Computational Tools and Resources Supporting CRISPR-Cas Experiments


Abstract
1. Introduction
2. Experimental Design—the Choice of a Genome Editing System
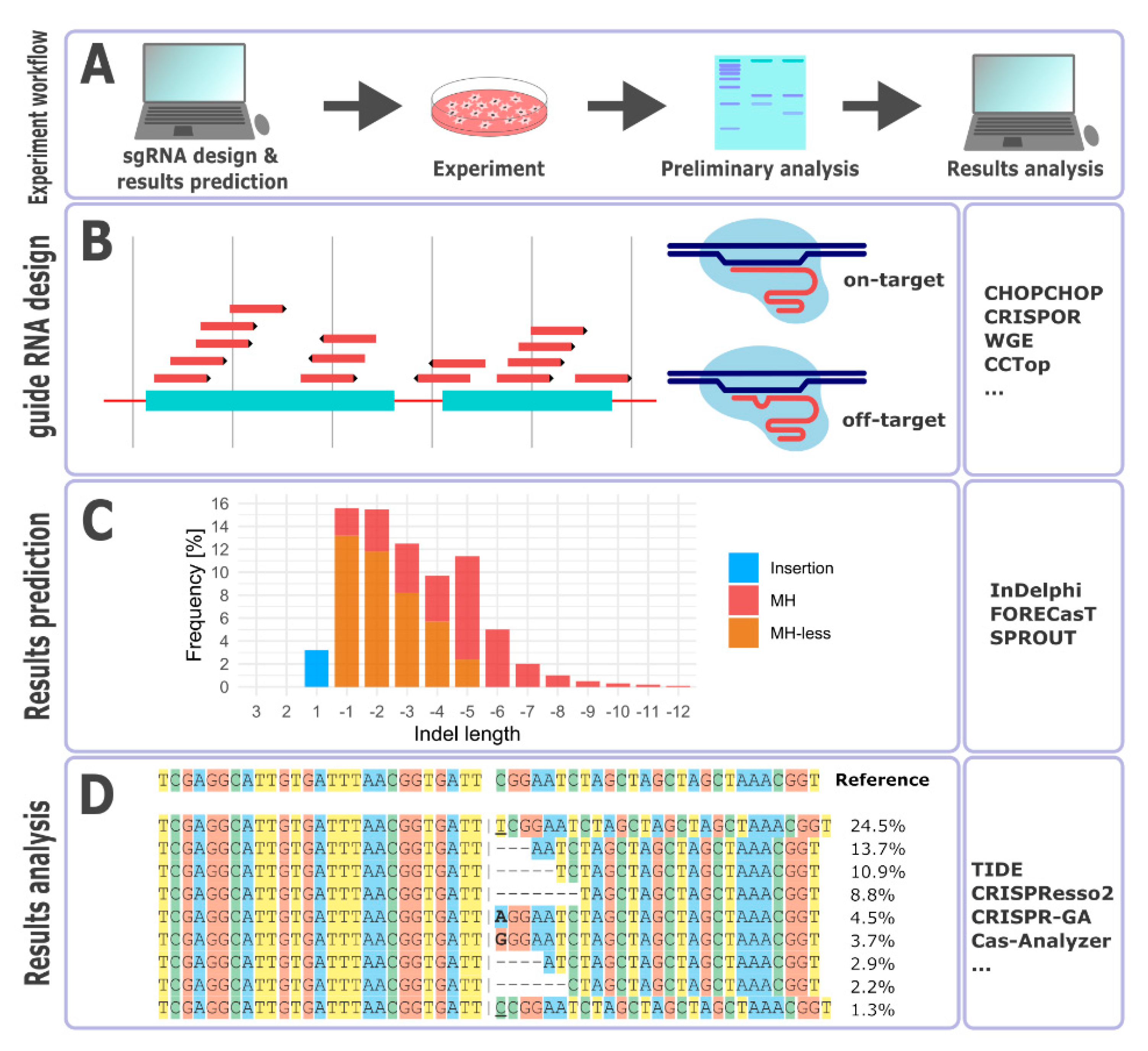
3. Guide RNA Design
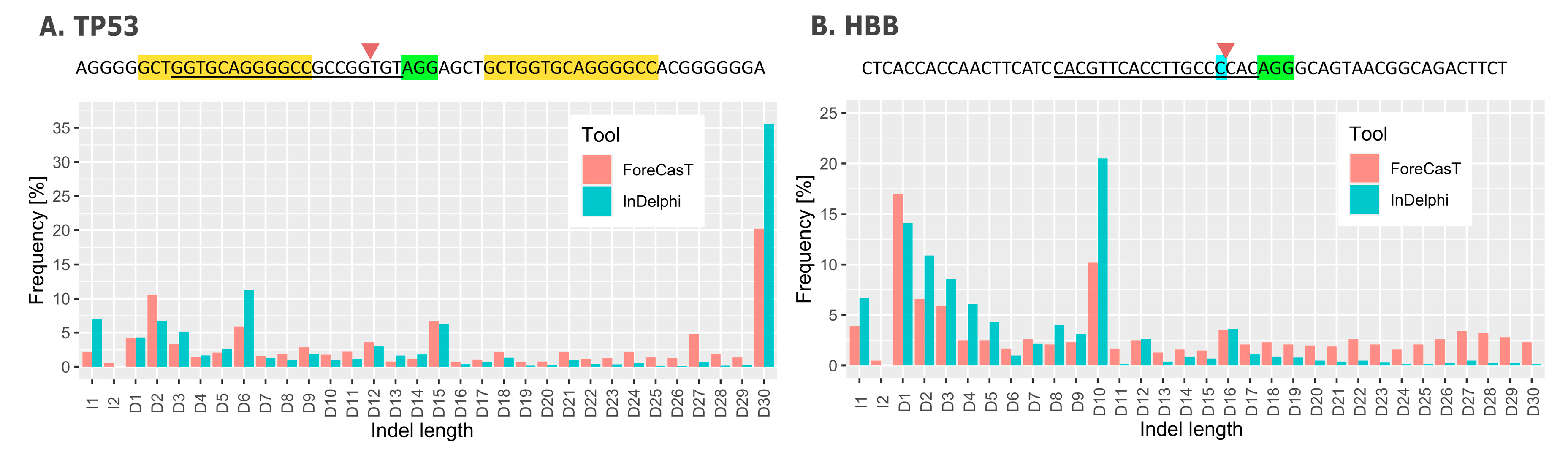
4. Repair Outcome Predictions
5. Outcomes Analysis
6. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019, 37, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.M.; Henser-Brownhill, T.; Monserrat, J.; Poetsch, A.R.; Luscombe, N.M.; Scaffidi, P. Target-Specific Precision of CRISPR-Mediated Genome Editing. Mol. Cell 2019, 73, 699–713.e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; McKenna, A.; Schreiber, J.; Haeussler, M.; Yin, Y.; Agarwal, V.; Noble, W.S.; Shendure, J. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 2019, 47, 7989–8003. [Google Scholar] [CrossRef] [PubMed]
- Van Overbeek, M.; Capurso, D.; Carter, M.M.; Thompson, M.S.; Frias, E.; Russ, C.; Reece-Hoyes, J.S.; Nye, C.; Gradia, S.; Vidal, B.; et al. DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 2016, 63, 633–646. [Google Scholar] [CrossRef]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Xu, H.; Xiao, T.; Chen, C.-H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef]
- Mendoza, B.J.; Trinh, C.T. Enhanced guide-RNA design and targeting analysis for precise CRISPR genome editing of single and consortia of industrially relevant and non-model organisms. Bioinformatics 2018, 34, 16–23. [Google Scholar] [CrossRef]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.-D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.; Zhang, Y.; Wang, H.; Ying, H.; Liu, M.; Li, D.; Lui, K.O.; Ding, Q. A Self-restricted CRISPR System to Reduce Off-target Effects. Mol. Ther. 2016, 24, 1508–1510. [Google Scholar] [CrossRef]
