Deficiency of GABARAP but Not Its Paralogs Causes Enhanced EGF-Induced EGFR Degradation

 ,
,  , , , ,
, , , ,  and
and
Abstract
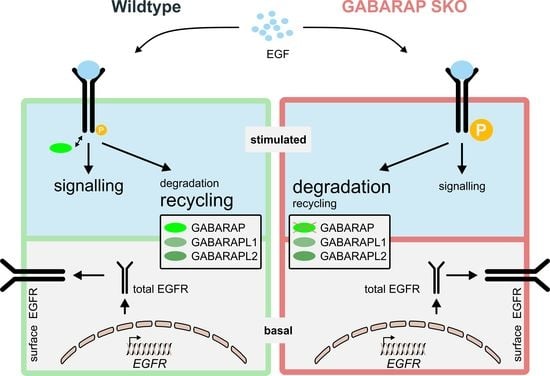
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. CRISPR/Cas9 Mediated Knockout Generation
2.4. Transient Transfection
2.5. Immunoblot
2.6. RT-PCR
2.7. EGF Uptake Assay
2.8. Confocal Laser Scanning Microscopy
2.9. EGFR Surface Expression Analysis
2.10. Spinning Disc Confocal Fluorescence Microscopy
2.11. Image Analysis
2.12. Bio-Layer Interferometry (BLI)
2.13. Co-immunoprecipitation
2.14. Statistical Analysis
3. Results
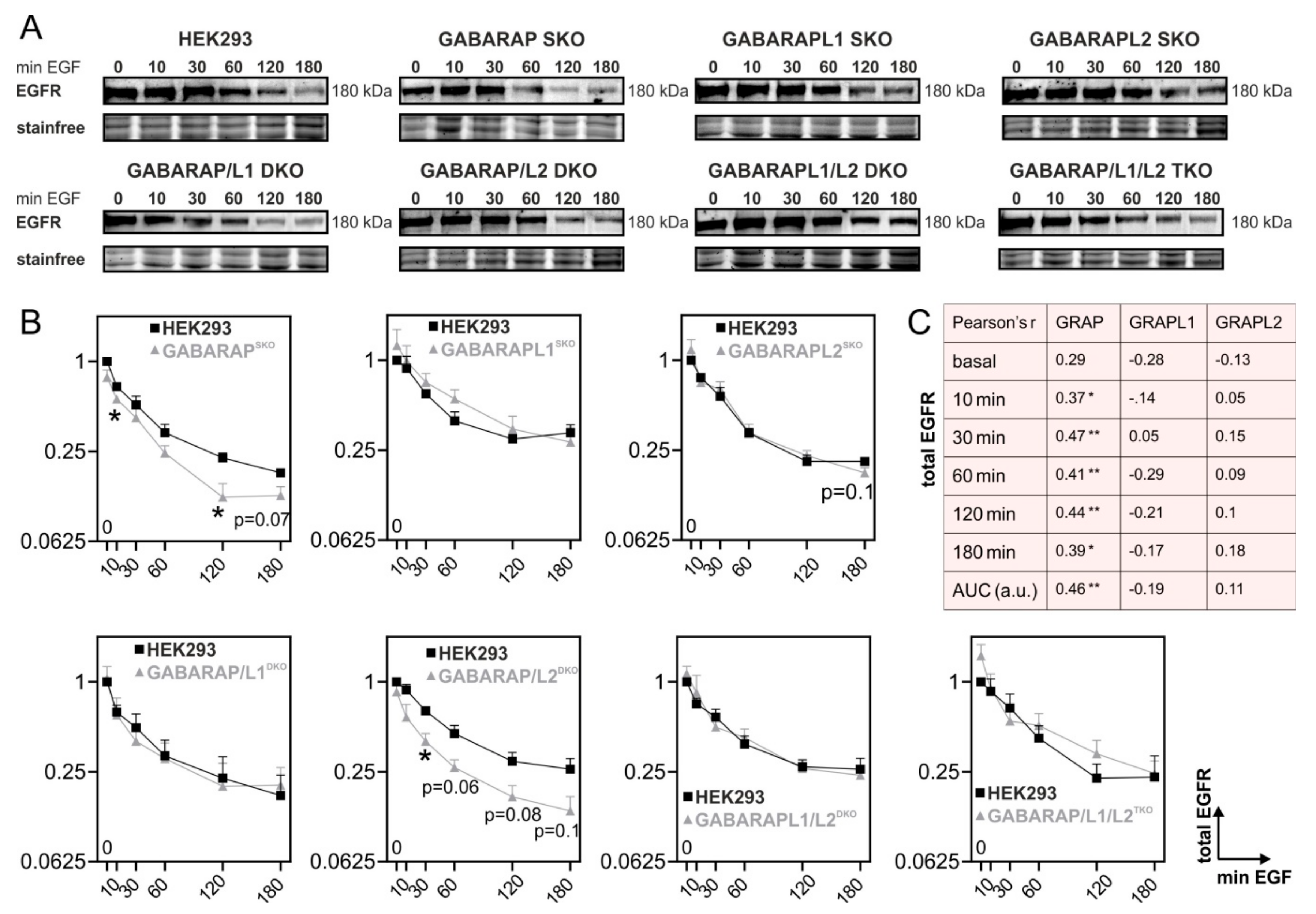
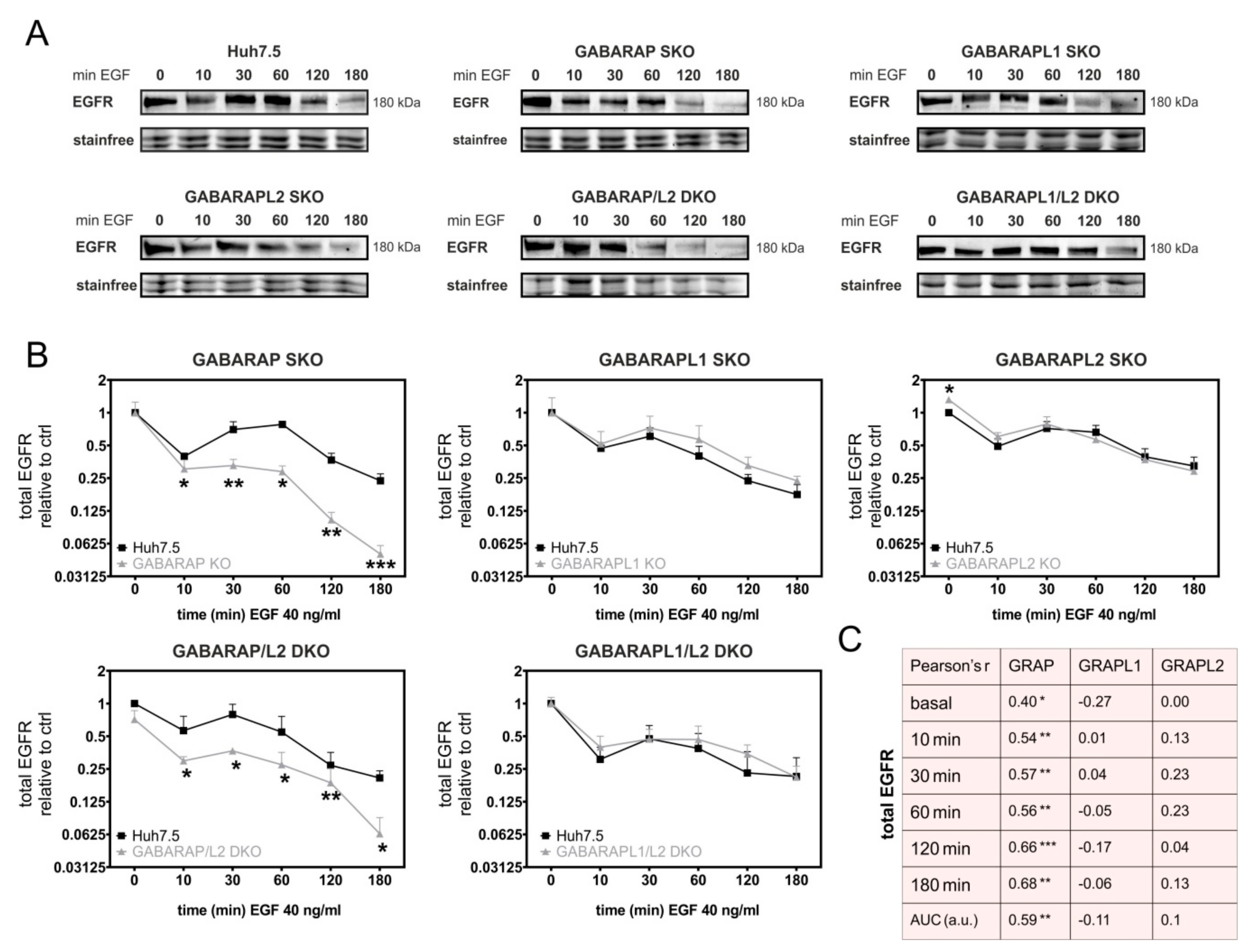
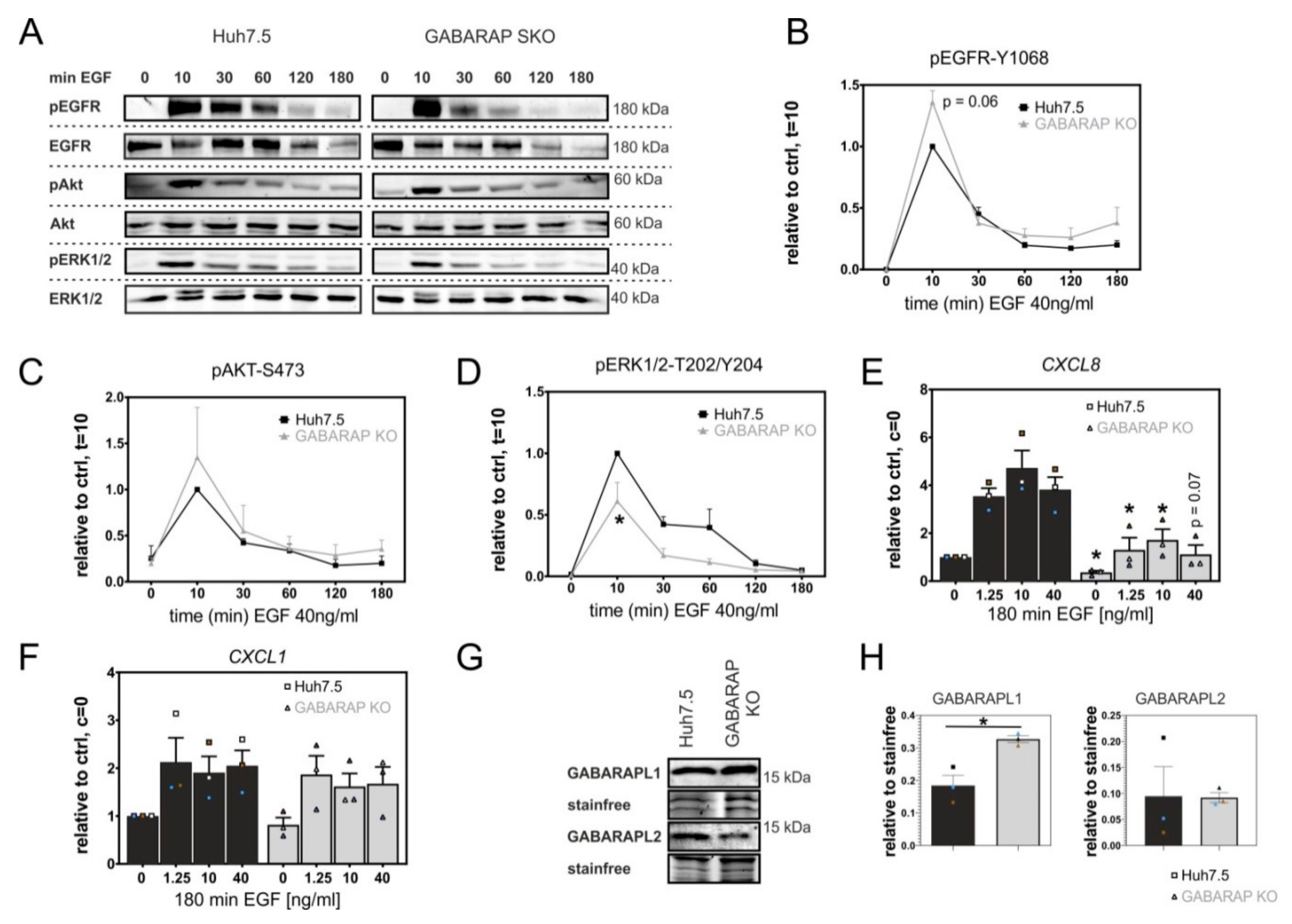
3.1. Deficiency for GABARAP, but Not GABARAPL1 or GABARAPL2 Is Associated with Increased Degradation of EGFR in Huh7.5 and HEK293 Cells
3.2. GABARAP Deficiency Alters EGFR Signaling on Protein and Gene Expression Levels and Increases GABARAPL1 but Not GABARAPL2 Protein Expression
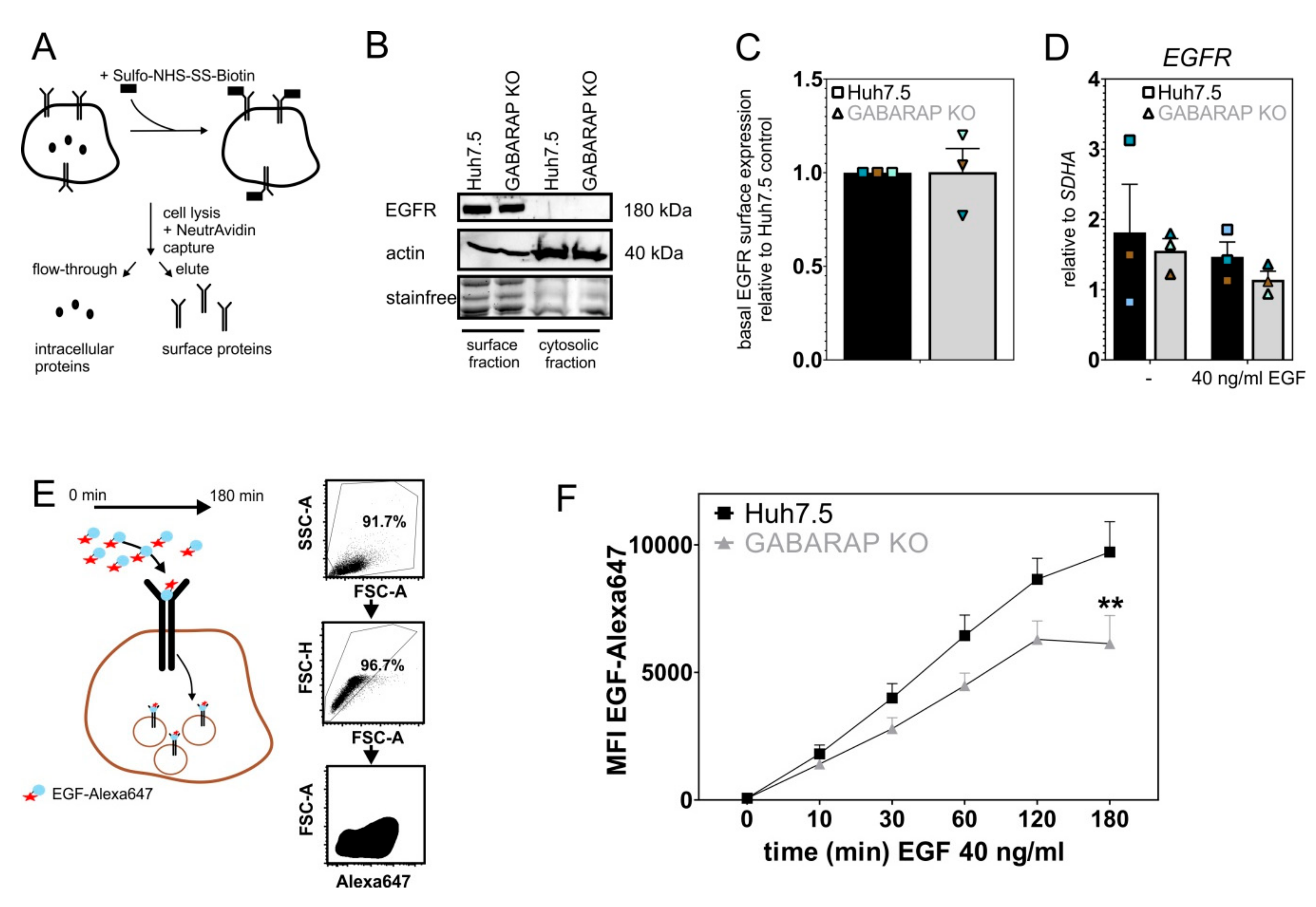
3.3. Basal EGFR Surface Expression Is Unaltered in GABARAP Deficient Cells, While EGF Uptake Capacity Is Impaired over Time
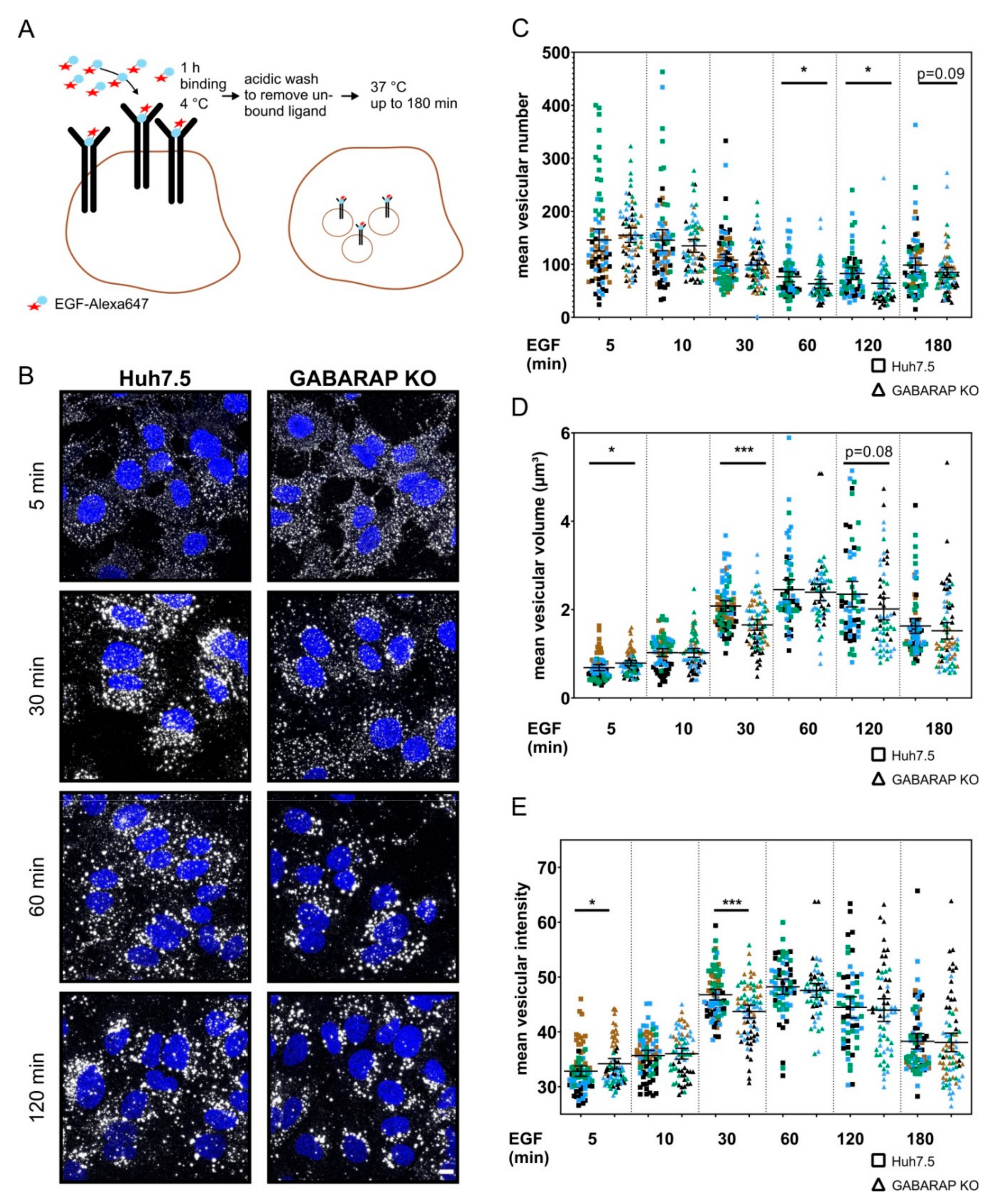
3.4. Tracking of Fluorescently-Labeled EGF Reveals Altered EGF Trafficking and Vesicular Composition in GABARAP Deficient Cells without Abrogation of General Endosomal Targeting
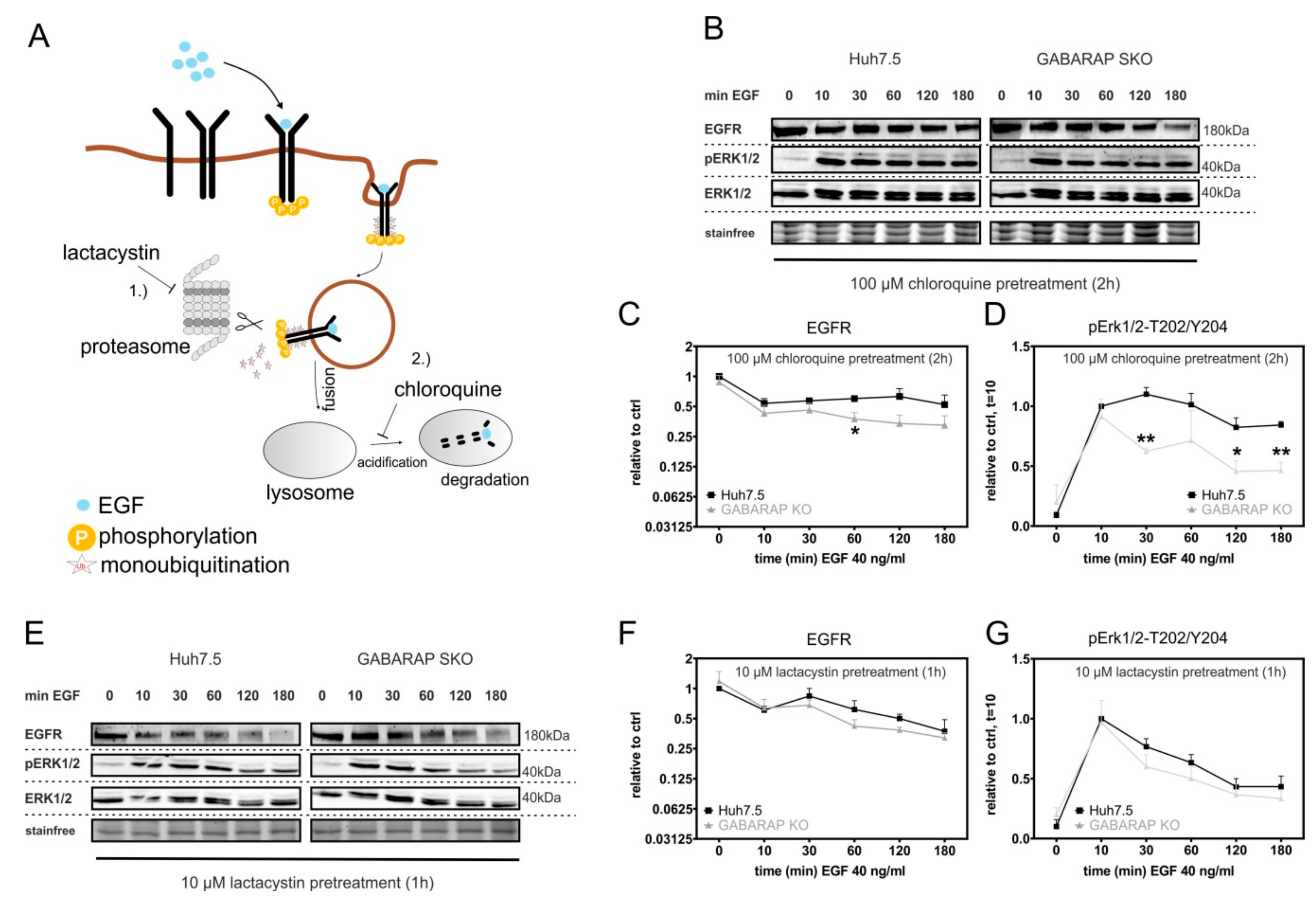
3.5. Accelerated EGFR Degradation in GABARAP Deficient Huh7.5 Cells Can Be Counteracted by Lysosomal and Proteasomal Inhibition
3.6. GABARAP and EGF Converge in Distinct Dynamic Vesicular Structures at Endogenous Expression Levels in HEK293 Cells
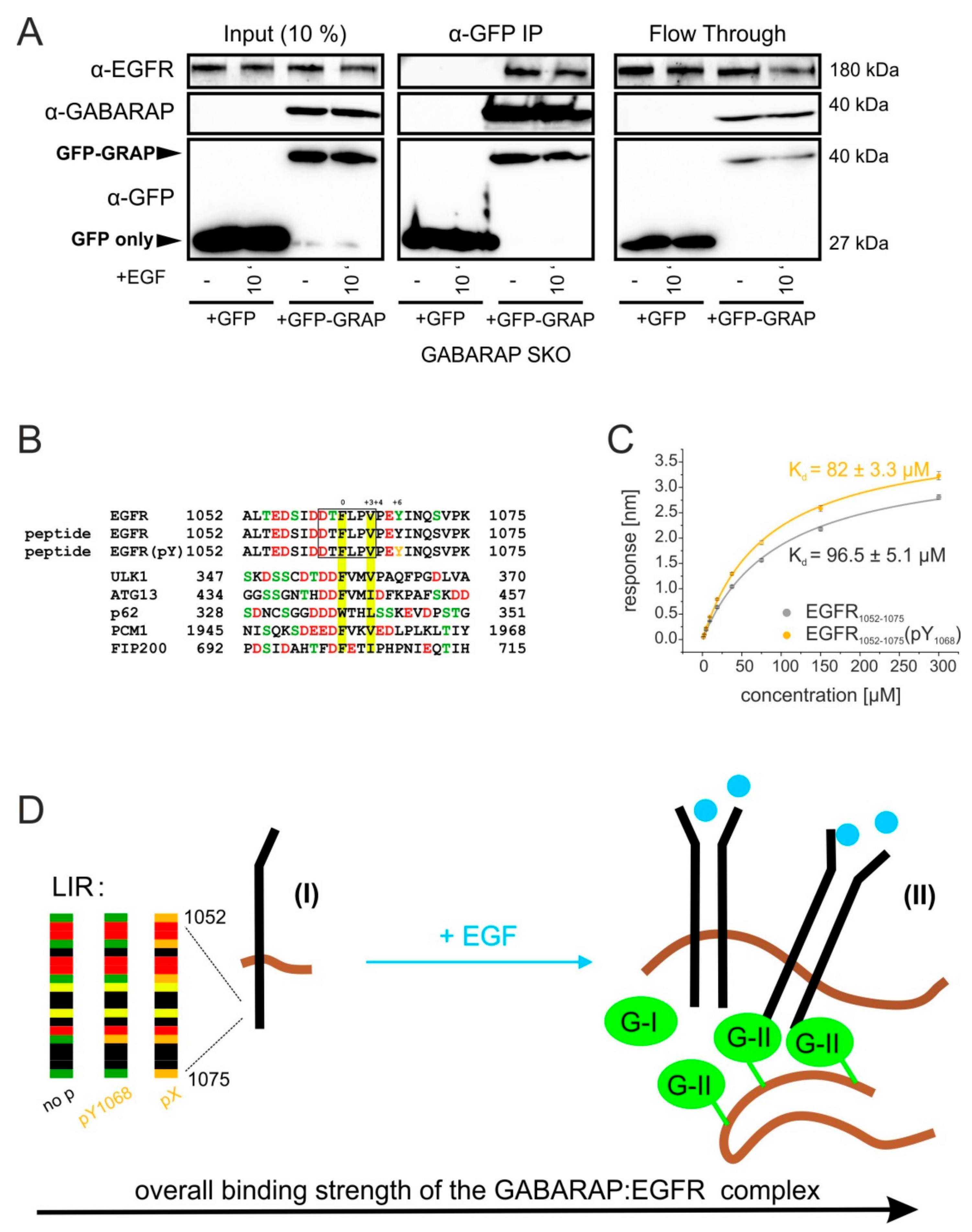
3.7. GABARAP Associates with EGFR during Co-Immunoprecipitation and Binds to Synthetic Peptides Derived from the EGFR Cytoplasmic Tail
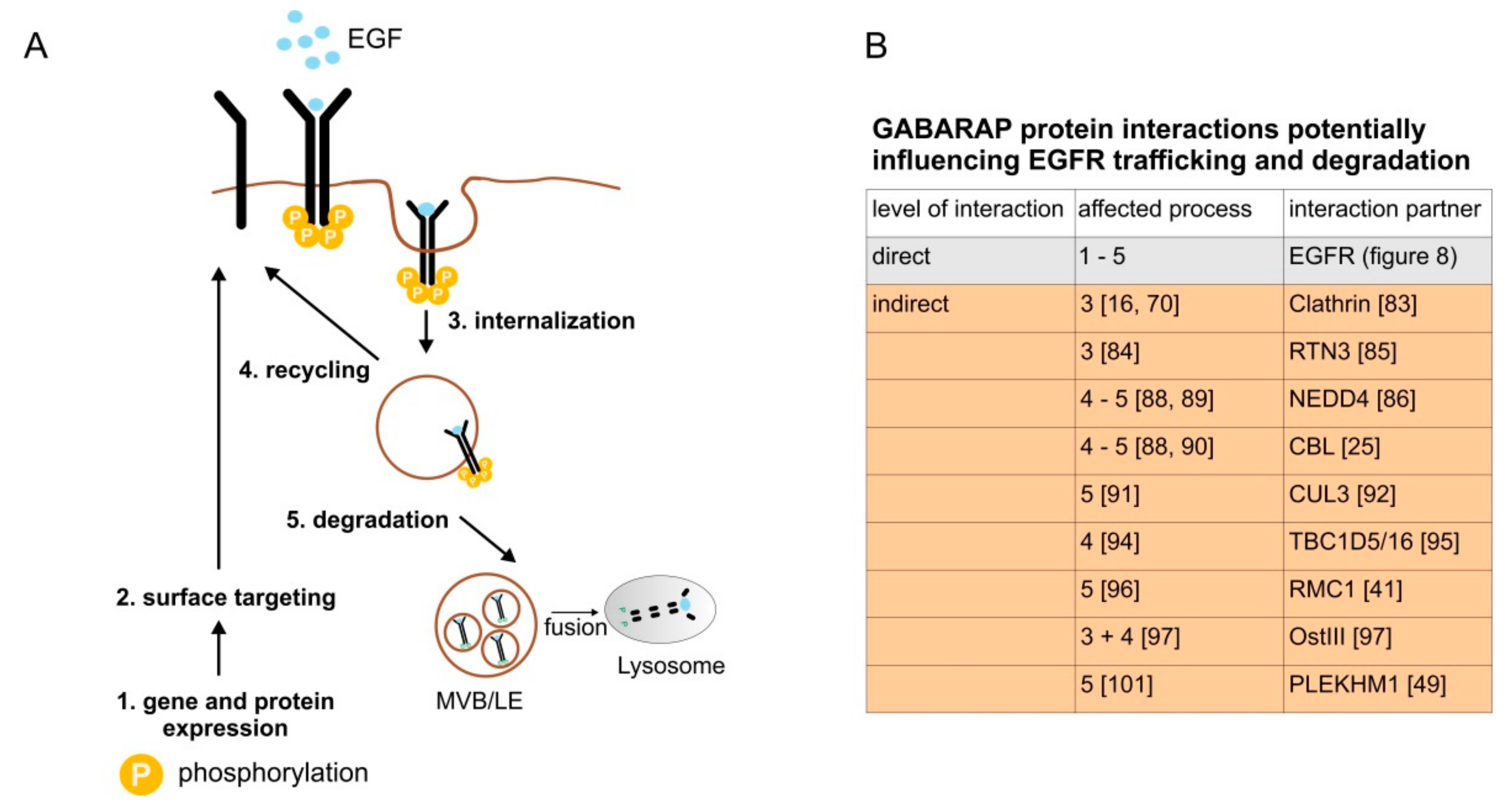
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Species | Catalog Number/Dilution-Application (Antigen Used for Antibody Production) |
|---|---|---|---|
| primary Abs: | |||
| anti-EGFR | Cell Signaling | rabbit | 2232/1:1000-IB (synthetic peptide corresponding to residues surrounding Tyr1068 of human EGFR) |
| anti-EGFR | Cell Signaling | rabbit | 4267/1:1000-IB (fusion protein containing the cytoplasmic domain of human EGFR) |
| anti-phospho-EGFR (Tyr1068) | Cell Signaling | rabbit | 2234/1:1000–IB (synthetic peptide corresponding to residues surrounding Tyr1068 of human EGFR) |
| anti-Akt (pan) | Cell Signaling | mouse | 2920/1:1000-IB (synthetic peptide at the carboxyterminal sequence of human Akt) |
| anti-phospho-Akt (Ser473) | Cell Signaling | rabbit | 4060/1:1000-IB (synthetic phosphopeptide corresponding to residues around Ser473 of human Akt) |
| anti-p44/42 MAPK (Erk1/2) | Cell Signaling | rabbit | 4695/1:1000-IB (synthetic peptide corresponding to residues near the C-terminus of rat p44 MAP kinase) |
| anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | mouse | 9106/1:1000-IB (synthetic phospho-peptide (KLH-coupled) corresponding to residues surrounding Thr202/Tyr204 of human p44 MAP kinase) |
| anti-beta actin | Abcam | mouse | ab6276/1:15 000-IB (DDDIAALVIDNGSGK) |
| anti-GABARAP | Cell Signaling | rabbit | 13733/1:1000-IB (synthetic peptide corresponding to residues surrounding Arg40 of human GABARAP) |
