al. Elafin Reverses Pulmonary Hypertension via Caveolin-1–Dependent Bone Morphogenetic Protein Signaling. Am. J. Respir. Crit. Care Med. 2015, 191, 1273–1286. [Google Scholar] [CrossRef]
- Kay, J.M.; Harris, P.; Heath, D. Pulmonary hypertension produced in rats by ingestion of Crotalaria spectabilis seeds. Thorax 1967, 22, 176–179. [Google Scholar] [CrossRef]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mahon, G.M.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef]
- Overbeek, M.J.; Mouchaers, K.T.B.; Niessen, H.M.; Hadi, A.M.; Kupreishvili, K.; Boonstra, A.; Voskuyl, A.E.; Belien, J.A.M.; Smit, E.F.; Dijkmans, B.C.; et al. Characteristics of Interstitial Fibrosis and Inflammatory Cell Infiltration in Right Ventricles of Systemic Sclerosis-Associated Pulmonary Arterial Hypertension. Available online: https://www.hindawi.com/journals/ijr/2010/604615/abs/ (accessed on 20 April 2019).
- De Raaf, M.A.; Schalij, I.; Gomez-Arroyo, J.; Rol, N.; Happé, C.; de Man, F.S.; Vonk-Noordegraaf, A.; Westerhof, N.; Voelkel, N.F.; Bogaard, H.J. SuHx rat model: Partly reversible pulmonary hypertension and progressive intima obstruction. Eur. Respir. J. 2014, 44, 160–168. [Google Scholar] [CrossRef]
- Handoko, M.L.; de Man, F.S.; Happé, C.M.; Schalij, I.; Musters, R.J.P.; Westerhof, N.; Postmus, P.E.; Paulus, W.J.; van der Laarse, W.J.; Vonk-Noordegraaf, A. Opposite Effects of Training in Rats with Stable and Progressive Pulmonary Hypertension. Circulation 2009, 120, 42–49. [Google Scholar] [CrossRef]
- Happé, C.M.; de Raaf, M.A.; Rol, N.; Schalij, I.; Vonk-Noordegraaf, A.; Westerhof, N.; Voelkel, N.F.; de Man, F.S.; Bogaard, H.J. Pneumonectomy combined with SU5416 induces severe pulmonary hypertension in rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L1088–L1097. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Rudarakanchana, N.; Flanagan, J.A.; Chen, H.; Upton, P.D.; Machado, R.; Patel, D.; Trembath, R.C.; Morrell, N.W. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum. Mol. Genet. 2002, 11, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Yang, X. Dysfunctional Smad Signaling Contributes to Abnormal Smooth Muscle Cell Proliferation in Familial Pulmonary Arterial Hypertension. Circ. Res. 2005, 96, 1053–1063. [Google Scholar] [CrossRef]
- Ramos, M.F.; Lamé, M.W.; Segall, H.J.; Wilson, D.W. Smad Signaling in the Rat Model of Monocrotaline Pulmonary Hypertension. Toxicol. Pathol. 2008, 36, 311–320. [Google Scholar] [CrossRef]
- Reynolds, A.M.; Holmes, M.D.; Danilov, S.M.; Reynolds, P.N. Targeted gene delivery of BMPR2 attenuates pulmonary hypertension. Eur. Respir. J. 2012, 39, 329–343. [Google Scholar] [CrossRef]
- Long, L.; Crosby, A.; Yang, X.; Southwood, M.; Upton, P.D.; Kim, D.-K.; Morrell, N.W. Altered Bone Morphogenetic Protein and Transforming Growth Factor- Signaling in Rat Models of Pulmonary Hypertension: Potential for Activin Receptor-Like Kinase-5 Inhibition in Prevention and Progression of Disease. Circulation 2009, 119, 566–576. [Google Scholar] [CrossRef]
- McMurtry, M.S.; Moudgil, R.; Hashimoto, K.; Bonnet, S.; Michelakis, E.D.; Archer, S.L. Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L872–L878. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Gräf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef]
- Harper, R.L.; Reynolds, A.M.; Bonder, C.S.; Reynolds, P.N. BMPR2 gene therapy for PAH acts via Smad and non-Smad signalling. Respirology 2016, 21, 727–733. [Google Scholar] [CrossRef]
- Al-Husseini, A.; Kraskauskas, D.; Mezzaroma, E.; Nordio, A.; Farkas, D.; Drake, J.I.; Abbate, A.; Felty, Q.; Voelkel, N.F. Vascular endothelial growth factor receptor 3 signaling contributes to angioobliterative pulmonary hypertension. Pulm. Circ. 2015, 5, 101–116. [Google Scholar] [CrossRef]
- Hwangbo, C.; Lee, H.-W.; Kang, H.; Ju, H.; Wiley, D.S.; Papangeli, I.; Han, J.; Kim, J.-D.; Dunworth, W.P.; Hu, X.; et al. Modulation of Endothelial Bone Morphogenetic Protein Receptor Type 2 Activity by Vascular Endothelial Growth Factor Receptor 3 in Pulmonary Arterial Hypertension. Circulation 2017, 135, 2288–2298. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-W.; Jin, S.-W. VEGFR3 as a novel modulator for PAH. Oncotarget 2017, 8, 84610–84611. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.-J.; Zwijsen, A.; ten Dijke, P.; Bailly, S. Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef] [PubMed]
