Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing



Abstract
1. Introduction
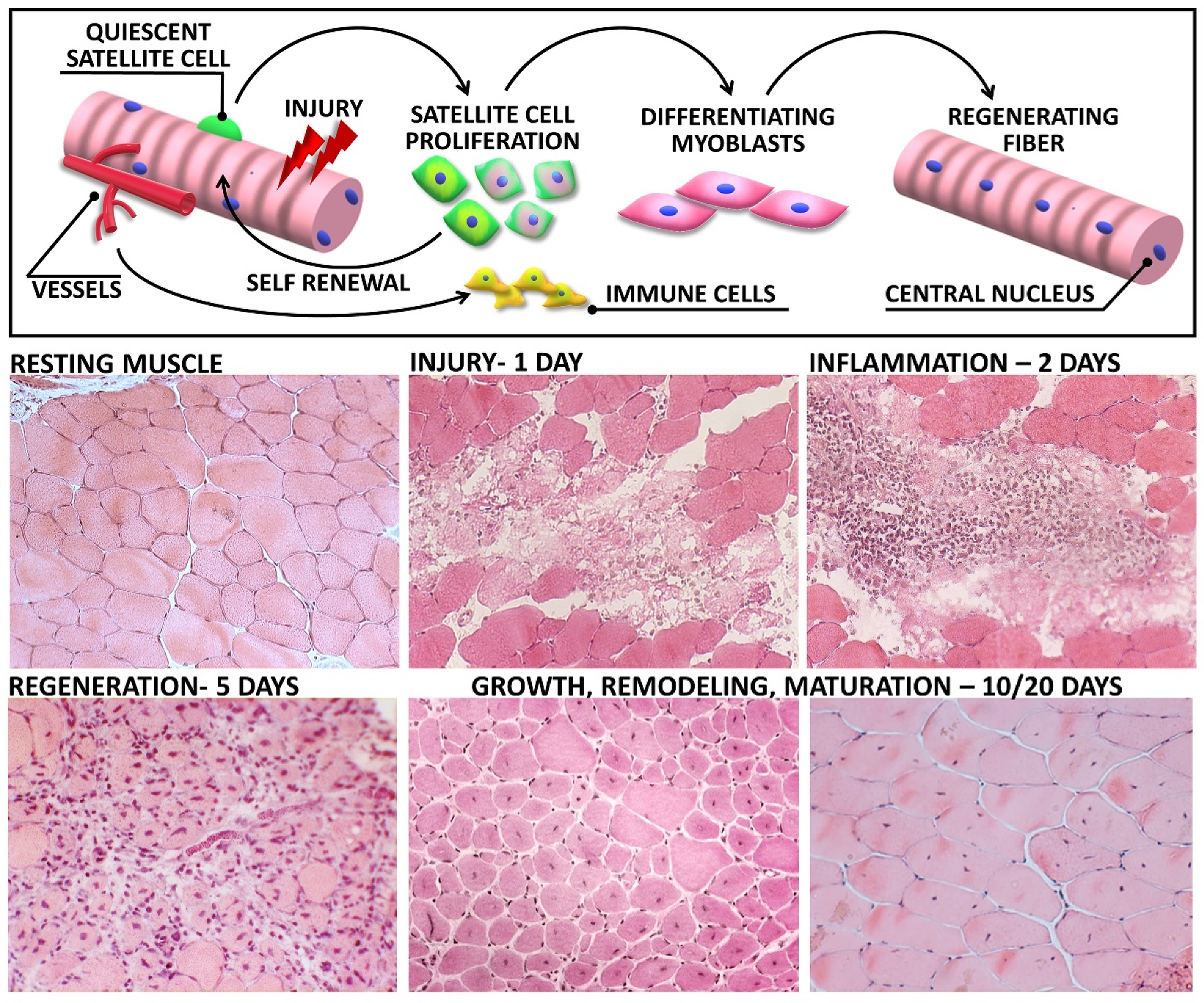
2. From Tissue Destruction to Recovery: Highlighting the Stages of Muscle Regeneration
2.1. Muscle Degeneration
2.2. Inflammatory Waves
2.3. Regeneration
2.3.1. The Role of Satellite Cells
2.3.2. The Role of “Non-Muscle” Stem Cells in Muscle Regeneration
2.4. Tissue Remodelling and Maturation
2.5. Re-Innervation and Functional Recovery
3. The Dynamic and the Regulation of Regenerative Phases are Altered in Pathologic Conditions: The Case of Muscular Dystrophy
4. Technical Approaches to Induce Experimental Muscle Damage and Regeneration
4.1. Models of Physical Injury
4.1.1. Freeze Injury
4.1.2. Crush Injury and Ischemia-Reperfusion Damage
4.2. Chemical Damage Induced by Myotoxic Agents
4.2.1. Cardiotoxin Injection
4.2.2. Notexin Injection
4.2.3. Bupivacaine Administration
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AchR | Acetylcholine receptor |
| Activ.SCs | Activated SCs |
| ADAM | A Disintegrin and Metalloproteinase |
| ADAM8 | A Disintegrin and Metalloproteinase Domain-Containing Protein 8 |
| ADP | Adenosine diphosphate |
| ALB | Albumin |
| BPVC | Bupivacaine |
| BrdU | 5-bromo-2′-deoxyuridine |
| BTX | α-Bungarotoxin |
| Cav1 | Caveolin 1 |
| CCR2 | Chemokine Receptor type 2 |
| CD11b | Cluster of Differentiation 11 b also known as Integrin Alpha M |
| CD133 | Cluster of Differentiation 133 |
| CD163 | Cluster of Differentiation 163 |
| CD206 | Cluster of Differentiation 206 also known as Mannose receptor C-type 1 |
| CD31 | Cluster of Differentiation 31 |
| CD34 | Cluster of Differentiation 34 |
| CD38 | Cluster of Differentiation 38 |
| CD45 | Cluster of Differentiation 45 |
| CD68 | Cluster of Differentiation 68 |
| CK | Creatine kinase |
| CK3 (CK-MM) | Creatine Kinase MM isoform |
| c-Met | Tyrosine-protein Kinase Met |
| c-Myc | MYC proto-oncogene, bHLH transcription factor |
| CS | Compartment Syndrome |
| CTR | Calcitonin Receptor |
| CTX | Cardiotoxin |
| CX3CR1 | C-X3-C Motif Chemokine Receptor 1 |
| CXCR4 | C-X-C Chemokine Receptor type 4 |
| DAMPs | Damage-Associated Molecular Patterns |
| DGC | Dystrophin-associated Glycoprotein Complex |
| Diff.SCs | Differentiating SCs |
| DMD | Duchenne Muscular Dystrophy |
| EBD | Evans Blue Dye |
| ECM | Extracellular Matrix |
| ED1 | Monoclonal antibody staining a single chain glycoprotein of 110 kDa on the lysosomal membrane of myeloid cells, i.e., the majority of tissue macrophages (being the rat homologue of human CD68) |
| ED2 | Monoclonal antibody reacting with a membrane antigen (175, 160, and 95 kDa) on resident rat macrophages such as monocytes and dendritic cells. ED2 discriminates between thymic cortical (positive for ED2) and medullary (negative for ED2) macrophages. The antigen is identical with CD163. |
| ED3 | Monoclonal antibody recognizing the rat CD169 cell surface antigen, a 185 kDa molecule expressed by macrophages in lymphoid organs (no monocytes or granulocytes). In the thymus, the antigen is expressed on clusters of dendritic cells (thymic nurse cells or TNC’s) in the (outer) cortex. |
| EDL | Extensor Digitorum Longus |
| EdU | 5-ethynyl-2′-deoxyuridine |
| Egr2 | Early growth response protein 2 |
| eMyHC | embryonal Myosin Heavy Chain |
| ENO3 | Enolase 3 |
| F3BA | Picrosirius red |
| F4/80 | Mouse macrophage marker. Also known as Ly71 and EMR1, the F4/80 antigen is part of the EGF-TM7 family. |
| FACS | Fluorescence-Activated Cell Sorting |
| FAPs | Fibroadipogenic Progenitors |
| FI | Freeze Injury |
| FoxK | Forkhead box protein K |
| Fpr2 | Formyl peptide receptor 2 |
| Gpr18 | G-protein coupled receptor 18 |
| Gr1 | Granulocyte antigen 1 |
| GSK3 | Glycogen Synthase Kinase 3 |
| H&E | Haematoxylin and Eosin staining |
| H2O2 | Hydrogen peroxide |
| Hey1 | Hairy/enhancer-of-split related with YRPW motif protein 1 |
| Heyl | Hairy/enhancer-of-split related with YRPW motif protein l |
| IFN-β | Interferon-β |
| IFN-γ | Interferon-γ |
| IGF-1 | Insulin-like Growth Factor 1 |
| IgG | Immunoglobulin G |
| IL-1 | Interleukin 1 |
| IL-1β | Interleukin-1β |
| IL-4 | Interleukin 4 |
| IL-6 | Interleukin 6 |
| IL6R | IL-6 receptor alpha |
| IL-8 | Interleukin 8; |
| Ki-67 protein (also known as MKI67) | marker of proliferation KI-67 |
| LDH | Lactate Dehydrogenase |
| Ly6C | Lymphocyte antigen 6 complex, locus C |
| Ly6G | Lymphocyte antigen 6 complex, locus G |
| Ly6G-PA-GFP | Ly6Gpos. cells with a photoactivatable GFP |
| MAC | Macrophages |
| Mcad | M-cadherin |
| MCK | Muscle Creatine Kinase |
| MCP-1 | Monocyte Chemoattractant Protein 1 |
| MFs | Myogenic Factors |
| MyHC | Myosin Heavy Chain |
| miRNAs | microRNAs |
| MPO | Myeloperoxidase |
| Mrf4 | Myogenic regulatory factor 4 |
| Myf-5 | Myogenic factor 5 |
| MyoD | Myoblast determination protein |
| myomiRs | microRNAs involved in the regulation of myocellular processes |
| nAChR | nicotinic Acetylcholine Receptor |
| NCAM | Neural Cell Adhesion Molecule |
| NEU | Neutrophils |
| NMJs | Neuromuscular Junctions |
| NTX | Notexin |
| P38 MAP kinases | P38 mitogen-activated protein kinases |
| Pax3 | Paired box transcription factor 3 |
| Pax7 | Paired box transcription factor 7 |
| PBS | Phosphate Buffered Saline |
| PCNA | Proliferating Cell Nuclear Antigen |
| PDGF-R-alpha | Platelet derived growth factor receptor alpha |
| Prolif.SCs | Proliferating SCs |
| Sca-1 | Stem cell antigen-1 |
| SCs | Satellite Cells |
| sIL6R | soluble Interleukin-6 Receptor alpha |