- Gao, Z.; Herrera-Carrillo, E.; Berkhout, B. A Single H1 Promoter Can Drive Both Guide RNA and Endonuclease Expression in the CRISPR-Cas9 System. Mol. Ther. Nucleic Acids 2019, 14, 32–40. [Google Scholar] [CrossRef]
- Liu, K.I.; Ramli, M.N.B.; Woo, C.W.A.; Wang, Y.; Zhao, T.; Zhang, X.; Yim, G.R.D.; Chong, B.Y.; Gowher, A.; Chua, M.Z.H.; et al. A chemical-inducible CRISPR–Cas9 system for rapid control of genome editing. Nat. Chem. Biol. 2016, 12, 980–987. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, J.; Cheng, M.; Liao, X.; Peng, S. Review of CRISPR/Cas9 sgRNA Design Tools. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 455–465. [Google Scholar] [CrossRef]
- Wilson, L.O.W.; O’Brien, A.R.; Bauer, D.C. The Current State and Future of CRISPR-Cas9 gRNA Design Tools. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Brazelton, V.A.; Zarecor, S.; Wright, D.A.; Wang, Y.; Liu, J.; Chen, K.; Yang, B.; Lawrence-Dill, C.J. A quick guide to CRISPR sgRNA design tools. GM Crop. Food 2015, 6, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Fonfara, I.; Le Rhun, A.; Chylinski, K.; Makarova, K.S.; Lécrivain, A.-L.; Bzdrenga, J.; Koonin, E.V.; Charpentier, E. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 2014, 42, 2577–2590. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.; Carlson-Stevermer, J.; Saha, K. Increasing the precision of gene editing in vitro, ex vivo, and in vivo. Curr. Opin. Biomed. Eng. 2018, 7, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef]
