N-Glycosylation and N-Glycan Processing in HBV Biology and Pathogenesis



Abstract
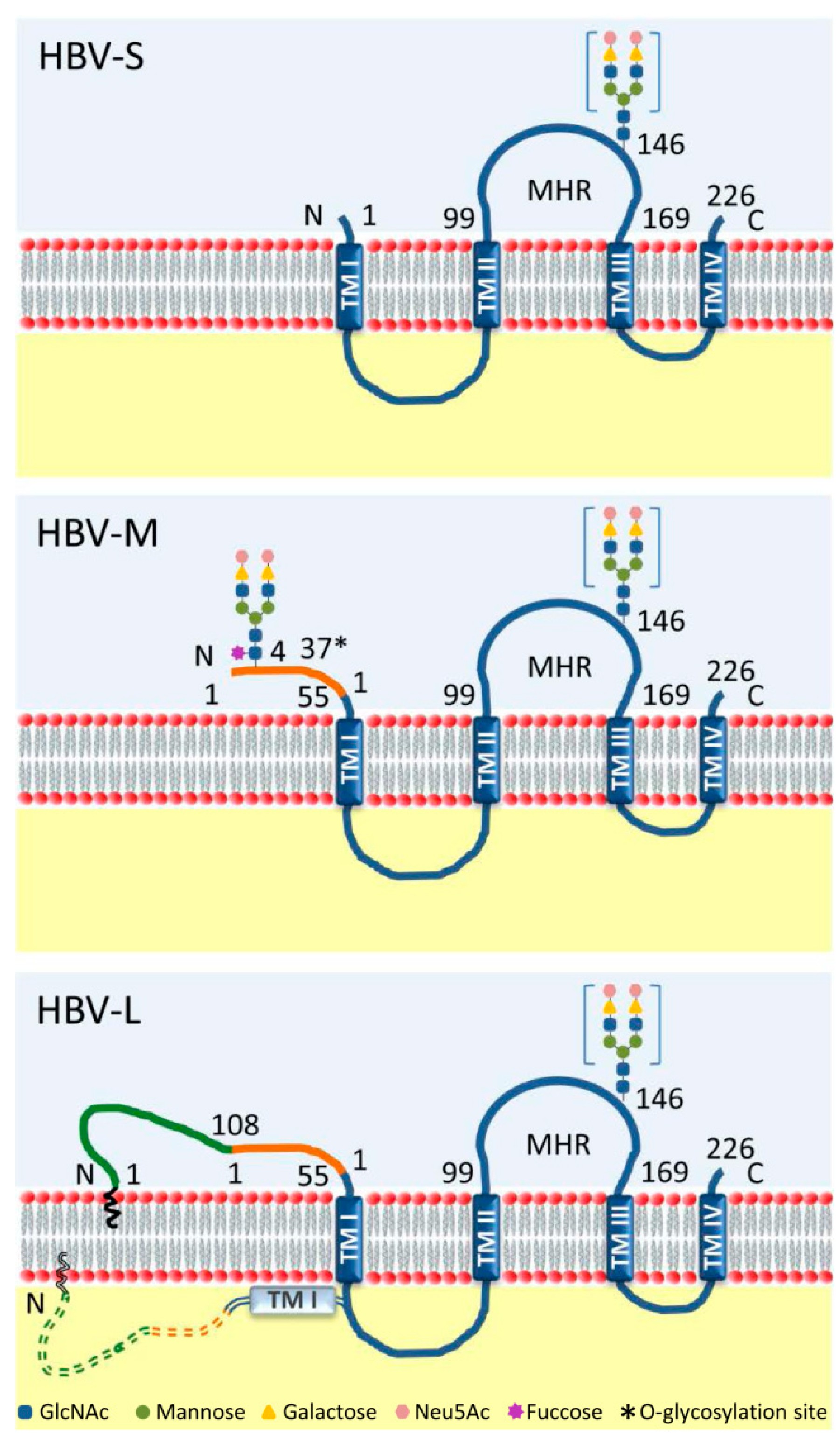
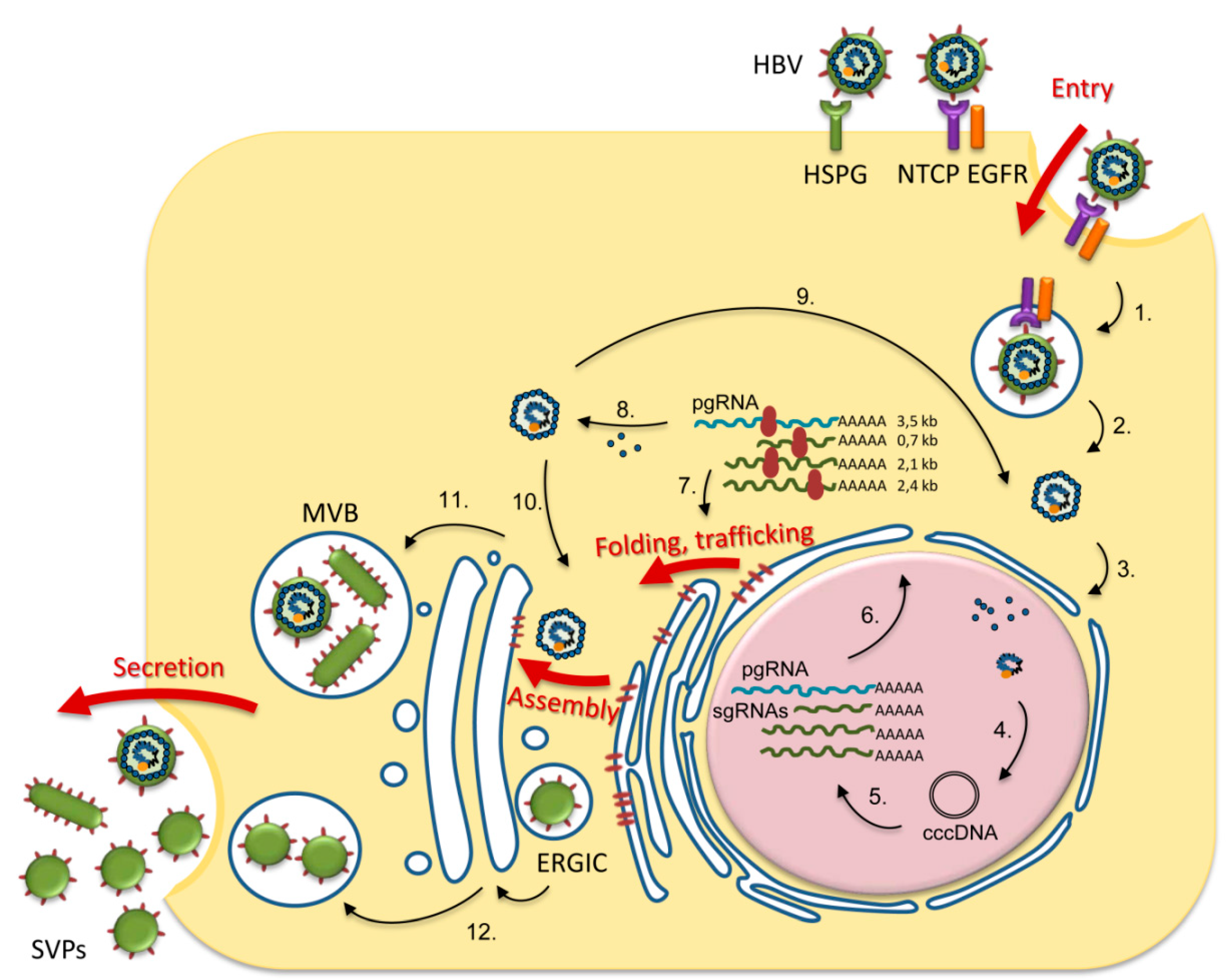
1. Introduction
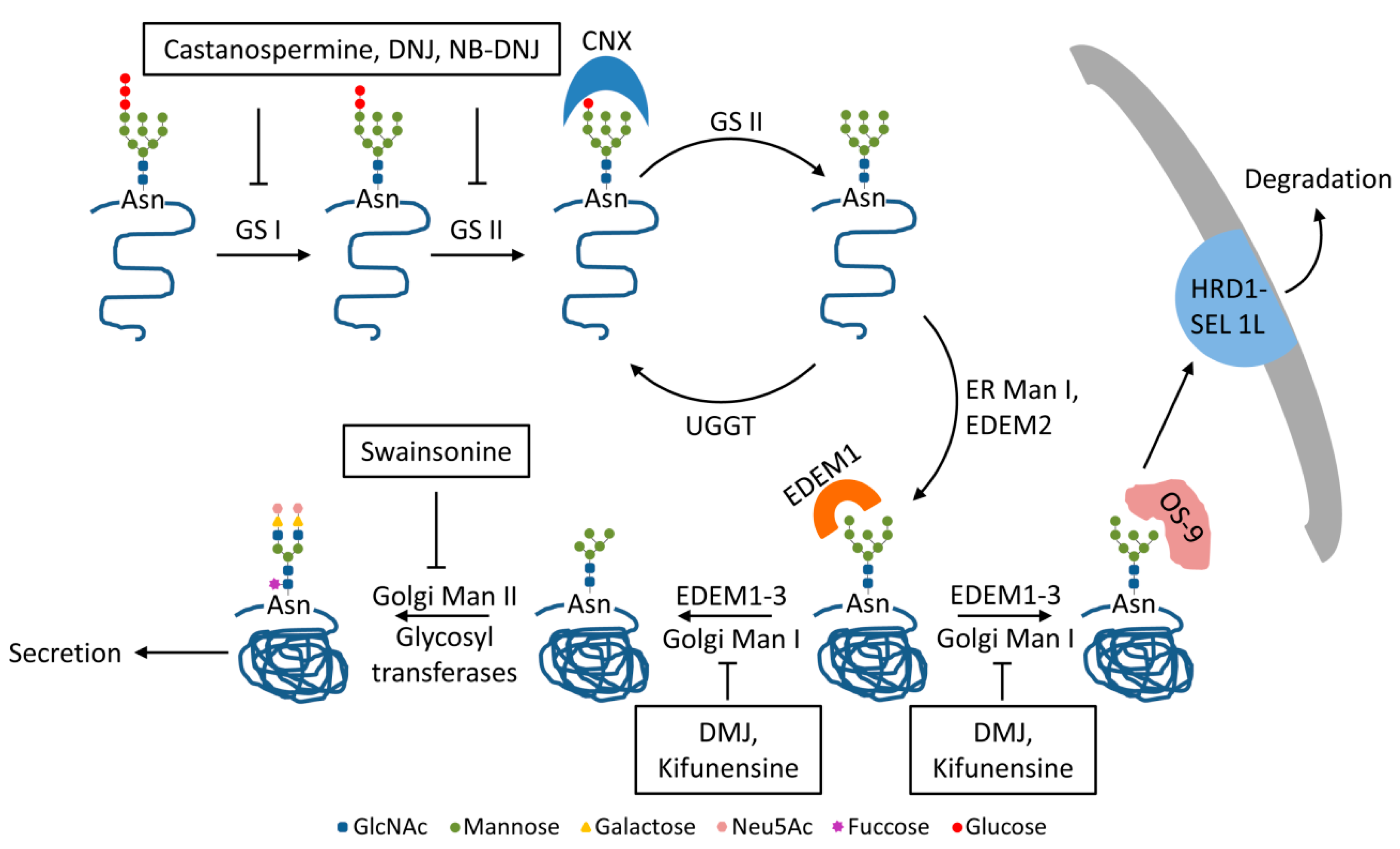
2. Trimming of HBV N-glycans by the ER α-glucosidases I and II: Asset or Vulnerability?
3. Mannose Trimming of HBV N-glycans: Crossroads between Protein Trafficking and Degradation
4. HBV N-glycosylation and the Interaction with the Host Immune System: Implications for Vaccine Development and Diagnosis
5. HBV N-glycosylation and the Carcinogenic Potential
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef] [PubMed]
- WHO. Hepatitis B; World Health Organization: Geneve, Switzerland, 2019. [Google Scholar]
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large surface proteins of hepatitis B virus containing the pre-s sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. Hepatitis B virus morphogenesis. Curr. Top. Microbiol. Immunol. 1996, 214, 297–337. [Google Scholar] [CrossRef] [PubMed]
- Patient, R.; Hourioux, C.; Roingeard, P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol. 2009, 11, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Bruss, V.; Vieluf, K. Functions of the internal pre-S domain of the large surface protein in hepatitis B virus particle morphogenesis. J. Virol. 1995, 69, 6652–6657. [Google Scholar] [CrossRef]
- Prange, R.; Streeck, R.E. Novel transmembrane topology of the hepatitis B virus envelope proteins. EMBO J. 1995, 14, 247–256. [Google Scholar] [CrossRef]
- Norder, H.; Courouce, A.M.; Magnius, L.O. Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology 1994, 198, 489–503. [Google Scholar] [CrossRef]
- Schmitt, S.; Glebe, D.; Alving, K.; Tolle, T.K.; Linder, M.; Geyer, H.; Linder, D.; Peter-Katalinic, J.; Gerlich, W.H.; Geyer, R. Analysis of the pre-S2 N- and O-linked glycans of the M surface protein from human hepatitis B virus. J. Biol. Chem. 1999, 274, 11945–11957. [Google Scholar] [CrossRef]
- Schmitt, S.; Glebe, D.; Tolle, T.K.; Lochnit, G.; Linder, D.; Geyer, R.; Gerlich, W.H. Structure of pre-S2 N- and O-linked glycans in surface proteins from different genotypes of hepatitis B virus. J. Gen. Virol. 2004, 85, 2045–2053. [Google Scholar] [CrossRef]
- Mehta, A.; Lu, X.; Block, T.M.; Blumberg, B.S.; Dwek, R.A. Hepatitis B virus (HBV) envelope glycoproteins vary drastically in their sensitivity to glycan processing: Evidence that alteration of a single N-linked glycosylation site can regulate HBV secretion. Proc. Natl. Acad. Sci. USA 1997, 94, 1822–1827. [Google Scholar] [CrossRef]