| Sox 15 | SRY-Box Transcription Factor 15 |
| Sox 8 | SRY-Box Transcription Factor 8 |
| TA | Tibialis Anterior muscle |
| Ter 119 or Ly76 | Lymphocyte antigen-76 |
| TGFβ | Transforming Growth Factor beta |
| TNF-α | Tumor Necrosis Factor alpha |
| VCAM1 | Vascular Cell Adhesion protein 1 |
| WNT | Wingless-related integration site |
References
- Forcina, L.; Miano, C.; Pelosi, L.; Musarò, A. An Overview About the Biology of Skeletal Muscle Satellite Cells. Curr. Genom. 2019, 20, 24–37. [Google Scholar] [CrossRef]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Blaauw, B.; Reggiani, C. The role of satellite cells in muscle hypertrophy. J. Muscle Res. Cell Motil. 2014, 35, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Xue, X.; Yu, P.; Ni, Y.; Chen, F. Evans Blue Dye: A Revisit of Its Applications in Biomedicine. Contrast Media Mol. Imaging 2018, 2018, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Rafael, J.A.; Chamberlain, J.S.; Campbell, K.P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997, 139, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Chen, F.; Ma, Z.; Dekeyzer, F.; Yu, J.; Xie, Y.; Cona, M.M.; Oyen, R.; Ni, Y. Towards stratifying ischemic components by cardiac MRI and multifunctional stainings in a rabbit model of myocardial infarction. Theranostics 2014, 4, 24–35. [Google Scholar] [CrossRef]
- Cona, M.M.; Koole, M.; Feng, Y.; Liu, Y.; Verbruggen, A.; Oyen, R.; Ni, Y. Biodistribution and radiation dosimetry of radioiodinated hypericin as a cancer therapeutic. Int. J. Oncol. 2014, 44, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Klyen, B.R.; Shavlakadze, T.; Radley-Crabb, H.G.; Grounds, M.D.; Sampson, D.D. Identification of muscle necrosis in the mdx mouse model of Duchenne muscular dystrophy using three-dimensional optical coherence tomography. J. Biomed. Opt. 2011, 16, 076013. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Zhang, G.; Griffin, J.B.; Hegge, J.O.; Huss, T.; Wolff, J.A. Use of Evans blue dye to compare limb muscles in exercised young and old mdx mice. Muscle Nerve 2010, 41, 487–499. [Google Scholar] [CrossRef]
- Hamer, P.W.; McGeachie, J.M.; Davies, M.J.; Grounds, M.D. Evans Blue Dye as an in vivo marker of myofibre damage: Optimising parameters for detecting initial myofibre membrane permeability. J. Anat. 2002, 200, 69–79. [Google Scholar] [CrossRef]
- Pelosi, L.; Berardinelli, M.G.; Forcina, L.; Spelta, E.; Rizzuto, E.; Nicoletti, C.; Camilli, C.; Testa, E.; Catizone, A.; De Benedetti, F.; et al. Increased levels of interleukin-6 exacerbate the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2015, 24, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A. The Basis of Muscle Regeneration. Adv. Biol. 2014, 2014, 1–16. [Google Scholar] [CrossRef]
- Matsuda, R.; Nishikawa, A.; Tanaka, H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with evans blue: Evidence of apoptosis in dystrophin-deficient muscle. J. Biochem. 1995, 118, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Lai, T.W. Optimization of Evans blue quantitation in limited rat tissue samples. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.E.; Prola, A.; Mariot, V.; Pini, V.; Meng, J.; Hourde, C.; Dumonceaux, J.; Conti, F.; Relaix, F.; Authier, F.J.; et al. Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat. Commun. 2018, 9, 3655. [Google Scholar] [CrossRef]
- Brancaccio, P.; Lippi, G.; Maffulli, N. Biochemical markers of muscular damage. Clin. Chem. Lab. Med. 2010, 48, 757–767. [Google Scholar] [CrossRef]
- Komulainen, J.; Kytola, J.; Vihko, V. Running-induced muscle injury and myocellular enzyme release in rats. J. Appl. Physiol. 1994, 77, 2299–2304. [Google Scholar] [CrossRef]
- Siracusa, J.; Koulmann, N.; Bourdon, S.; Goriot, M.E.; Banzet, S. Circulating miRNAs as Biomarkers of Acute Muscle Damage in Rats. Am. J. Pathol. 2016, 186, 1313–1327. [Google Scholar] [CrossRef]
- Nishimura, D.; Sakai, H.; Sato, T.; Sato, F.; Nishimura, S.; Toyama-Sorimachi, N.; Bartsch, J.W.; Sehara-Fujisawa, A. Roles of ADAM8 in elimination of injured muscle fibers prior to skeletal muscle regeneration. Mech. Dev. 2015, 135, 58–67. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef]
- Yang, W.; Hu, P. Skeletal muscle regeneration is modulated by inflammation. J. Orthop. Transl. 2018, 13, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion During Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Orekhova, V.A.; Nikiforov, N.G.; Myasoedova, V.A.; Grechko, A.V.; Romanenko, E.B.; Zhang, D.; Chistiakov, D.A. Monocyte differentiation and macrophage polarization. Vessel Plus 2019, 3, 10. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.J.; Donners, M.M.P.C. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; de Dios Ruiz-Rosado, J.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [PubMed]
- Kastenschmidt, J.M.; Avetyan, I.; Armando Villalta, S. Characterization of the inflammatory response in dystrophic muscle using flow cytometry. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; pp. 43–56. [Google Scholar] [CrossRef]
- Crane, M.J.; Daley, J.M.; van Houtte, O.; Brancato, S.K.; Henry, W.L.; Albina, J.E. The Monocyte to Macrophage Transition in the Murine Sterile Wound. PLoS ONE 2014, 9, e86660. [Google Scholar] [CrossRef]
- Gnocchi, V.F.; White, R.B.; Ono, Y.; Ellis, J.A.; Zammit, P.S. Further Characterisation of the Molecular Signature of Quiescent and Activated Mouse Muscle Satellite Cells. PLoS ONE 2009, 4, e5205. [Google Scholar] [CrossRef]