- Williams, D.J.; Puhl, H.L.; Ikeda, S.R. A Simple, Highly Efficient Method for Heterologous Expression in Mammalian Primary Neurons Using Cationic Lipid-mediated mRNA Transfection. Front. Neurosci. 2010, 4, 181. [Google Scholar] [CrossRef]
- Lattanzi, A.; Meneghini, V.; Pavani, G.; Amor, F.; Ramadier, S.; Felix, T.; Antoniani, C.; Masson, C.; Alibeu, O.; Lee, C.; et al. Optimization of CRISPR/Cas9 Delivery to Human Hematopoietic Stem and Progenitor Cells for Therapeutic Genomic Rearrangements. Mol. Ther. 2019, 27, 137–150. [Google Scholar] [CrossRef]
- Xu, X.; Wan, T.; Xin, H.; Li, D.; Pan, H.; Wu, J.; Ping, Y. Delivery of CRISPR/Cas9 for therapeutic genome editing. J. Gene Med. 2019, 21, e3107. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- User Guides & Protocols. Available online: https://eu.idtdna.com/pages/support/guides-and-protocols (accessed on 19 February 2020).
- Addgene: CRISPR References and Information. Available online: https://www.addgene.org/crispr/reference/#protocols (accessed on 19 February 2020).
- CRISPR Protocols and Methods | Springer Nature Experiments. Available online: https://experiments.springernature.com/techniques/crispr (accessed on 19 February 2020).
- Protocols for the CRISPR/Cas Technology_Bio-protocol. Available online: https://bio-protocol.org/Special_Issue_info.aspx?siid=11 (accessed on 19 February 2020).