| anti-GABARAP | In-house | rat | In-house/undiluted-IF Institute for Diabetes and Obesity, Monoclonal Antibody Core Facility, GST-hGABARAP_aa1-117 [65] |
| anti-GABARAPL1 | Cell Signaling | rabbit | 26632/1:1000-IB (synthetic peptide corresponding to residues near the amino terminus of human GABARAPL1) |
| anti-GABARAPL2 | Cell Signaling | rabbit | 14256/1:1000-IB (synthetic peptide corresponding to residues near the carboxy terminus of human GABARAPL2) |
| anti-GFP (HRP-coupled) | Miltenyi Biotech | mouse | 130-091-833/1:2000-IB (proprietary) |
| anti-RAB5 | Cell Signaling | rabbit | 3547S/1:100-IF (synthetic peptide corresponding to residues surrounding Gly190 of human Rab5A protein) |
| anti-RAB7 | Cell Signaling | rabbit | 9367S/1:100-IF (synthetic peptide corresponding to residues surrounding Glu188 of human Rab7 protein) |
| anti-RAB11 | Cell Signaling | rabbit | 5589S/1:100-IF (synthetic peptide corresponding to residues near the amino terminus of human Rab11 protein. |
| secondary Abs: | |||
| anti-Mouse IgG H&L (Alexa Fluor® 488) | Abcam | goat | 150117/1:250–IF or 1:5000-IB (mouse IgG) |
| anti-Rabbit IgG H&L (Alexa Fluor® 647) | Abcam | goat | 150083/1:250–IF or 1:5000-IB (rabbit IgG) |
| anti-rat IgG+IgM H&L (Alexa Fluor® 488) | Jackson ImmunoResearch | goat | 112-545-068/1:250–IF (rat IgG + IgM) |
| anti-rabbit IgG H&L (Alexa Fluor® 555 preadsorbed) | Abcam | goat | 150086/1:250–IF (rabbit IgG) |
| Sequence (5′ → 3′) | |
|---|---|
| qPCR | |
| CXCL8 forward | AGAAGTTTTTGAAGAGGGCTGAGA |
| CXCL8 reverse | CAGACCCACACAATACATGAAGTG |
| CXCL1 forward | CTGGCGGATCCAAGCAAAT |
| CXCL1 reverse | CATTCCCCTGCCTTCACAAT |
| SDHA forward | AGATGTGGTGTCTCGGTCGAT |
| SDHA reverse | CGTGATCTTTCTCAGGGCCA |
| EGFR forward | CATCCAGTGGCGGGACATAG |
| EGFR reverse | GGGACAGCTTGGATCACACT |
| Genotyping | |
| GABARAP forward | GGGTTGGTGAATAGGGAAGTGG |
| GABARAP reverse | CACTCCTTTCATCCTGGGTCC |
| GABARAPL1 forward | TGGGAACCTGATCCAAGACTC |
| GABARAPL1 reverse | GCCAGGAAGCTAGTCCAAAAC |
| GABARAPL2 forward | CTTGCTGGGAGCTAGTAGGG |
| GABARAPL2 reverse | TGAGGCACCCTGAACAGCA |
References
- Cohen, S.; Carpenter, G.; King, L. Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J. Biol. Chem. 1980, 255, 4834–4842. [Google Scholar] [PubMed]
- Chen, J.; Zeng, F.; Forrester, S.J.; Eguchi, S.; Zhang, M.-Z.; Harris, R.C. Expression and Function of the Epidermal Growth Factor Receptor in Physiology and Disease. Physiol. Rev. 2016, 96, 1025–1069. [Google Scholar] [CrossRef]
- Roberts, A.B.; Lamb, L.C.; Newton, D.L.; Sporn, M.B.; De Larco, J.E.; Todaro, G.J. Transforming growth factors: Isolation of polypeptides from virally and chemically transformed cells by acid/ethanol extraction. Proc. Natl. Acad. Sci. USA 1980, 77, 3494–3498. [Google Scholar] [CrossRef] [PubMed]
- Shoyab, M.; McDonald, V.L.; Bradley, J.G.; Todaro, G.J. Amphiregulin: A bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc. Natl. Acad. Sci. USA 2006, 85, 6528–6532. [Google Scholar] [CrossRef]
- Cohen, S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 1962, 237, 1555–1562. [Google Scholar]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Honegger, A.M.; Kris, R.M.; Ullrich, A.; Schlessinger, J. Evidence that autophosphorylation of solubilized receptors for epidermal growth factor is mediated by intermolecular cross-phosphorylation. Proc. Natl. Acad. Sci. USA 1989, 86, 925–929. [Google Scholar] [CrossRef]