- Fukada, S.I.; Yamaguchi, M.; Kokubo, H.; Ogawa, R.; Uezumi, A.; Yoneda, T.; Matev, M.M.; Motohashi, N.; Ito, T.; Zolkiewska, A.; et al. Hesr1 and Hesr3 are essential to generate undifferentiated quiescent satellite cells and to maintain satellite cell numbers. Development 2011, 138, 4609–4619. [Google Scholar] [CrossRef]
- Maesner, C.C.; Almada, A.E.; Wagers, A.J. Established cell surface markers efficiently isolate highly overlapping populations of skeletal muscle satellite cells by fluorescence-activated cell sorting. Skelet. Muscle 2016, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Montarras, D.; Zaffran, S.; Gayraud-Morel, B.; Rocancourt, D.; Tajbakhsh, S.; Mansouri, A.; Cumano, A.; Buckingham, M. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 2006, 172, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Mechtersheimer, G.; Staudter, M.; Möller, P. Expression of the natural killer cell-associated antigens CD56 and CD57 in human neural and striated muscle cells and in their tumors. Cancer Res. 1991, 51, 1300–1307. [Google Scholar] [PubMed]
- Tatsumi, R.; Anderson, J.E.; Nevoret, C.J.; Halevy, O.; Allen, R.E. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev. Biol. 1998, 194, 114–128. [Google Scholar] [CrossRef]
- Jesse, T.L.; LaChance, R.; Iademarco, M.F.; Dean, D.C. Interferon regulatory factor-2 is a transcriptional activator in muscle where it regulates expression of vascular cell adhesion molecule-1. J. Cell Biol. 1998, 140, 1265–1276. [Google Scholar] [CrossRef]
- Cornelison, D.D.W.; Filla, M.S.; Stanley, H.M.; Rapraeger, A.C.; Olwin, B.B. Syndecan-3 and syndecan-4 specifically mark skeletal muscle satellite cells and are implicated in satellite cell maintenance and muscle regeneration. Dev. Biol. 2001, 239, 79–94. [Google Scholar] [CrossRef]
- Sherwood, R.I.; Christensen, J.L.; Conboy, I.M.; Conboy, M.J.; Rando, T.A.; Weissman, I.L.; Wagers, A.J. Isolation of adult mouse myogenic progenitors: Functional heterogeneity of cells within and engrafting skeletal muscle. Cell 2004, 119, 543–554. [Google Scholar] [CrossRef]
- Volonte, D.; Liu, Y.; Galbiati, F. The modulation of caveolin-1 expression controls satellite cell activation during muscle repair. FASEB J. 2005, 19, 1–36. [Google Scholar] [CrossRef]
- Chang, N.C.; Sincennes, M.C.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segalés, J.; Muñoz-Cánoves, P.; Ming, H.; Rudnicki, M.A. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem Cell 2018, 22, 755.e6–768.e6. [Google Scholar] [CrossRef]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Hak, K.K.; Yong, S.L.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Uezumi, A.; Fukada, S.I.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, S.E.; Bishop, J.E.; McGrouther, G.; Laurent, G.J. Mechanisms of tissue repair: From wound healing to fibrosis. Int. J. Biochem. Cell Biol. 1997, 29, 5–17. [Google Scholar] [CrossRef]
- Dunn, A.; Marcinczyk, M.; Talovic, M.; Patel, K.; Haas, G.; Garg, K. Role of Stem Cells and Extracellular Matrix in the Regeneration of Skeletal Muscle. In Muscle Cell and Tissue—Current Status of Research Field; Sakuma, P.K., Ed.; InTechOpen: London, UK, 2018. [Google Scholar]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef]
- Kjær, M. Role of Extracellular Matrix in Adaptation of Tendon and Skeletal Muscle to Mechanical Loading. Physiol. Rev. 2004, 84, 649–698. [Google Scholar] [CrossRef]
- Garg, K.; Boppart, M.D. Influence of exercise and aging on extracellular matrix composition in the skeletal muscle stem cell niche. J. Appl. Physiol. 2016, 121, 1053–1058. [Google Scholar] [CrossRef]
- Sanes, J.R. The basement membrane/basal lamina of skeletal muscle. J. Biol. Chem. 2003, 278, 12601–12604. [Google Scholar] [CrossRef]
- Tu, H.; Zhang, D.; Corrick, R.M.; Muelleman, R.L.; Wadman, M.C.; Li, Y.L. Morphological regeneration and functional recovery of neuromuscular junctions after tourniquet-induced injuries in mouse hindlimb. Front. Physiol. 2017, 8, 207. [Google Scholar] [CrossRef]
- Kirby, T.J.; Chaillou, T.; McCarthy, J.J. The role of microRNAs in skeletal muscle health and disease. Front. Biosci. (Landmark Ed.) 2016, 20, 37–77. [Google Scholar]
- Pelosi, L.; Coggi, A.; Forcina, L.; Musarò, A. MicroRNAs modulated by local mIGF-1 expression in mdx dystrophic mice. Front. Aging Neurosci. 2015, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, G.; Zhang, P.; Fan, J. Programmed cell death and its role in inflammation. Mil. Med. Res. 2015, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M. Phagocytosis of necrotic muscle in muscle isografts is influenced by the strain age and sex of host mice. J. Pathol. 1987, 153, 71–82. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol. 2007, 578, 327–336. [Google Scholar] [CrossRef]
- Summan, M.; Warren, G.L.; Mercer, R.R.; Chapman, R.; Hulderman, T.; Van Rooijen, N.; Simeonova, P.P. Macrophages and skeletal muscle regeneration: A clodronate-containing liposome depletion study. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1488–R1495. [Google Scholar] [CrossRef]
- Tidball, J.G. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R345–R353. [Google Scholar] [CrossRef]
- Teixeira, C.F.P.; Zamunér, S.R.; Zuliani, J.P.; Fernandes, C.M.; Cruz-Hofling, M.A.; Fernandes, I.; Chaves, F.; Gutiérrez, J.M. Neutrophils do not contribute to local tissue damage, but play a key role in skeletal muscle regeneration, in mice injected with Bothrops asper snake venom. Muscle Nerve 2003, 28, 449–459. [Google Scholar] [CrossRef]