- Welply, J.K.; Shenbagamurthi, P.; Lennarz, W.J.; Naider, F. Substrate recognition by oligosaccharyltransferase. Studies on glycosylation of modified Asn-X-Thr/Ser tripeptides. J. Biol. Chem. 1983, 258, 11856–11863. [Google Scholar] [PubMed]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef] [PubMed]
- Datema, R.; Olofsson, S.; Romero, P.A. Inhibitors of protein glycosylation and glycoprotein processing in viral systems. Pharmacol. Ther. 1987, 33, 221–286. [Google Scholar] [CrossRef]
- Hebert, D.N.; Foellmer, B.; Helenius, A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 1995, 81, 425–433. [Google Scholar] [CrossRef]
- Werr, M.; Prange, R. Role for calnexin and N-linked glycosylation in the assembly and secretion of hepatitis B virus middle envelope protein particles. J. Virol. 1998, 72, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Bruss, V.; Yen, T.S. Formation of intracellular particles by hepatitis B virus large surface protein. J. Virol. 1997, 71, 5487–5494. [Google Scholar] [CrossRef]
- Lazar, C.; Uta, M.; Petrescu, S.M.; Branza-Nichita, N. Novel function of the endoplasmic reticulum degradation-enhancing alpha-mannosidase-like proteins in the human hepatitis B virus life cycle, mediated by the middle envelope protein. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Wu, J.; Dong, M.; Shen, Z.; Lin, Y.; Li, F.; Zhang, Y.; Mao, R.; Lu, M.; et al. Host Gene SEL1L Involved in Endoplasmic Reticulum-Associated Degradation Pathway Could Inhibit Hepatitis B Virus at RNA, DNA, and Protein Levels. Front. Microbiol. 2019, 10, 2869. [Google Scholar] [CrossRef]
- Block, T.M.; Lu, X.; Platt, F.M.; Foster, G.R.; Gerlich, W.H.; Blumberg, B.S.; Dwek, R.A. Secretion of human hepatitis B virus is inhibited by the imino sugar N-butyldeoxynojirimycin. Proc. Natl. Acad. Sci. USA 1994, 91, 2235–2239. [Google Scholar] [CrossRef]
- Lu, X.; Mehta, A.; Dwek, R.; Butters, T.; Block, T. Evidence that N-linked glycosylation is necessary for hepatitis B virus secretion. Virology 1995, 213, 660–665. [Google Scholar] [CrossRef]
- Patzer, E.J.; Nakamura, G.R.; Yaffe, A. Intracellular transport and secretion of hepatitis B surface antigen in mammalian cells. J. Virol. 1984, 51, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mehta, A.; Dadmarz, M.; Dwek, R.; Blumberg, B.S.; Block, T.M. Aberrant trafficking of hepatitis B virus glycoproteins in cells in which N-glycan processing is inhibited. Proc. Natl. Acad. Sci. USA 1997, 94, 2380–2385. [Google Scholar] [CrossRef]