- Honegger, A.M.; Schmidt, A.; Ullrich, A.; Schlessinger, J. Evidence for epidermal growth factor (EGF)-induced intermolecular autophosphorylation of the EGF receptors in living cells. Mol. Cell. Biol. 1990, 10, 4035–4044. [Google Scholar] [CrossRef]
- Xie, H.; Pallero, M.A.; Gupta, K.; Chang, P.; Ware, M.F.; Witke, W.; Kwiatkowski, D.J.; Lauffenburger, D.A.; Murphy-Ullrich, J.E.; Wells, A. EGF receptor regulation of cell motility: EGF induces disassembly of focal adhesions independently of the motility-associated PLCgamma signaling pathway. J. Cell Sci. 1998, 111 Pt 5, 615–624. [Google Scholar]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar]
- Uemura, T.; Kametaka, S.; Waguri, S. GGA2 interacts with EGFR cytoplasmic domain to stabilize the receptor expression and promote cell growth. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Cohen, S. 125I-Labeled human epidermal growth factor: Binding, Internalization, and Degradation in Human Fibroblasts Antiserum to hEGF 1251-hEGF Binding Assay Iodination o f hEGF and Antiserum Autoradiography. J. Cell Biol. 1976, 71, 159–171. [Google Scholar] [CrossRef]
- Haigler, H.T.; McKanna, J.A.; Cohen, S. Direct visualization of the binding and internalization of a ferritin conjugate of epidermal growth factor in human carcinoma cells A-431. J. Cell Biol. 1979, 81, 382–395. [Google Scholar] [CrossRef]
- Hanover, J.A.; Willingham, M.C.; Pastan, I. Kinetics of transit of transferrin and epidermal growth factor through clathrin-coated membranes. Cell 1984, 39, 283–293. [Google Scholar] [CrossRef]
- Sigismund, S.; Woelk, T.; Puri, C.; Maspero, E.; Tacchetti, C.; Transidico, P.; Di Fiore, P.P.; Polo, S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. USA 2005, 102, 2760–2765. [Google Scholar] [CrossRef]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The EGFR odyssey—From activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Klionsky, D.J. Xenophagy: A battlefield between host and microbe, and a possible avenue for cancer treatment. Autophagy 2017, 13, 223–224. [Google Scholar] [CrossRef]
- Sandilands, E.; Serrels, B.; Wilkinson, S.; Frame, M.C. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep. 2012, 13, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, E.; Serrels, B.; McEwan, D.G.; Morton, J.P.; MacAgno, J.P.; McLeod, K.; Stevens, C.; Brunton, V.G.; Langdon, W.Y.; Vidal, M.; et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat. Cell Biol. 2012, 14, 51–60. [Google Scholar] [CrossRef]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef]
- Claude-Taupin, A.; Jia, J.; Mudd, M.; Deretic, V. Autophagy’s secret life: Secretion instead of degradation. Essays Biochem. 2017, 61, 637–647. [Google Scholar]
- Matsuura, A.; Tsukada, M.; Wada, Y.; Ohsumi, Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 1997, 192, 245–250. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Weiergräber, O.; Mohrlüder, J.; Willbold, D. Atg8 Family Proteins—Autophagy and Beyond. In Autophagy—A Double-Edged Sword—Cell Survival or Death? InTech: Rijeka, Croatia, 2013; pp. 267–322. [Google Scholar]