- Frenette, J.; Cai, B.; Tidball, J.G. Complement activation promotes muscle inflammation during modified muscle use. Am. J. Pathol. 2000, 156, 2103–2110. [Google Scholar] [CrossRef]
- Kishimoto, T.K.; Rothlein, R. Integrins, ICAMs, and Selectins: Role and Regulation of Adhesion Molecules in Neutrophil Recruitment to Inflammatory Sites. Adv. Pharmacol. 1994, 25, 117–169. [Google Scholar] [CrossRef]
- Muller, W.A. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003, 24, 326–333. [Google Scholar] [CrossRef]
- Walzog, B.; Gaehtgens, P. Adhesion Molecules: The Path to a New Understanding of Acute Inflammation. News Physiol. Sci. 2000, 15, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sixt, M.; Hallmann, R.; Wendler, O.; Scharffetter-Kochanek, K.; Sorokin, L.M. Cell adhesion and migration properties of β2-integrin negative polymorphonuclear granulocytes on defined extracellular matrix molecules: Relevance for leukocyte extravasation. J. Biol. Chem. 2001, 276, 18878–18887. [Google Scholar] [CrossRef] [PubMed]
- Pizza, F.X.; Peterson, J.M.; Baas, J.H.; Koh, T.J. Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J. Physiol. 2005, 562, 899–913. [Google Scholar] [CrossRef]
- Chazaud, B.; Brigitte, M.; Yacoub-Youssef, H.; Arnold, L.; Gherardi, R.; Sonnet, C.; Lafuste, P.; Chretien, F. Dual and beneficial roles of macrophages during skeletal muscle regeneration. Exerc. Sport Sci. Rev. 2009, 37, 18–22. [Google Scholar] [CrossRef]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef]
- McLennan, I.S. Resident macrophages (ED2- and ED3-positive) do not phagocytose degenerating rat skeletal muscle fibres. Cell Tissue Res. 1993, 272, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; Van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Silva, M.T. When two is better than one: Macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J. Leukoc. Biol. 2010, 87, 93–106. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yue, F.; Kuang, S. Muscle Histology Characterization Using H&E Staining and Muscle Fiber Type Classification Using Immunofluorescence Staining. Bio Protoc. 2017, 7, e2279. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musarò, A.; Rosenthal, N. Monocyte/macrophage-derived IGF-1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol. Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Scharner, J.; Zammit, P.S. The muscle satellite cell at 50: The formative years. Skelet. Muscle 2011, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Creuzet, S.; Lescaudron, L.; Li, Z.; Fontaine-Pérus, J. MyoD, myogenin, and desmin-nls-lacZ transgene emphasize the distinct patterns of satellite cell activation in growth and regeneration. Exp. Cell Res. 1998, 243, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Yablonka-Reuveni, Z.; Rivera, A.J. Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Dev. Biol. 1994, 164, 588–603. [Google Scholar] [CrossRef]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Mead, T.J.; Lefebvre, V. Proliferation assays (BrdU and EdU) on skeletal tissue sections. Methods Mol. Biol. 2014, 1130, 233–243. [Google Scholar] [CrossRef]
- Ikeda, T.; Ichii, O.; Otsuka-Kanazawa, S.; Nakamura, T.; Elewa, Y.H.A.; Kon, Y. Degenerative and regenerative features of myofibers differ among skeletal muscles in a murine model of muscular dystrophy. J. Muscle Res. Cell Motil. 2016, 37, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Suzuki, M.; Nakagawa, H.; Ninagawa, N.; Torihashi, S. Switching of actin isoforms in skeletal muscle differentiation using mouse ES cells. Histochem. Cell Biol. 2009, 132, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Agarwal, M.; Kumar, A.; Kumar, P.; Saini, M.; Kardon, G.; Mathew, S.J. Myosin Heavy Chain-embryonic is a crucial regulator of skeletal muscle development and differentiation. bioRxiv 2018, 261685. [Google Scholar] [CrossRef]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef]
- Guiraud, S.; Edwards, B.; Squire, S.E.; Moir, L.; Berg, A.; Babbs, A.; Ramadan, N.; Wood, M.J.; Davies, K.E. Embryonic myosin is a regeneration marker to monitor utrophin-based therapies for DMD. Hum. Mol. Genet. 2019, 28, 307–319. [Google Scholar] [CrossRef]
- De Angelis, L.; Berghella, L.; Coletta, M.; Lattanzi, L.; Zanchi, M.; Cusella-De Angelis, M.G.; Ponzetto, C.; Cossu, G. Skeletal myogenic progenitors originating from embryonic dorsal aorta coexpress endothelial and myogenic markers and contribute to postnatal muscle growth and regeneration. J. Cell Biol. 1999, 147, 869–877. [Google Scholar] [CrossRef]
- Kuang, S.; Chargé, S.B.; Seale, P.; Huh, M.; Rudnicki, M.A. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J. Cell Biol. 2006, 172, 103–113. [Google Scholar] [CrossRef]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef]
- Asakura, A.; Seale, P.; Girgis-Gabardo, A.; Rudnicki, M.A. Myogenic specification of side population cells in skeletal muscle. J. Cell Biol. 2002, 159, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Gussoni, E.; Soneoka, Y.; Strickland, C.D.; Buzney, E.A.; Khan, M.K.; Flint, A.F.; Kunkel, L.M.; Mulligan, R.C. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 1999, 401, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Messina, G.; Biressi, S.; Cossu, G. Non Muscle Stem Cells and Muscle Regeneration. In Skeletal Muscle Repair and Regeneration; Springer: Dordrecht, The Netherlands, 2008; pp. 65–84. [Google Scholar]