- Vouillot, L.; Thélie, A.; Pollet, N. Comparison of T7E1 and Surveyor Mismatch Cleavage Assays to Detect Mutations Triggered by Engineered Nucleases. G3: Genes Genomes Genet. 2015, 5, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, M.; Czubak, K.; Juzwa, W.; Krzyzosiak, W.J.; Olejniczak, M.; Kozlowski, P. qEva-CRISPR: A method for quantitative evaluation of CRISPR/Cas-mediated genome editing in target and off-target sites. Nucleic Acids Res. 2018, 46, e101. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Magor, G.W.; Gillinder, K.R.; Perkins, A.C. A high-throughput screening strategy for detecting CRISPR-Cas9 induced mutations using next-generation sequencing. BMC Genom. 2014, 15, 1002. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Yang, Z.; Steentoft, C.; Hauge, C.; Hansen, L.; Thomsen, A.L.; Niola, F.; Vester-Christensen, M.B.; Frödin, M.; Clausen, H.; Wandall, H.H.; et al. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015, 43, e59. [Google Scholar] [CrossRef]
- Findlay, S.D.; Vincent, K.M.; Berman, J.R.; Postovit, L.-M. A Digital PCR-Based Method for Efficient and Highly Specific Screening of Genome Edited Cells. PLoS ONE 2016, 11, e0153901. [Google Scholar] [CrossRef]
- Lomov, N.A.; Viushkov, V.S.; Petrenko, A.P.; Syrkina, M.S.; Rubtsov, M.A. Methods of Evaluating the Efficiency of CRISPR/Cas Genome Editing. Mol. Biol. 2019, 53, 862–875. [Google Scholar] [CrossRef]
- Sentmanat, M.F.; Peters, S.T.; Florian, C.P.; Connelly, J.P.; Pruett-Miller, S.M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Germini, D.; Tsfasman, T.; Zakharova, V.V.; Sjakste, N.; Lipinski, M.; Vassetzky, Y. A Comparison of Techniques to Evaluate the Effectiveness of Genome Editing. Trends Biotechnol. 2018, 36, 147–159. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, G.; Li, J.; Zhang, X.; Huang, S.; Xiang, S.; Hu, X.; Liu, C. CRISPRlnc: A manually curated database of validated sgRNAs for lncRNAs. Nucleic Acids Res. 2019, 47, D63–D68. [Google Scholar] [CrossRef] [PubMed]
- Varshney, G.K.; Zhang, S.; Pei, W.; Adomako-Ankomah, A.; Fohtung, J.; Schaffer, K.; Carrington, B.; Maskeri, A.; Slevin, C.; Wolfsberg, T.; et al. CRISPRz: A database of zebrafish validated sgRNAs. Nucleic Acids Res. 2016, 44, D822–D826. [Google Scholar] [CrossRef] [PubMed]
- Addgene: Validated gRNA Sequences. Available online: https://www.addgene.org/crispr/reference/grna-sequence/ (accessed on 19 February 2020).
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Torres-Perez, R.; Garcia-Martin, J.A.; Montoliu, L.; Oliveros, J.C.; Pazos, F. WeReview: CRISPR Tools—Live Repository of Computational Tools for Assisting CRISPR/Cas Experiments. Bioengineering 2019, 6, 63. [Google Scholar] [CrossRef]
- Chari, R.; Mali, P.; Moosburner, M.; Church, G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods 2015, 12, 823–826. [Google Scholar] [CrossRef]
- Wong, N.; Liu, W.; Wang, X. WU-CRISPR: Characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015, 16, 218. [Google Scholar] [CrossRef]
- Labuhn, M.; Adams, F.F.; Ng, M.; Knoess, S.; Schambach, A.; Charpentier, E.M.; Schwarzer, A.; Mateo, J.L.; Klusmann, J.-H.; Heckl, D. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR–Cas9 applications. Nucleic Acids Res. 2018, 46, 1375–1385. [Google Scholar] [CrossRef]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.-B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef]
- Lee, C.M.; Davis, T.H.; Bao, G. Examination of CRISPR/Cas9 design tools and the effect of target site accessibility on Cas9 activity. Exp. Physiol. 2018, 103, 456–460. [Google Scholar] [CrossRef]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Gagnon, J.A.; Thyme, S.B.; Valen, E. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016, 44, W272–W276. [Google Scholar] [CrossRef] [PubMed]