- Zhang, Z.; Xu, X.; Ma, J.; Wu, J.; Wang, Y.; Zhou, R.; Han, J. Gene Deletion of Gabarap Enhances Nlrp3 Inflammasome-Dependent Inflammatory Responses. J. Immunol. 2013, 190, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Keulers, T.G.; Schaaf, M.B.E.; Rouschop, K.M.A. Autophagy-dependent secretion: Contribution to tumor progression. Front. Oncol. 2016, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Yan, Y.; Zhu, C.; Wang, S.; Yan, X.; Lu, L.; Li, W.; Hu, J.; Wei, W.; Mu, Y.; et al. Borna Disease Virus P Protein Affects Neural Transmission through Interactions with Gamma-Aminobutyric Acid Receptor-Associated Protein. J. Virol. 2008, 82, 12487–12497. [Google Scholar] [CrossRef]
- Chen, R.Y.; Shen, K.L.; Chen, Z.; Fan, W.W.; Xie, X.L.; Meng, C.; Chang, X.J.; Zheng, L.B.; Jeswin, J.; Li, C.H.; et al. White spot syndrome virus entry is dependent on multiple endocytic routes and strongly facilitated by Cq-GABARAP in a CME-dependent manner. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Boeske, A.; Schwarten, M.; Ma, P.; Tusche, M.; Mötter, J.; Möller, C.; Neudecker, P.; Hoffmann, S.; Willbold, D. Direct binding to GABARAP family members is essential for HIV-1 Nef plasma membrane localization. Sci. Rep. 2017, 7, 5797. [Google Scholar] [CrossRef]
- Mohrlüder, J.; Schwarten, M.; Willbold, D. Structure and potential function of γ-aminobutyrate type A receptor-associated protein. FEBS J. 2009, 276, 4989–5005. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GAB ARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef]
- Vaites, L.P.; Paulo, J.A.; Huttlin, E.L.; Harper, J.W. Systematic Analysis of Human Cells Lacking ATG8 Proteins Uncovers Roles for GABARAPs and the CCZ1/MON1 Regulator C18orf8/RMC1 in Macroautophagic and Selective Autophagic Flux. Mol. Cell. Biol. 2017, 38, e00392-17. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Rozenknop, A.; Rogov, V.V.; Rogova, N.Y.; Löhr, F.; Güntert, P.; Dikic, I.; Dötsch, V. Characterization of the interaction of GABARAPL-1 with the LIR motif of NBR1. J. Mol. Biol. 2011, 410, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Suzuki, H.; Fiskin, E.; Wild, P.; Kniss, A.; Rozenknop, A.; Kato, R.; Kawasaki, M.; McEwan, D.G.; Löhr, F.; et al. Structural basis for phosphorylation-triggered autophagic clearance of Salmonella. Biochem. J. 2013, 454, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef]
- Abdollahzadeh, I.; Schwarten, M.; Gensch, T.; Willbold, D.; Weiergräber, O.H. The Atg8 family of proteins-modulating shape and functionality of autophagic membranes. Front. Genet. 2017, 8, 109. [Google Scholar] [CrossRef]
- Lystad. Simonsen Mechanisms and Pathophysiological Roles of the ATG8 Conjugation Machinery. Cells 2019, 8, 973. [Google Scholar] [CrossRef]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Löhr, F.; Wakatsuki, S.; Dötsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2018, 19, e47268. [Google Scholar] [CrossRef]
- Wirth, M.; Zhang, W.; Razi, M.; Nyoni, L.; Joshi, D.; O’Reilly, N.; Johansen, T.; Tooze, S.A.; Mouilleron, S. Molecular determinants regulating selective binding of autophagy adapters and receptors to ATG8 proteins. Nat. Commun. 2019, 10, 2055. [Google Scholar] [CrossRef]
- Marshall, R.S.; Hua, Z.; Mali, S.; McLoughlin, F.; Vierstra, R.D. ATG8-Binding UIM Proteins Define a New Class of Autophagy Adaptors and Receptors. Cell 2019, 177, 766–781. [Google Scholar] [CrossRef]
- Wang, H.; Bedford, F.K.; Brandon, N.J.; Moss, S.J.; Olsen, R.W. GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature 1999, 397, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, H.; Vicini, S.; Olsen, R.W. The gamma -aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc. Natl. Acad. Sci. USA 2000, 97, 11557–11562. [Google Scholar] [CrossRef] [PubMed]