- Hautefort, A.; Mendes-Ferreira, P.; Sabourin, J.; Manaud, G.; Bertero, T.; Rucker-Martin, C.; Riou, M.; Adão, R.; Manoury, B.; Lambert, M.; et al. Bmpr2 Mutant Rats Develop Pulmonary and Cardiac Characteristics of Pulmonary Arterial Hypertension. Circulation 2019, 139, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Yang, X.; Southwood, M.; Lu, J.; Marciniak, S.J.; Dunmore, B.J.; Morrell, N.W. Chloroquine Prevents Progression of Experimental Pulmonary Hypertension via Inhibition of Autophagy and Lysosomal Bmpr-II Degradation. Circ. Res. 2013, 112, 1159–1170. [Google Scholar] [CrossRef]
- Dunmore, B.J.; Drake, K.M.; Upton, P.D.; Toshner, M.R.; Aldred, M.A.; Morrell, N.W. The lysosomal inhibitor, chloroquine, increases cell surface BMPR-II levels and restores BMP9 signalling in endothelial cells harbouring BMPR-II mutations. Hum. Mol. Genet. 2013, 22, 3667–3679. [Google Scholar] [CrossRef]
- Yang, J.; Li, X.; Al-Lamki, R.S.; Wu, C.; Weiss, A.; Berk, J.; Schermuly, R.T.; Morrell, N.W. Sildenafil Potentiates Bone Morphogenetic Protein Signaling in Pulmonary Arterial Smooth Muscle Cells and in Experimental Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 34–42. [Google Scholar] [CrossRef]
- Yung, L.-M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A Selective TGFβ Ligand Trap Attenuates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1140–1151. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; Del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Zamanian, R.T. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602449. [Google Scholar] [CrossRef]
- Orriols, M.; Gomez-Puerto, M.C.; ten Dijke, P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell. Mol. Life Sci. 2017, 74, 2979–2995. [Google Scholar] [CrossRef]
- Kurakula, K.; Sun, X.-Q.; Happé, C.; da Bos, D.S.G.; Szulcek, R.; Schalij, I.; Wiesmeijer, K.C.; Lodder, K.; Tu, L.; Guignabert, C.; et al. Prevention of Progression of Pulmonary Hypertension by the Nur77 Agonist 6-mercaptopurine: Role of BMP Signalling. Eur. Respir. J. 2019, 54, 1802400. [Google Scholar] [CrossRef] [PubMed]
- Botros, L.; Szulcek, R.; Jansen, S.M.; Kurakula, K.; Goumans, M.T.; van Kuilenburg, A.B.P.; Vonk Noordegraaf, A.; de Man, F.S.; Aman, J.; Bogaard, H.J. The Effects of Mercaptopurine on Pulmonary Vascular Resistance and BMPR2 Expression in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Van der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.; Szulcek, R.; Bourgeois, A.; Lampron, M.-C.; Habbout, K.; Martineau, S.; et al. Multicenter Preclinical Validation of BET Inhibition for the Treatment of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Happé, C.; Kurakula, K.; Sun, X.-Q.; da Silva Goncalves Bos, D.; Rol, N.; Guignabert, C.; Tu, L.; Schalij, I.; Wiesmeijer, K.C.; Tura-Ceide, O.; et al. The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells 2020, 9, 1422. https://doi.org/10.3390/cells9061422
Happé C, Kurakula K, Sun X-Q, da Silva Goncalves Bos D, Rol N, Guignabert C, Tu L, Schalij I, Wiesmeijer KC, Tura-Ceide O, et al. The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells. 2020; 9(6):1422. https://doi.org/10.3390/cells9061422
Chicago/Turabian StyleHappé, Chris, Kondababu Kurakula, Xiao-Qing Sun, Denielli da Silva Goncalves Bos, Nina Rol, Christophe Guignabert, Ly Tu, Ingrid Schalij, Karien C. Wiesmeijer, Olga Tura-Ceide, and et al. 2020. "The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient" Cells 9, no. 6: 1422. https://doi.org/10.3390/cells9061422
APA StyleHappé, C., Kurakula, K., Sun, X.-Q., da Silva Goncalves Bos, D., Rol, N., Guignabert, C., Tu, L., Schalij, I., Wiesmeijer, K. C., Tura-Ceide, O., Vonk Noordegraaf, A., de Man, F. S., Bogaard, H. J., & Goumans, M.-J. (2020). The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells, 9(6), 1422. https://doi.org/10.3390/cells9061422