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.; Fuller, C.K.; Donohoue, P.D.; Jones, B.N.; Thompson, M.S.; Carter, M.M.; Gradia, S.; Vidal, B.; Garner, E.; Slorach, E.M.; et al. Mapping the genomic landscape of CRISPR–Cas9 cleavage. Nat. Methods 2017, 14, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.-S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef]
- Listgarten, J.; Weinstein, M.; Kleinstiver, B.P.; Sousa, A.A.; Joung, J.K.; Crawford, J.; Gao, K.; Hoang, L.; Elibol, M.; Doench, J.G.; et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2018, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Abadi, S.; Yan, W.X.; Amar, D.; Mayrose, I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017, 13, e1005807. [Google Scholar] [CrossRef]
- Singh, R.; Kuscu, C.; Quinlan, A.; Qi, Y.; Adli, M. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015, 43, e118. [Google Scholar] [CrossRef]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 80. [Google Scholar] [CrossRef]
- Zhang, D.; Hurst, T.; Duan, D.; Chen, S.-J. Unified energetics analysis unravels SpCas9 cleavage activity for optimal gRNA design. Proc. Natl. Acad. Sci. USA 2019, 116, 8693–8698. [Google Scholar] [CrossRef]
- Bae, S.; Kweon, J.; Kim, H.S.; Kim, J.-S. Microhomology-based choice of Cas9 nuclease target sites. Nat. Methods 2014, 11, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Graf, R.; Li, X.; Chu, V.T.; Rajewsky, K. sgRNA Sequence Motifs Blocking Efficient CRISPR/Cas9-Mediated Gene Editing. Cell Rep. 2019, 26, 1098–1103.e3. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2017, 803–805, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef]
- Taheri-Ghahfarokhi, A.; Taylor, B.J.M.; Nitsch, R.; Lundin, A.; Cavallo, A.-L.; Madeyski-Bengtson, K.; Karlsson, F.; Clausen, M.; Hicks, R.; Mayr, L.M.; et al. Decoding non-random mutational signatures at Cas9 targeted sites. Nucleic Acids Res. 2018, 46, 8417–8434. [Google Scholar] [CrossRef] [PubMed]
- Lemos, B.R.; Kaplan, A.C.; Bae, J.E.; Ferrazzoli, A.E.; Kuo, J.; Anand, R.P.; Waterman, D.P.; Haber, J.E. CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. Proc. Natl. Acad. Sci. USA 2018, 115, E2040–E2047. [Google Scholar] [CrossRef]
- Leenay, R.T.; Aghazadeh, A.; Hiatt, J.; Tse, D.; Hultquist, J.F.; Krogan, N.; Wu, Z.; Marson, A.; May, A.P.; Zou, J. Systematic characterization of genome editing in primary T cells reveals proximal genomic insertions and enables machine learning prediction of CRISPR-Cas9 DNA repair outcomes. bioRxiv 2018, 404947. [Google Scholar] [CrossRef]
- Cradick, T.J.; Fine, E.J.; Antico, C.J.; Bao, G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Kousholt, A.N.; Harmsen, T.; Leemans, C.; Chen, T.; Jonkers, J.; van Steensel, B. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 2018, 46, e58. [Google Scholar] [CrossRef]
- Lee, H.; Chang, H.Y.; Cho, S.W.; Ji, H.P. CRISPRpic: Fast and precise analysis for CRISPR-induced mutations via prefixed index counting. Nar. Genom. Bioinform. 2020, 2. [Google Scholar] [CrossRef]
- Connelly, J.P.; Pruett-Miller, S.M. CRIS.py: A Versatile and High-throughput Analysis Program for CRISPR-based Genome Editing. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, H.; Burger, A.; Biyong, B.; Felker, A.; Hess, C.; Zaugg, J.; Chiavacci, E.; Anders, C.; Jinek, M.; Mosimann, C.; et al. CrispRVariants charts the mutation spectrum of genome engineering experiments. Nat. Biotechnol. 2016, 34, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lim, K.; Kim, J.-S.; Bae, S. Cas-analyzer: An online tool for assessing genome editing results using NGS data. Bioinformatics 2017, 33, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Guell, M.; Yang, L.; Church, G.M. Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics 2014, 30, 2968–2970. [Google Scholar] [CrossRef] [PubMed]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E.; et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef]