- Green, F.; O’Hare, T.; Blackwell, A.; Enns, C.A. Association of human transferrin receptor with GABARAP. FEBS Lett. 2002, 518, 101–106. [Google Scholar] [CrossRef]
- Laínez, S.; Valente, P.; Ontoria-Oviedo, I.; Estévez-Herrera, J.; Camprubí-Robles, M.; Ferrer-Montiel, A.; Planells-Cases, R. GABAA receptor associated protein (GABARAP) modulates TRPV1 expression and channel function and desensitization. FASEB J. 2010, 24, 1958–1970. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.L.; Re, R.N.; DeHaro, D.L.; Abadie, J.M.; Peters, M.; Alam, J. The trafficking protein GABARAP binds to and enhances plasma membrane expression and function of the angiotensin II type 1 receptor. Circ. Res. 2008, 102, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Reining, S.C.; Liesegang, A.; Betz, H.; Biber, J.; Murer, H.; Hernando, N. Expression of renal and intestinal Na/Pi cotransporters in the absence of GABARAP. Pflug. Arch. Eur. J. Physiol. 2010, 460, 207–217. [Google Scholar] [CrossRef]
- Dutta, P.; Dargahi, L.; O’Connell, K.E.; Bolia, A.; Ozkan, B.; Sailer, A.W.; Dev, K.K. A novel modelling mechanism of PAEL receptor and GABARAPL2 interaction involved in Parkinson’s disease. Neurosci. Lett. 2018, 673, 12–18. [Google Scholar] [CrossRef]
- Mansuy, V.; Boireau, W.; Fraichard, A.; Schlick, J.L.; Jouvenot, M.; Delage-Mourroux, R. GEC1, a protein related to GABARAP, interacts with tubulin and GABA A receptor. Biochem. Biophys. Res. Commun. 2004, 325, 639–648. [Google Scholar] [CrossRef]
- Chen, C.; Li, J.G.; Chen, Y.; Huang, P.; Wang, Y.; Liu-Chen, L.Y. GEC1 interacts with the κ opioid receptor and enhances expression of the receptor. J. Biol. Chem. 2006, 281, 7983–7993. [Google Scholar] [CrossRef]
- Keulers, T.G.; Schaaf, M.B.E.; Peeters, H.J.M.; Savelkouls, K.G.M.; Vooijs, M.A.; Bussink, J.; Jutten, B.; Rouschop, K.M.A. GABARAPL1 is required for increased EGFR membrane expression during hypoxia. Radiother. Oncol. 2015, 116, 417–422. [Google Scholar] [CrossRef]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly Permissive Cell Lines for Subgenomic and Genomic Hepatitis C Virus RNA Replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, S.; Fox, D.T.; Wahl, G.M. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 1991, 251, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.C.; Graham, F.L.; Smiley, J.; Nairn, R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J. Gen. Virol. 1977, 36, 59–72. [Google Scholar]
- Simons, I.M.; Mohrlüder, J.; Feederle, R.; Kremmer, E.; Zobel, T.; Dobner, J.; Bleffert, N.; Hoffmann, S.; Willbold, D. The highly GABARAP specific rat monoclonal antibody 8H5 visualizes GABARAP in immunofluorescence imaging at endogenous levels. Sci. Rep. 2019, 9, 526. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Gutiérrez, B.; Anzola, A.; Martínez-Augustin, O.; De Medina, F.S. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal. Biochem. 2014, 467, 1–3. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Möckel, C.; Kubiak, J.; Schillinger, O.; Kühnemuth, R.; Della Corte, D.; Schröder, G.F.; Willbold, D.; Strodel, B.; Seidel, C.A.M.; Neudecker, P. Integrated NMR, Fluorescence, and Molecular Dynamics Benchmark Study of Protein Mechanics and Hydrodynamics. J. Phys. Chem. B 2019, 123, 1453–1480. [Google Scholar] [CrossRef]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; Di Fiore, P.P. Clathrin-Mediated Internalization Is Essential for Sustained EGFR Signaling but Dispensable for Degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef]