- Simsek, E.; Mehta, A.; Zhou, T.; Dwek, R.A.; Block, T. Hepatitis B virus large and middle glycoproteins are degraded by a proteasome pathway in glucosidase-inhibited cells but not in cells with functional glucosidase enzyme. J. Virol. 2005, 79, 12914–12920. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, J.; Protzer, U.; Siddiqui, A. Revisiting Hepatitis B Virus: Challenges of Curative Therapies. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Himmelsbach, K.; Ren, H.; Boller, K.; Hildt, E. Subviral Hepatitis B Virus Filaments, like Infectious Viral Particles, Are Released via Multivesicular Bodies. J. Virol. 2015, 90, 3330–3341. [Google Scholar] [CrossRef]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef]
- Lazar, C.; Durantel, D.; Macovei, A.; Zitzmann, N.; Zoulim, F.; Dwek, R.A.; Branza-Nichita, N. Treatment of hepatitis B virus-infected cells with alpha-glucosidase inhibitors results in production of virions with altered molecular composition and infectivity. Antivir. Res. 2007, 76, 30–37. [Google Scholar] [CrossRef]
- Sureau, C.; Fournier-Wirth, C.; Maurel, P. Role of N glycosylation of hepatitis B virus envelope proteins in morphogenesis and infectivity of hepatitis delta virus. J. Virol. 2003, 77, 5519–5523. [Google Scholar] [CrossRef][Green Version]
- Chang, J.; Block, T.M.; Guo, J.T. Antiviral therapies targeting host ER alpha-glucosidases: Current status and future directions. Antivir. Res. 2013, 99, 251–260. [Google Scholar] [CrossRef]
- Block, T.M.; Lu, X.; Mehta, A.S.; Blumberg, B.S.; Tennant, B.; Ebling, M.; Korba, B.; Lansky, D.M.; Jacob, G.S.; Dwek, R.A. Treatment of chronic hepadnavirus infection in a woodchuck animal model with an inhibitor of protein folding and trafficking. Nat. Med. 1998, 4, 610–614. [Google Scholar] [CrossRef]
- Barker, M.K.; Rose, D.R. Specificity of Processing alpha-glucosidase I is guided by the substrate conformation: Crystallographic and in silico studies. J. Biol. Chem. 2013, 288, 13563–13574. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.T.; Alonzi, D.S.; Marti, L.; Reca, I.B.; Kiappes, J.L.; Struwe, W.B.; Cross, A.; Basu, S.; Lowe, E.D.; Darlot, B.; et al. Structures of mammalian ER alpha-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proc. Natl. Acad. Sci. USA 2016, 113, E4630–E4638. [Google Scholar] [CrossRef] [PubMed]
- Olivari, S.; Molinari, M. Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 2007, 581, 3658–3664. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef]
- Ninagawa, S.; Okada, T.; Sumitomo, Y.; Kamiya, Y.; Kato, K.; Horimoto, S.; Ishikawa, T.; Takeda, S.; Sakuma, T.; Yamamoto, T.; et al. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 2014, 206, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gao, B.; Ye, L.; Han, X.; Wang, W.; Kong, L.; Fang, X.; Zeng, Y.; Zheng, H.; Li, S.; et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007, 124, 44–49. [Google Scholar] [CrossRef]