- Joe, A.W.B.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M.V. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Wosczyna, M.N.; Konishi, C.T.; Perez, E.E.; Gan, Q.; Wagner, M.W.; Rando, T.A.; Perez Carbajal, E.E.; Wang, T.T.; Walsh, R.A. Mesenchymal Stromal Cells Are Required for Regeneration and Homeostatic Maintenance of Skeletal Muscle In Brief Article Mesenchymal Stromal Cells Are Required for Regeneration and Homeostatic Maintenance of Skeletal Muscle. Cell Rep. 2019, 27, 2029.e5–2035.e5. [Google Scholar] [CrossRef] [PubMed]
- Fiore, D.; Judson, R.N.; Low, M.; Lee, S.; Zhang, E.; Hopkins, C.; Xu, P.; Lenzi, A.; Rossi, F.M.V.; Lemos, D.R. Pharmacological blockage of fibro/adipogenic progenitor expansion and suppression of regenerative fibrogenesis is associated with impaired skeletal muscle regeneration. Stem Cell Res. 2016, 17, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Lukjanenko, L.; Karaz, S.; Stuelsatz, P.; Gurriaran-Rodriguez, U.; Michaud, J.; Dammone, G.; Sizzano, F.; Mashinchian, O.; Ancel, S.; Migliavacca, E.; et al. Aging Disrupts Muscle Stem Cell Function by Impairing Matricellular WISP1 Secretion from Fibro-Adipogenic Progenitors. Cell Stem Cell 2019, 24, 433.e7–446.e7. [Google Scholar] [CrossRef]
- Davis, M.E.; Gumucio, J.P.; Sugg, K.B.; Bedi, A.; Mendias, C.L. MMP inhibition as a potential method to augment the healing of skeletal muscle and tendon extracellular matrix. J. Appl. Physiol. 2013, 115, 884–891. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J. Matrix metalloproteinase and tissue inhibitor of metalloproteinase responses to muscle damage after eccentric exercise. J. Exerc. Rehabil. 2016, 12, 260–265. [Google Scholar] [CrossRef]
- Arecco, N.; Clarke, C.J.; Jones, F.K.; Simpson, D.M.; Mason, D.; Beynon, R.J.; Pisconti, A. Elastase levels and activity are increased in dystrophic muscle and impair myoblast cell survival, proliferation and differentiation. Sci. Rep. 2016, 6, 1–20. [Google Scholar] [CrossRef]
- Grounds, M.D. Complexity of Extracellular Matrix and Skeletal Muscle Regeneration. In Skeletal Muscle Repair and Regeneration; Springer: Amsterdam, The Netherlands, 2008; pp. 269–302. [Google Scholar]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef]
- Garg, K.; Corona, B.T.; Walters, T.J. Therapeutic strategies for preventing skeletal muscle fibrosis after injury. Front. Pharmacol. 2015, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Gillies, A.R.; Chapman, M.A.; Bushong, E.A.; Deerinck, T.J.; Ellisman, M.H.; Lieber, R.L. High resolution three-dimensional reconstruction of fibrotic skeletal muscle extracellular matrix. J. Physiol. 2017, 595, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Dey, P. Basic and Advanced Laboratory Techniques in Histopathology and Cytology; Springer: Singapore, 2018. [Google Scholar]
- Junqueira, L.C.U.; Bignolas, G.; Brentani, R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 1979, 11, 447–455. [Google Scholar] [CrossRef]
- Montes, G.S.; Junqueira, L.C. The use of the Picrosirius-polarization method for the study of the biopathology of collagen. Mem. Inst. Oswaldo Cruz 1991, 86, 1–11. [Google Scholar] [CrossRef]
- Lattouf, R.; Younes, R.; Lutomski, D.; Naaman, N.; Godeau, G.; Senni, K.; Changotade, S. Picrosirius Red Staining: A Useful Tool to Appraise Collagen Networks in Normal and Pathological Tissues. J. Histochem. Cytochem. 2014, 62, 751–758. [Google Scholar] [CrossRef]
- Séguier, S.; Godeau, G.; Brousse, N. Collagen Fibers and Inflammatory Cells in Healthy and Diseased Human Gingival Tissues: A Comparative and Quantitative Study by Immunohistochemistry and Automated Image Analysis. J. Periodontol. 2000, 71, 1079–1085. [Google Scholar] [CrossRef]
- Tatsumi, R.; Sankoda, Y.; Anderson, J.E.; Sato, Y.; Mizunoya, W.; Shimizu, N.; Suzuki, T.; Yamada, M.; Rhoads, R.P.; Ikeuchi, Y.; et al. Possible implication of satellite cells in regenerative motoneuritogenesis: HGF upregulates neural chemorepellent Sema3A during myogenic differentiation Possible implication of satellite cells in regenerative motoneuritogenesis: HGF upregulates neural che-morepellent Sema3A during myogenic differentiation. Am. J. Physiol. Cell Physiol. 2009, 297, 238–252. [Google Scholar] [CrossRef]
- Ling, S.C.; Dastidar, S.G.; Tokunaga, S.; Ho, W.Y.; Lim, K.; Ilieva, H.; Parone, P.A.; Tyan, S.H.; Tse, T.M.; Chang, J.C.; et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. Elife 2019, 8, e40811. [Google Scholar] [CrossRef]
- Rizzuto, E.; Pisu, S.; Nicoletti, C.; Del Prete, Z.; Musarò, A. Measuring neuromuscular junction functionality. J. Vis. Exp. 2017, 2017, 55227. [Google Scholar] [CrossRef]
- Rizzuto, E.; Pisu, S.; Musarò, A.; Del Prete, Z. Measuring Neuromuscular Junction Functionality in the SOD1G93A Animal Model of Amyotrophic Lateral Sclerosis. Ann. Biomed. Eng. 2015, 43, 2196–2206. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, Z.; Musarò, A.; Rizzuto, E. Measuring mechanical properties, including isotonic fatigue, of fast and slow MLC/mIgf-1 transgenic skeletal muscle. Ann. Biomed. Eng. 2008, 36, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Radley, H.G.; Lynch, G.S.; Nagaraju, K.; De Luca, A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol. Dis. 2008, 31, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lagrota-Candido, J.; Vasconcellos, R.; Cavalcanti, M.; Bozza, M.; Savino, W.; Quirico-Santos, T. Resolution of skeletal muscle inflammation in mdx dystrophic mouse is accompanied by increased immunoglobulin and interferon-γ production. Int. J. Exp. Pathol. 2002, 83, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Pelosi, L.; Miano, C.; Musarò, A. Insights into the Pathogenic Secondary Symptoms Caused by the Primary Loss of Dystrophin. J. Funct. Morphol. Kinesiol. 2017, 2, 44. [Google Scholar] [CrossRef]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’Amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; De Benedetti, F.; et al. Functional and Morphological Improvement of Dystrophic Muscle by Interleukin 6 Receptor Blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef]