- Gu, Y.; Princely Abudu, Y.; Kumar, S.; Bissa, B.; Choi, S.W.; Jia, J.; Lazarou, M.; Eskelinen, E.; Johansen, T.; Deretic, V. Mammalian Atg8 Proteins Regulate Lysosome and Autolysosome Biogenesis Through SNAREs. EMBO J. 2019, 38, e101994. [Google Scholar] [CrossRef]
- Groepper, C.; Rufinatscha, K.; Schröder, N.; Stindt, S.; Ehlting, C.; Albrecht, U.; Bock, H.H.; Bartenschlager, R.; Häussinger, D.; Bode, J.G. HCV modifies EGF signalling and upregulates production of CXCR2 ligands: Role in inflammation and antiviral immune response. J. Hepatol. 2018, 69, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Salova, A.V.; Belyaeva, T.N.; Leontieva, E.A.; Kornilova, E.S. EGF receptor lysosomal degradation is delayed in the cells stimulated with EGF-Quantum dot bioconjugate but earlier key events of endocytic degradative pathway are similar to that of native EGF. Oncotarget 2017, 8, 44335–44350. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sorkin, A.; Carter, R. Endocytosis of Functional Epidermal Growth Factor Receptor-Green Fluorescent Protein Chimera. J. Biol. Chem. 1998, 273, 35000–35007. [Google Scholar]
- Ettenberg, S.A.; Magnifico, A.; Cuello, M.; Nau, M.M.; Rubinstein, Y.R.; Yarden, Y.; Weissman, A.M.; Lipkowitz, S. Cbl-b-dependent Coordinated Degradation of the Epidermal Growth Factor Receptor Signaling Complex. J. Biol. Chem. 2001, 276, 27677–27684. [Google Scholar] [CrossRef] [PubMed]
- Alwan, H.A.J.; Van Zoelen, E.J.J.; Van Leeuwen, J.E.M. Ligand-induced lysosomal epidermal growth factor receptor (EGFR) degradation is preceded by proteasome-dependent EGFR de-ubiquitination. J. Biol. Chem. 2003, 278, 35781–35790. [Google Scholar] [CrossRef]
- Mayle, K.M.; Le, A.M.; Kamei, D.T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 264–281. [Google Scholar] [CrossRef] [PubMed]
- Mesaki, K.; Tanabe, K.; Obayashi, M.; Oe, N.; Takei, K. Fission of tubular endosomes triggers endosomal acidification and movement. PLoS ONE 2011, 6, e19764. [Google Scholar] [CrossRef]
- Jacomin, A.C.; Samavedam, S.; Promponas, V.; Nezis, I.P. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy 2016, 12, 1945–1953. [Google Scholar] [CrossRef]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef]
- Capuani, F.; Conte, A.; Argenzio, E.; Marchetti, L.; Priami, C.; Polo, S.; Di Fiore, P.P.; Sigismund, S.; Ciliberto, A. Quantitative analysis reveals how EGFR activation and downregulation are coupled in normal but not in cancer cells. Nat. Commun. 2015, 6, 7999. [Google Scholar] [CrossRef]
- Freed, D.M.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Mohrlüder, J.; Hoffmann, Y.; Stangler, T.; Hänel, K.; Willbold, D. Identification of clathrin heavy chain as a direct interaction partner for the γ-aminobutyric acid type A receptor associated protein. Biochemistry 2007, 46, 14537–14543. [Google Scholar] [CrossRef] [PubMed]
- Caldieri, G.; Barbieri, E.; Nappo, G.; Raimondi, A.; Bonora, M.; Conte, A.; Verhoef, L.G.G.C.; Confalonieri, S.; Malabarba, M.G.; Bianchi, F.; et al. Reticulon 3-Dependent ER-PM contact sites control EGFR nonclathrin endocytosis. Science 2017, 356, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Morozzi, G.; Hölper, S.; Mari, M.; Harwardt, M.L.I.E.; Yan, R.; Müller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 2017, 6, e25555. [Google Scholar] [CrossRef]