- Lazar, C.; Macovei, A.; Petrescu, S.; Branza-Nichita, N. Activation of ERAD pathway by human hepatitis B virus modulates viral and subviral particle production. PLoS ONE 2012, 7, e34169. [Google Scholar] [CrossRef]
- Hauri, H.; Appenzeller, C.; Kuhn, F.; Nufer, O. Lectins and traffic in the secretory pathway. FEBS Lett. 2000, 476, 32–37. [Google Scholar] [CrossRef]
- Reitter, J.N.; Means, R.E.; Desrosiers, R.C. A role for carbohydrates in immune evasion in AIDS. Nat. Med. 1998, 4, 679–684. [Google Scholar] [CrossRef]
- Van Vliet, S.J.; den Dunnen, J.; Gringhuis, S.I.; Geijtenbeek, T.B.; van Kooyk, Y. Innate signaling and regulation of Dendritic cell immunity. Curr. Opin. Immunol. 2007, 19, 435–440. [Google Scholar] [CrossRef]
- Figdor, C.G.; van Kooyk, Y.; Adema, G.J. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol. 2002, 2, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Moris, A.; Pajot, A.; Blanchet, F.; Guivel-Benhassine, F.; Salcedo, M.; Schwartz, O. Dendritic cells and HIV-specific CD4+ T cells: HIV antigen presentation, T-cell activation, and viral transfer. Blood 2006, 108, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Martin-Moreno, A.; Munoz-Fernandez, M.A. Dendritic Cells, the Double Agent in the War Against HIV-1. Front. Immunol. 2019, 10, 2485. [Google Scholar] [CrossRef] [PubMed]
- Op den Brouw, M.L.; de Jong, M.A.; Ludwig, I.S.; van der Molen, R.G.; Janssen, H.L.; Geijtenbeek, T.B.; Woltman, A.M. Branched oligosaccharide structures on HBV prevent interaction with both DC-SIGN and L-SIGN. J. Viral Hepat. 2008, 15, 675–683. [Google Scholar] [CrossRef]
- Wang, M.; Zou, X.; Tian, D.; Hu, S.; Jiang, L. Role of Dendritic Cell-Specific ICAM-3-Grabbing Nonintegrin on Dendritic Cells in the Recognition of Hepatitis B Virus. Viral Immunol. 2015, 28, 331–338. [Google Scholar] [CrossRef]
- Mishra, H.; Mishra, D.; Mishra, P.K.; Nahar, M.; Dubey, V.; Jain, N.K. Evaluation of solid lipid nanoparticles as carriers for delivery of hepatitis B surface antigen for vaccination using subcutaneous route. J. Pharm. Pharm. Sci. 2010, 13, 495–509. [Google Scholar] [CrossRef]
- Hyakumura, M.; Walsh, R.; Thaysen-Andersen, M.; Kingston, N.J.; La, M.; Lu, L.; Lovrecz, G.; Packer, N.H.; Locarnini, S.; Netter, H.J. Modification of Asparagine-Linked Glycan Density for the Design of Hepatitis B Virus Virus-Like Particles with Enhanced Immunogenicity. J. Virol. 2015, 89, 11312–11322. [Google Scholar] [CrossRef]