- Klein, S.M.; Prantl, L.; Geis, S.; Felthaus, O.; Dolderer, J.; Anker, A.M.; Zeitler, K.; Alt, E.; Vykoukal, J. Circulating serum CK level vs. muscle impairment for in situ monitoring burden of disease in Mdx-mice. Clin. Hemorheol. Microcirc. 2017, 65, 327–334. [Google Scholar] [CrossRef]
- Liu, N.; Williams, A.H.; Maxeiner, J.M.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Investig. 2012, 122, 2054–2065. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef]
- Vignier, N.; Amor, F.; Fogel, P.; Duvallet, A.; Poupiot, J.; Charrier, S.; Arock, M.; Montus, M.; Nelson, I.; Richard, I.; et al. Distinctive Serum miRNA Profile in Mouse Models of Striated Muscular Pathologies. PLoS ONE 2013, 8, e55281. [Google Scholar] [CrossRef]
- Jeanson-Leh, L.; Lameth, J.; Krimi, S.; Buisset, J.; Amor, F.; Le Guiner, C.; Barthélémy, I.; Servais, L.; Blot, S.; Voit, T.; et al. Serum profiling identifies novel muscle miRNA and cardiomyopathy-related miRNA biomarkers in golden retriever muscular dystrophy dogs and duchenne muscular dystrophy patients. Am. J. Pathol. 2014, 184, 2885–2898. [Google Scholar] [CrossRef] [PubMed]
- Zaharieva, I.T.; Calissano, M.; Scoto, M.; Preston, M.; Cirak, S.; Feng, L.; Collins, J.; Kole, R.; Guglieri, M.; Straub, V.; et al. Dystromirs as Serum Biomarkers for Monitoring the Disease Severity in Duchenne Muscular Dystrophy. PLoS ONE 2013, 8, e80263. [Google Scholar] [CrossRef] [PubMed]
- Perfetti, A.; Greco, S.; Bugiardini, E.; Cardani, R.; Gaia, P.; Gaetano, C.; Meola, G.; Martelli, F. Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromuscul. Disord. 2014, 24, 509–515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McDouall, R.M.; Dunn, M.J.; Dubowitz, V. Nature of the mononuclear infiltrate and the mechanism of muscle damage in juvenile dermatomyositis and Duchenne muscular dystrophy. J. Neurol. Sci. 1990, 99, 199–217. [Google Scholar] [CrossRef]
- Spencer, M.J.; Walsh, C.M.; Dorshkind, K.A.; Rodriguez, E.M.; Tidball, J.G. Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin- mediated cytotoxicity. J. Clin. Investig. 1997, 99, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Wehling, M.; Spencer, M.J.; Tidball, J.G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 2001, 155, 123–131. [Google Scholar] [CrossRef]
- Madaro, L.; Torcinaro, A.; De Bardi, M.; Contino, F.F.; Pelizzola, M.; Diaferia, G.R.; Imeneo, G.; Bouchè, M.; Puri, P.L.; De Santa, F. Macrophages fine tune satellite cell fate in dystrophic skeletal muscle of mdx mice. PLoS Genet. 2019, 15, e1008408. [Google Scholar] [CrossRef]
- Turk, R.; Sterrenburg, E.; de Meijer, E.J.; van Ommen, G.J.B.; den Dunnen, J.T.; ’t Hoen, P.A.C. Muscle regeneration in dystrophin-deficient mdx mice studied by gene expression profiling. BMC Genom. 2005, 6, 98. [Google Scholar] [CrossRef]
- Pelosi, M.; De Rossi, M.; Barberi, L.; Musarò, A. IL-6 Impairs Myogenic Differentiation by Downmodulation of p90RSK/eEF2 and mTOR/p70S6K Axes, without Affecting AKT Activity. Biomed Res. Int. 2014, 2014, 206026. [Google Scholar] [CrossRef]
- Kurosaka, M.; Machida, S. Interleukin-6-induced satellite cell proliferation is regulated by induction of the JAK2/STAT3 signalling pathway through cyclin D1 targeting. Cell Prolif. 2013, 46, 365–373. [Google Scholar] [CrossRef]
- Wada, E.; Tanihata, J.; Iwamura, A.; Takeda, S.; Hayashi, Y.K.; Matsuda, R. Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. Skelet. Muscle 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Reggio, A.; Rosina, M.; Palma, A.; Cerquone Perpetuini, A.; Lisa Petrilli, L.; Gargioli, C.; Fuoco, C.; Micarelli, E.; Giuliani, G.; Cerretani, M.; et al. Adipogenesis of skeletal muscle fibro/adipogenic progenitors is affected by the WNT5a/GSK3/β-catenin axis. Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cullen, M.; Mastaglia, F. Pathological reactions of skeletal muscle. In Skeletal Muscle Pathology; Mastaglia, F.L., Walton, J., Eds.; Churchill Livingstone: London, UK, 1982; pp. 88–139. [Google Scholar]
- Harris, J.B. Myotoxic phospholipases A2 and the regeneration of skeletal muscles. Toxicon 2003, 42, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.; Besnard, A.; Latil, M.; Jouvion, G.; Briand, D.; Thépenier, C.; Pascal, Q.; Guguin, A.; Gayraud-Morel, B.; Cavaillon, J.M.; et al. Comparative Study of Injury Models for Studying Muscle Regeneration in Mice. PLoS ONE 2016, 11, e0147198. [Google Scholar] [CrossRef]
- Warren, G.L.; Hulderman, T.; Mishra, D.; Gao, X.; Millecchia, L.; O’Farrell, L.; Kuziel, W.A.; Simeonova, P.P. Chemokine receptor CCR2 involvement in skeletal muscle regeneration. FASEB J. 2005, 19, 1–23. [Google Scholar] [CrossRef]
- Le, G.; Lowe, D.A.; Kyba, M. Freeze injury of the tibialis anterior muscle. Methods Mol. Biol. 2016, 1460, 33–41. [Google Scholar] [CrossRef]
- Forcina, L.; Miano, C.; Musarò, A. The physiopathologic interplay between stem cells and tissue niche in muscle regeneration and the role of IL-6 on muscle homeostasis and diseases. Cytokine Growth Factor Rev. 2018, 41, 1–9. [Google Scholar] [CrossRef]