- Sun, A.; Wei, J.; Childress, C.; Shaw, J.H.; Peng, K.; Shao, G.; Yang, W.; Lin, Q. The E3 ubiquitin ligase NEDD4 is an LC3-interactive protein and regulates autophagy. Autophagy 2017, 13, 522–537. [Google Scholar] [CrossRef]
- Haglund, K.; Sigismund, S.; Polo, S.; Szymkiewicz, I.; Di Fiore, P.P.; Dikic, I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 2003, 5, 461–466. [Google Scholar] [CrossRef]
- Critchley, W.; Pellet-Many, C.; Ringham-Terry, B.; Harrison, M.; Zachary, I.; Ponnambalam, S. Receptor Tyrosine Kinase Ubiquitination and De-Ubiquitination in Signal Transduction and Receptor Trafficking. Cells 2018, 7, 22. [Google Scholar] [CrossRef]
- Lin, Q.; Wang, J.; Childress, C.; Sudol, M.; Carey, D.J.; Yang, W. HECT E3 Ubiquitin Ligase Nedd4-1 Ubiquitinates ACK and Regulates Epidermal Growth Factor (EGF)-Induced Degradation of EGF Receptor and ACK. Mol. Cell. Biol. 2010, 30, 1541–1554. [Google Scholar] [CrossRef]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 122–139. [Google Scholar] [CrossRef]
- Huotari, J.; Meyer-Schaller, N.; Hubner, M.; Stauffer, S.; Katheder, N.; Horvath, P.; Mancini, R.; Helenius, A.; Peter, M. Cullin-3 regulates late endosome maturation. Proc. Natl. Acad. Sci. USA 2012, 109, 823–828. [Google Scholar] [CrossRef]
- Genau, H.M.; Huber, J.; Baschieri, F.; Akutsu, M.; Dötsch, V.; Farhan, H.; Rogov, V.; Behrends, C. CUL3-KBTBD6/KBTBD7Ubiquitin Ligase Cooperates with GABARAP Proteins to Spatially Restrict TIAM1-RAC1 Signaling. Mol. Cell 2015, 57, 995–1010. [Google Scholar] [CrossRef] [PubMed]
- Wandinger-Ness, A.; Zerial, M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 2014, 6, a022616. [Google Scholar] [CrossRef] [PubMed]
- Goueli, B.S.; Powell, M.; Finger, E.C.; Pfeffer, S.R. TBC1D16 is a Rab4A GTPase activating protein that regulates receptor recycling and EGF receptor signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 15787–15792. [Google Scholar] [CrossRef] [PubMed]
- Popovic, D.; Akutsu, M.; Novak, I.; Harper, J.W.; Behrends, C.; Dikic, I. Rab GTPase-Activating Proteins in Autophagy: Regulation of Endocytic and Autophagy Pathways by Direct Binding to Human ATG8 Modifiers. Mol. Cell. Biol. 2012, 32, 1733–1744. [Google Scholar] [CrossRef]
- Nordmann, M.; Cabrera, M.; Perz, A.; Bröcker, C.; Ostrowicz, C.; Engelbrecht-Vandré, S.; Ungermann, C. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr. Biol. 2010, 20, 1654–1659. [Google Scholar] [CrossRef]
- Ieguchi, K.; Ueda, S.; Kataoka, T.; Satoh, T. Role of the guanine nucleotide exchange factor Ost in negative regulation of receptor endocytosis by the small GTPase Rac. J. Biol. Chem. 2007, 282, 23296–23305. [Google Scholar] [CrossRef]
- Longva, K.E.; Blystad, F.D.; Stang, E.; Larsen, A.M.; Johannessen, L.E.; Madshus, I.H. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol. 2002, 156, 843–854. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Oksvold, M.P.; Skarpen, E.; Wierød, L.; Paulsen, R.E.; Huitfeldt, H.S. Re-localization of activated EGF receptor and its signal transducers to multivesicular compartments downstream of early endosomes in response to EGF. Eur. J. Cell Biol. 2001, 80, 285–294. [Google Scholar] [CrossRef]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; MirandadeStegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef]