- Rottinghaus, S.T.; Poland, G.A.; Jacobson, R.M.; Barr, L.J.; Roy, M.J. Hepatitis B DNA vaccine induces protective antibody responses in human non-responders to conventional vaccination. Vaccine 2003, 21, 4604–4608. [Google Scholar] [CrossRef]
- Xing, Y.; Huang, Z.; Lin, Y.; Li, J.; Chou, T.H.; Lu, S.; Wang, S. The ability of Hepatitis B surface antigen DNA vaccine to elicit cell-mediated immune responses, but not antibody responses, was affected by the deglysosylation of S antigen. Vaccine 2008, 26, 5145–5152. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Jia, Y.; Li, J.; Huang, Z.; Lu, S.; Xing, Y. N-Linked Glycosylation at an Appropriate Position in the Pre-S2 Domain Is Critical for Cellular and Humoral Immunity against Middle HBV Surface Antigen. Tohoku J. Exp. Med. 2015, 236, 131–138. [Google Scholar] [CrossRef]
- Simsek, E.; Sinnathamby, G.; Block, T.M.; Liu, Y.; Philip, R.; Mehta, A.S.; Norton, P.A. Inhibition of cellular alpha-glucosidases results in increased presentation of hepatitis B virus glycoprotein-derived peptides by MHC class I. Virology 2009, 384, 12–15. [Google Scholar] [CrossRef]
- Yu, D.M.; Li, X.H.; Mom, V.; Lu, Z.H.; Liao, X.W.; Han, Y.; Pichoud, C.; Gong, Q.M.; Zhang, D.H.; Zhang, Y.; et al. N-glycosylation mutations within hepatitis B virus surface major hydrophilic region contribute mostly to immune escape. J. Hepatol. 2014, 60, 515–522. [Google Scholar] [CrossRef]
- Chen, Y.; Qian, F.; Yuan, Q.; Li, X.; Wu, W.; Guo, X.; Li, L. Mutations in hepatitis B virus DNA from patients with coexisting HBsAg and anti-HBs. J. Clin. Virol. 2011, 52, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Kohno, H.; Inoue, T.; Tsuda, F.; Okamoto, H.; Akahane, Y. Mutations in the envelope gene of hepatitis B virus variants co-occurring with antibody to surface antigen in sera from patients with chronic hepatitis B. J. Gen. Virol. 1996, 77 Pt 8, 1825–1831. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, X.; Tian, Y.; Song, J.; Yang, D.; Roggendorf, M.; Lu, M.; Chen, X. Biological significance of amino acid substitutions in hepatitis B surface antigen (HBsAg) for glycosylation, secretion, antigenicity and immunogenicity of HBsAg and hepatitis B virus replication. J. Gen. Virol. 2010, 91, 483–492. [Google Scholar] [CrossRef]
- Kang, Y.; Li, F.; Guo, H.; Yang, S.; Zhang, Y.; Zhu, H.; Wang, J.; Mao, R.; Qin, Y.; Xu, J.; et al. Amino acid substitutions Q129N and T131N/M133T in hepatitis B surface antigen (HBsAg) interfere with the immunogenicity of the corresponding HBsAg or viral replication ability. Virus Res. 2018, 257, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Salpini, R.; Piermatteo, L.; Battisti, A.; Colagrossi, L.; Aragri, M.; Yu La Rosa, K.; Bertoli, A.; Saccomandi, P.; Lichtner, M.; Marignani, M.; et al. A Hyper-Glycosylation of HBV Surface Antigen Correlates with HBsAg-Negativity at Immunosuppression-Driven HBV Reactivation in Vivo and Hinders HBsAg Recognition in Vitro. Viruses 2020, 12, 251. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, Y.; Zhao, J.; Xu, Z.; Chen, R.; Si, L.; Lu, S.; Li, X.; Wang, S.; Zhang, K.; et al. Characterization of Novel Hepatitis B Virus PreS/S-Gene Mutations in a Patient with Occult Hepatitis B Virus Infection. PLoS ONE 2016, 11, e0155654. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Qin, Y.; Guarnieri, M.; Garcia, T.; Kwei, K.; Mizokami, M.; Zhang, J.; Li, J.; Wands, J.R.; Tong, S. Impairment of hepatitis B virus virion secretion by single-amino-acid substitutions in the small envelope protein and rescue by a novel glycosylation site. J. Virol. 2010, 84, 12850–12861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chang, L.; Laperche, S.; Ji, H.; Zhao, J.; Jiang, X.; Wang, L.; Candotti, D. Occult HBV infection in Chinese blood donors: Role of N-glycosylation mutations and amino acid substitutions in S protein transmembrane domains. Emerg. Microbes Infect. 2019, 8, 1337–1346. [Google Scholar] [CrossRef]
- Hoofnagle, J.H. Reactivation of hepatitis B. Hepatology 2009, 49, S156–S165. [Google Scholar] [CrossRef] [PubMed]
- Salpini, R.; Colagrossi, L.; Bellocchi, M.C.; Surdo, M.; Becker, C.; Alteri, C.; Aragri, M.; Ricciardi, A.; Armenia, D.; Pollicita, M.; et al. Hepatitis B surface antigen genetic elements critical for immune escape correlate with hepatitis B virus reactivation upon immunosuppression. Hepatology 2015, 61, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, G.; Locarnini, S.; Pollicino, T.; Levrero, M.; Zoulim, F.; Lok, A.S.; Allain, J.P.; Berg, T.; Bertoletti, A.; Brunetto, M.R.; et al. Update of the statements on biology and clinical impact of occult hepatitis B virus infection. J. Hepatol. 2019, 71, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Lafaro, K.J.; Demirjian, A.N.; Pawlik, T.M. Epidemiology of hepatocellular carcinoma. Surg. Oncol. Clin. N. Am. 2015, 24, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Coppola, N.; Onorato, L.; Iodice, V.; Starace, M.; Minichini, C.; Farella, N.; Liorre, G.; Filippini, P.; Sagnelli, E.; de Stefano, G. Occult HBV infection in HCC and cirrhotic tissue of HBsAg-negative patients: A virological and clinical study. Oncotarget 2016, 7, 62706–62714. [Google Scholar] [CrossRef]