- Forcina, L.; Miano, C.; Scicchitano, B.M.; Rizzuto, E.; Berardinelli, M.G.; De Benedetti, F.; Pelosi, L.; Musarò, A. Increased Circulating Levels of Interleukin-6 Affect the Redox Balance in Skeletal Muscle. Oxid. Med. Cell. Longev. 2019, 2019, 3018584. [Google Scholar] [CrossRef]
- Schultz, E.; Jaryszak, D.L.; Gibson, M.C.; Albright, D.J. Absence of exogenous satellite cell contribution to regeneration of frozen skeletal muscle. J. Muscle Res. Cell Motil. 1986, 7, 361–367. [Google Scholar] [CrossRef]
- Torrente, Y.; Belicchi, M.; Sampaolesi, M.; Pisati, F.; Meregalli, M.; D’Antona, G.; Tonlorenzi, R.; Porretti, L.; Gavina, M.; Mamchaoui, K.; et al. Human circulating AC133+ stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. J. Clin. Investig. 2004, 114, 182–195. [Google Scholar] [CrossRef]
- Reis, N.D.; Better, O.S. Mechanical muscle-crush injury and acute muscle-crush compartment syndrome. J. Bone Joint Surg. Br. 2005, 87, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Dobek, G.L.; Fulkerson, N.D.; Nicholas, J.; Schneider, B.S.P. Mouse model of muscle crush injury of the legs. Comp. Med. 2013, 63, 227–232. [Google Scholar] [PubMed]
- Crisco, J.J.; Jokl, P.; Heinen, G.T.; Connell, M.D.; Panjabi, M.M. A Muscle Contusion Injury Model: Biomechanics, Physiology, and Histology. Am. J. Sports Med. 1994, 22, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Kerkweg, U.; Schmitz, D.; de Groot, H. Screening for the Formation of Reactive Oxygen Species and of NO in Muscle Tissue and Remote Organs upon Mechanical Trauma to the Mouse Hind Limb. Eur. Surg. Res. 2006, 38, 83–89. [Google Scholar] [CrossRef]
- McBrier, N.M.; Neuberger, T.; Denegar, C.R.; Sharkey, N.A.; Webb, A.G. Magnetic resonance imaging of acute injury in rats and the effects of buprenorphine on limb volume. J. Am. Assoc. Lab. Anim. Sci. 2009, 48, 147–151. [Google Scholar]
- Stratos, I.; Graff, J.; Rotter, R.; Mittlmeier, T.; Vollmar, B. Open blunt crush injury of different severity determines nature and extent of local tissue regeneration and repair. J. Orthop. Res. 2010, 28, 950–957. [Google Scholar] [CrossRef]
- Takagi, R.; Fujita, N.; Arakawa, T.; Kawada, S.; Ishii, N.; Miki, A. Influence of icing on muscle regeneration after crush injury to skeletal muscles in rats. J. Appl. Physiol. 2011, 110, 382–388. [Google Scholar] [CrossRef]
- Criswell, T.L.; Corona, B.T.; Ward, C.L.; Miller, M.; Patel, M.; Wang, Z.; Christ, G.J.; Soker, S. Compression-induced muscle injury in rats that mimics compartment syndrome in humans. Am. J. Pathol. 2012, 180, 787–797. [Google Scholar] [CrossRef]
- Ciciliot, S.; Schiaffino, S. Regeneration of Mammalian Skeletal Muscle: Basic Mechanisms and Clinical Implications. Curr. Pharm. Des. 2010, 16, 906–914. [Google Scholar] [CrossRef]
- Oyster, N.; Witt, M.; Gharaibeh, B.; Poddar, M.; Schneppendahl, J.; Huard, J. Characterization of a compartment syndrome-like injury model. Muscle Nerve 2015, 51, 750–758. [Google Scholar] [CrossRef]
- Lømo, T. What controls the position, number, size, and distribution of neuromuscular junctions on rat muscle fibers? J. Neurocytol. 2003, 32, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Klein-Ogus, C.; Harris, J.B. Preliminary observations of satellite cells in undamaged fibres of the rat soleus muscle assaulted by a snake-venom toxin. Cell Tissue Res. 1983, 230, 671–776. [Google Scholar] [CrossRef] [PubMed]
- Benoit, P.W.; Belt, W.D. Destruction and regeneration of skeletal muscle after treatment with a local anaesthetic, bupivacaine (Marcaine). J. Anat. 1970, 107, 547–556. [Google Scholar]
- Gutiérrez, J.M.; Ownby, C.L. Skeletal muscle degeneration induced by venom phospholipases A 2: Insights into the mechanisms of local and systemic myotoxicity. Toxicon 2003, 42, 915–931. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.B.; Grubb, B.D.; Maltin, C.A.; Dixon, R. The neurotoxicity of the venom phospholipases A2, notexin and taipoxin. Exp. Neurol. 2000, 161, 517–526. [Google Scholar] [CrossRef]
- Harris, J.B.; Vater, R.; Wilson, M.; Cullen, M.J. Muscle fibre breakdown in venom-induced muscle degeneration. J. Anat. 2003, 202, 363–372. [Google Scholar] [CrossRef]
- Rosenblatt, J.D. A time course study of the isometric contractile properties of rat extensor digitorum longus muscle injected with bupivacaine. Comp. Biochem. Physiol. Comp. Physiol. 1992, 101, 361–367. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Escalante, T.; Hernández, R.; Gastaldello, S.; Saravia-Otten, P.; Rucavado, A. Why is skeletal muscle regeneration impaired after myonecrosis induced by viperid snake venoms? Toxins (Basel) 2018, 10, 182. [Google Scholar] [CrossRef]
- Hodges, S.J.; Agbaji, A.S.; Harvey, A.L.; Hider, R.C. Cobra cardiotoxins. Purification, effects on skeletal muscle and structure/activity relationships. Eur. J. Biochem. 1987, 165, 373–383. [Google Scholar] [CrossRef]
- Guardiola, O.; Andolfi, G.; Tirone, M.; Iavarone, F.; Brunelli, S.; Minchiotti, G. Induction of acute skeletal muscle regeneration by cardiotoxin injection. J. Vis. Exp. 2017, 2017, 54515. [Google Scholar] [CrossRef]
- Czerwinska, A.M.; Streminska, W.; Ciemerych, M.A.; Grabowska, I. Mouse gastrocnemius muscle regeneration after mechanical or cardiotoxin injury. Folia Histochem. Cytobiol. 2012, 50, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Masuda, S.; Tamura, T.; Kai, K.; Ojima, K.; Fukase, A.; Motoyoshi, K.; Kamakura, K.; Miyagoe-Suzuki, Y.; Takeda, S. Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: A role for osteopontin. Am. J. Pathol. 2003, 163, 203–215. [Google Scholar] [CrossRef]