- Ariotti, N.; Liang, H.; Xu, Y.; Zhang, Y.; Yonekubo, Y.; Inder, K.; Du, G.; Parton, R.G.; Hancock, J.F.; Plowman, S.J. Epidermal Growth Factor Receptor Activation Remodels the Plasma Membrane Lipid Environment To Induce Nanocluster Formation. Mol. Cell. Biol. 2010, 30, 3795–3804. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.I.; van Lengerich, B.; Eichel, K.; Cha, M.; Patterson, D.M.; Yoon, T.Y.; von Zastrow, M.; Jura, N.; Gartner, Z.J. Phosphorylated EGFR Dimers Are Not Sufficient to Activate Ras. Cell Rep. 2018, 22, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Needham, S.R.; Roberts, S.K.; Arkhipov, A.; Mysore, V.P.; Tynan, C.J.; Zanetti-Domingues, L.C.; Kim, E.T.; Losasso, V.; Korovesis, D.; Hirsch, M.; et al. EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat. Commun. 2016, 7, 13307. [Google Scholar] [CrossRef] [PubMed]
- Heukers, R.; Vermeulen, J.F.; Fereidouni, F.; Bader, A.N.; Voortman, J.; Roovers, R.C.; Gerritsen, H.C.; Van Bergen En Henegouwen, P.M.P. Endocytosis of EGFR requires its kinase activity and N-terminal transmembrane dimerization motif. J. Cell Sci. 2013, 126, 4900–4912. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, V.; Ma, P.; Thielmann, Y.; Hartmann, R.; Weiergräber, O.H.; Mohrlüder, J.; Willbold, D. Assessment of GABARAP self-association by its diffusion properties. J. Biomol. NMR 2010, 48, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kuma, A.; Sugiura, Y.; Ichimura, Y.; Obata, M.; Kitamura, H.; Okuda, S.; Lee, H.C.; Ikeda, K.; Kanegae, Y.; et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019, 10, 1567. [Google Scholar] [CrossRef]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-Mediated Beclin 1 Phosphorylation in Autophagy Suppression, Tumor Progression, and Tumor Chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef]
- Winer, H.; Fraiberg, M.; Abada, A.; Dadosh, T.; Tamim-Yecheskel, B.C.; Elazar, Z. Autophagy differentially regulates TNF receptor Fn14 by distinct mammalian Atg8 proteins. Nat. Commun. 2018, 9, 3744. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobner, J.; Simons, I.M.; Rufinatscha, K.; Hänsch, S.; Schwarten, M.; Weiergräber, O.H.; Abdollahzadeh, I.; Gensch, T.; Bode, J.G.; Hoffmann, S.; et al. Deficiency of GABARAP but Not Its Paralogs Causes Enhanced EGF-Induced EGFR Degradation. Cells 2020, 9, 1296. https://doi.org/10.3390/cells9051296
Dobner J, Simons IM, Rufinatscha K, Hänsch S, Schwarten M, Weiergräber OH, Abdollahzadeh I, Gensch T, Bode JG, Hoffmann S, et al. Deficiency of GABARAP but Not Its Paralogs Causes Enhanced EGF-Induced EGFR Degradation. Cells. 2020; 9(5):1296. https://doi.org/10.3390/cells9051296
Chicago/Turabian StyleDobner, Jochen, Indra M. Simons, Kerstin Rufinatscha, Sebastian Hänsch, Melanie Schwarten, Oliver H. Weiergräber, Iman Abdollahzadeh, Thomas Gensch, Johannes G. Bode, Silke Hoffmann, and et al. 2020. "Deficiency of GABARAP but Not Its Paralogs Causes Enhanced EGF-Induced EGFR Degradation" Cells 9, no. 5: 1296. https://doi.org/10.3390/cells9051296
APA StyleDobner, J., Simons, I. M., Rufinatscha, K., Hänsch, S., Schwarten, M., Weiergräber, O. H., Abdollahzadeh, I., Gensch, T., Bode, J. G., Hoffmann, S., & Willbold, D. (2020). Deficiency of GABARAP but Not Its Paralogs Causes Enhanced EGF-Induced EGFR Degradation. Cells, 9(5), 1296. https://doi.org/10.3390/cells9051296