- Chen, X.; Wu, F.; Liu, Y.; Lou, J.; Zhu, B.; Zou, L.; Chen, W.; Gong, J.; Wang, Y.; Zhong, R. The contribution of serum hepatitis B virus load in the carcinogenesis and prognosis of hepatocellular carcinoma: Evidence from two meta-analyses. Oncotarget 2016, 7, 49299–49309. [Google Scholar] [CrossRef]
- Jang, J.S.; Kim, H.S.; Kim, H.J.; Shin, W.G.; Kim, K.H.; Lee, J.H.; Kim, H.Y.; Kim, D.J.; Lee, M.S.; Park, C.K.; et al. Association of concurrent hepatitis B surface antigen and antibody to hepatitis B surface antigen with hepatocellular carcinoma in chronic hepatitis B virus infection. J. Med. Virol. 2009, 81, 1531–1538. [Google Scholar] [CrossRef]
- Seo, S.I.; Choi, H.S.; Choi, B.Y.; Kim, H.S.; Kim, H.Y.; Jang, M.K. Coexistence of hepatitis B surface antigen and antibody to hepatitis B surface may increase the risk of hepatocellular carcinoma in chronic hepatitis B virus infection: A retrospective cohort study. J. Med. Virol. 2014, 86, 124–130. [Google Scholar] [CrossRef]
- Qiao, Y.; Lu, S.; Xu, Z.; Li, X.; Zhang, K.; Liu, Y.; Zhao, L.; Chen, R.; Si, L.; Lin, S.; et al. Additional N-glycosylation mutation in the major hydrophilic region of hepatitis B virus S gene is a risk indicator for hepatocellular carcinoma occurrence in patients with coexistence of HBsAg/anti-HBs. Oncotarget 2017, 8, 61719–61730. [Google Scholar] [CrossRef]
- Wang, H.C.; Huang, W.; Lai, M.D.; Su, I.J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mutation | Glycosylation | Properties | Reference |
|---|---|---|---|
| T123N | Yes | Poor recognition by HBsAg antibodies (Abs) Triggers anti-HBsAg humoral response Strongly reduces assembly of HBV (genotype C) | [56] |
| Significantly reduced reactivity with commercial HBsAg ELISA kits Able to induce protective HBsAg Abs titers Normal viral replication | [57] | ||
| Isolated from patients with HBV reactivation Reduced reactivity with commercial HBsAg ELISA kits HBsAg negativity at reactivation No effect on HBV replication (genotype D) No effect on HBsAg secretion | [58] | ||
| T115N T117N 114N-insertion S113N+T131N+M133T * | Yes | Isolated from patients with HBV reactivation Reduced reactivity with commercial HBsAg ELISA kits HBsAg negativity at reactivation No effect on HBV replication (genotype D) No effect on HBsAg secretion | [58] |
| K160N | Yes | Poor recognition by HBsAg Abs Triggers anti-HBsAg humoral response Enhanced cellular immune response Compensates for the negative effect of A159G mutation on HBV production | [56] |
| G130N ** | NE | Weak recognition by a conformational HBsAg Abs | [55] |
| Q129N | Yes | Decreased binding to HBsAg Abs Increased HBV production in trans-complementary studies | [53] |
| Impaired recognition by HBsAg Abs Poor immunogenicity, still protective HBsAg Abs titers Impairs viral replication but not HBsAg secretion | [57] | ||