- El Andalousi, R.B.; Daussin, P.A.; Micallef, J.P.; Roux, C.; Nougues, J.; Chammas, M.; Reyne, Y.; Bacou, F. Changes in mass and performance in rabbit muscles after muscle damage with or without transplantation of primary satellite cells. Cell Transplant. 2002, 11, 169–180. [Google Scholar] [CrossRef]
- Sambasivan, R.; Yao, R.; Kissenpfennig, A.; van Wittenberghe, L.; Paldi, A.; Gayraud-Morel, B.; Guenou, H.; Malissen, B.; Tajbakhsh, S.; Galy, A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 2011, 138, 3647–3656. [Google Scholar] [CrossRef]
- Ho, A.T.V.; Palla, A.R.; Blake, M.R.; Yucel, N.D.; Wang, Y.X.; Magnusson, K.E.G.; Holbrook, C.A.; Kraft, P.E.; Delp, S.L.; Blau, H.M. Prostaglandin E2 is essential for efficacious skeletal muscle stem-cell function, augmenting regeneration and strength. Proc. Natl. Acad. Sci. USA 2017, 114, 6675–6684. [Google Scholar] [CrossRef] [PubMed]
- Siles, L.; Ninfali, C.; Cortés, M.; Darling, D.S.; Postigo, A. ZEB1 protects skeletal muscle from damage and is required for its regeneration. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.-T.; Cheung, K.-K.; Au, S.W.N.; Yeung, S.S.; Yeung, E.W. Overexpression of Mechano-Growth Factor Modulates Inflammatory Cytokine Expression and Macrophage Resolution in Skeletal Muscle Injury. Front. Physiol. 2018, 9, 999. [Google Scholar] [CrossRef] [PubMed]
- Mirtschin, P.; Davis, R. Dangerous Snakes of Australia. An Illustrated Guide to Australia’s Most Venomous Snakes; Rigby Publishers: Adelaide, Australia, 1982. [Google Scholar]
- Plant, D.R.; Colarossi, F.E.; Lynch, G.S. Notexin causes greater myotoxic damage and slower functional repair in mouse skeletal muscles than bupivacaine. Muscle Nerve 2006, 34, 577–585. [Google Scholar] [CrossRef]
- Vignaud, A.; Hourdé, C.; Butler-Browne, G.; Ferry, A. Differential recovery of neuromuscular function after nerve/muscle injury induced by crude venom from Notechis scutatus, cardiotoxin from Naja atra and bupivacaine treatments in mice. Neurosci. Res. 2007, 58, 317–323. [Google Scholar] [CrossRef]
- Gayraud-Morel, B.; Chrétien, F.; Flamant, P.; Gomès, D.; Zammit, P.S.; Tajbakhsh, S. A role for the myogenic determination gene Myf5 in adult regenerative myogenesis. Dev. Biol. 2007, 312, 13–28. [Google Scholar] [CrossRef]
- Baghdadi, M.B.; Tajbakhsh, S. Regulation and phylogeny of skeletal muscle regeneration. Dev. Biol. 2018, 433, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Zink, W.; Seif, C.; Bohl, J.R.E.; Hacke, N.; Braun, P.M.; Sinner, B.; Martin, E.; Fink, R.H.A.; Graf, B.M. The acute myotoxic effects of bupivacaine and ropivacaine after continuous peripheral nerve blockades. Anesth. Analg. 2003, 97, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Zink, W.; Graf, B.M.; Sinner, B.; Martin, E.; Fink, R.H.A.; Kunst, G. Differential effects of bupivacaine on intracellular Ca 2+ regulation: Potential mechanisms of its myotoxicity. Anesthesiology 2002, 97, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Hirata, S.; Miyasaka, N.; Kawahata, K.; Kohsaka, H. Injury and subsequent regeneration of muscles for activation of local innate immunity to facilitate the development and relapse of autoimmune myositis in C57BL/6 mice. Arthritis Rheumatol. 2015, 67, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Dobrowolny, G.; Sica, G.; Musaro, A. Molecular Insights into Muscle Homeostasis, Atrophy and Wasting. Curr. Genom. 2018, 19, 356–369. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Stage | Markers | Recognition | References |
|---|---|---|---|
| Degeneration | Serum CK, LDH, troponin, miR-378a-3p, miR-434-3p | Muscle damage | [16,18] |
| Albumin, IgG fiber uptake | Myofiber permeability | [5,15] | |
| Inflammation | CD11bpos./Ly6Gpos./Ly6Cneg. | Neutrophils | [19,20] |
| Ly6Chigh/CCR2pos./CX3CR1low | Pro-inflammatory monocytes | [21,22,23,24,25,26] | |
| Ly6Clow/CCR2neg./CX3CR1high | Patrolling monocytes | ||
| CD11b, Ly6C, F4/80, CD68, CD38, Gpr18, Fpr2 | M1 Macrophages | [27,28,29] | |
| CD206, CD11c, CD163, Arginase1, Egr2, c-Myc | M2 Macrophages | ||
| Regeneration | Pax3, Pax7, CD34, NCAM, VCAM-1, Cav1, Mcad, Syndecan 3-4, Sox8-15, Integrin α7-β1, CTR, Emerin, Hey1, Heyl | Quiescent SCs | [1,12,30,31,32,33,34,35,36,37,38,39] |
| Pax7high/MyoDlow, DGC, p38γ | Proliferating/Self renewing SCs | [1,12,40,41,42,43] | |
| Pax7low/MyoDhigh, Myf-5, p38α-β | Committed SCs | ||
| MyoD, Myogenin, Mrf4, miR206, miR486 | Differentiating SCs | ||
| CD45neg./CD31neg./ α7 intneg./Scapos./PDGFR αpos. | FAPs | [1,11,44,45] | |
| Collagen I–III–IV, laminin, fibronectin, proteoglicans | ECM | [46,47,48,49,50] | |
| Remodeling, Maturation and Functional retrieval | eMyHC | Regenerating Myofibers | [12] |
| AchRs/Synaptohysin/ Neurofilament markers | NMJs | [51] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forcina, L.; Cosentino, M.; Musarò, A. Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing. Cells 2020, 9, 1297. https://doi.org/10.3390/cells9051297
Forcina L, Cosentino M, Musarò A. Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing. Cells. 2020; 9(5):1297. https://doi.org/10.3390/cells9051297
Chicago/Turabian StyleForcina, Laura, Marianna Cosentino, and Antonio Musarò. 2020. "Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing" Cells 9, no. 5: 1297. https://doi.org/10.3390/cells9051297
APA StyleForcina, L., Cosentino, M., & Musarò, A. (2020). Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing. Cells, 9(5), 1297. https://doi.org/10.3390/cells9051297