| Decreased binding to HBsAg Abs (by 70.3%) Increased HBV secretion in trans-complementary studies | *** [59] | ||
| T131N/M133T # | Yes | Impaired recognition by HBsAg Abs Poor immunogenicity, still protective HBsAg Abs titers Normal viral replication | [57] |
| Decreased binding to HBsAg Abs (by 84.5%) Increased HBV secretion in trans-complementary studies | *** [59] | ||
| T131N # | Yes | Decreased binding to anti-HBsAg Abs Increased HBV production in trans-complementary studies | [53] |
| “RPCMNCTI” insertion between residues 126−127 | Yes | Moderate decrease of HBsAg Abs binding (by 23.2%) Normal HBV secretion in trans-complementary studies | *** [59] |
| 3 aa insertion between residues 114-115 | Yes | Decreased binding to anti-HBsAg Abs Increased HBV production in trans-complementary studies | [53] |
| M133T # | Yes | Increased virion secretion Restores virion secretion of G119E, G145R, I110M, R169P immune-escape mutants but not recognition by the neutralizing Abs. | [60] |
| T116N (genotype B) | Yes | No significant change of HBsAg antigenicity | *** [61] |
| TCT123-125NFT (genotype B) TCT123-125NCT (genotypes B and C) | Yes | Low reactivity against HBsAg Abs Impaired HBsAg secretion | *** [61] |
| TSM131-133NSS (genotype B) TSM131-133NST/NYT (genotypes B and C) | Yes | No significant change of HBsAg antigenicity | *** [61] |
| NCT146-148SCT/YCT (genotype C) NCT146-148DCT (genotypes B and C) | No Removes the conserved N146 glycosylation site | No significant change of HBsAg antigenicity -Normal HBsAg secretion | *** [61] |
| GTS130-132NTS (genotypes B and C) | Yes | No significant change of HBsAg antigenicity | *** [61] |
| GSS112-114NAT (genotype B) TTS115-117NTS (genotype C) | No | No significant change of HBsAg antigenicity | *** [61] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobrica, M.-O.; Lazar, C.; Branza-Nichita, N. N-Glycosylation and N-Glycan Processing in HBV Biology and Pathogenesis. Cells 2020, 9, 1404. https://doi.org/10.3390/cells9061404
Dobrica M-O, Lazar C, Branza-Nichita N. N-Glycosylation and N-Glycan Processing in HBV Biology and Pathogenesis. Cells. 2020; 9(6):1404. https://doi.org/10.3390/cells9061404
Chicago/Turabian StyleDobrica, Mihaela-Olivia, Catalin Lazar, and Norica Branza-Nichita. 2020. "N-Glycosylation and N-Glycan Processing in HBV Biology and Pathogenesis" Cells 9, no. 6: 1404. https://doi.org/10.3390/cells9061404
APA StyleDobrica, M.-O., Lazar, C., & Branza-Nichita, N. (2020). N-Glycosylation and N-Glycan Processing in HBV Biology and Pathogenesis. Cells, 9(6), 1404. https://doi.org/10.3390/cells9061404