- Pinello, L.; Canver, M.C.; Hoban, M.D.; Orkin, S.H.; Kohn, D.B.; Bauer, D.E.; Yuan, G.-C. Analyzing CRISPR genome editing experiments with CRISPResso. Nat. Biotechnol. 2016, 34, 695–697. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813.e6. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef]
- Hart, T.; Moffat, J. BAGEL: A computational framework for identifying essential genes from pooled library screens. BMC Bioinform. 2016, 17, 164. [Google Scholar] [CrossRef]
- Jeong, H.-H.; Kim, S.Y.; Rousseaux, M.W.C.; Zoghbi, H.Y.; Liu, Z. CRISPRCloud2: A cloud-based platform for deconvolving CRISPR screen data. bioRxiv 2018. [Google Scholar] [CrossRef]
- Spahn, P.N.; Bath, T.; Weiss, R.J.; Kim, J.; Esko, J.D.; Lewis, N.E.; Harismendy, O. PinAPL-Py: A comprehensive web-application for the analysis of CRISPR/Cas9 screens. Sci. Rep. 2017, 7, 15854. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Schwering, M.; Pelz, O.; Rauscher, B.; Zhan, T.; Heigwer, F.; Boutros, M. CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. bioRxiv 2017. [Google Scholar] [CrossRef]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.-J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wu, X.; Wang, J.; Wang, Y.; Qiu, Z.; Chang, T.; Huang, H.; Lin, R.-J.; Yee, J.-K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015, 33, 175–178. [Google Scholar] [CrossRef]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Wyman, S.K.; Richardson, C.D.; Yeh, C.D.; Akcakaya, P.; Porritt, M.J.; Morlock, M.; Vu, J.T.; Kazane, K.R.; Watry, H.L.; et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 2019, 364, 286–289. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef]
- Horlbeck, M.A.; Witkowsky, L.B.; Guglielmi, B.; Replogle, J.M.; Gilbert, L.A.; Villalta, J.E.; Torigoe, S.E.; Tjian, R.; Weissman, J.S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. eLife 2016, 5, e12677. [Google Scholar] [CrossRef]
- Isaac, R.S.; Jiang, F.; Doudna, J.A.; Lim, W.A.; Narlikar, G.J.; Almeida, R. Nucleosome breathing and remodeling constrain CRISPR-Cas9 function. eLife 2016, 5, e13450. [Google Scholar] [CrossRef]
- Yarrington, R.M.; Verma, S.; Schwartz, S.; Trautman, J.K.; Carroll, D. Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, 9351–9358. [Google Scholar] [CrossRef]
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. Acs. Synth. Biol. 2017, 6, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Fløe, L.; Petersen, T.S.; Huang, J.; Xu, F.; Bolund, L.; Luo, Y.; Lin, L. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017, 591, 1892–1901. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Mäkelä, M.I.E.; Barker, H.R.; Bäuerlein, C.A.; Häkkinen, T.; Nykter, M.; Rämet, M. Chromatin accessibility is associated with CRISPR-Cas9 efficiency in the zebrafish (Danio rerio). PLoS ONE 2018, 13, e0196238. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Rinsma, M.; Janssen, J.M.; Liu, J.; Maggio, I.; Gonçalves, M.A.F.V. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016, 44, 6482–6492. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Joung, J.K.; Pinello, L. Impact of Genetic Variation on CRISPR-Cas Targeting. Cris. J. 2018, 1, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Lessard, S.; Francioli, L.; Alfoldi, J.; Tardif, J.-C.; Ellinor, P.T.; MacArthur, D.G.; Lettre, G.; Orkin, S.H.; Canver, M.C. Human genetic variation alters CRISPR-Cas9 on- and off-targeting specificity at therapeutically implicated loci. Proc. Natl. Acad. Sci. USA 2017, 114, E11257–E11266. [Google Scholar] [CrossRef]
- Scott, D.A.; Zhang, F. Implications of human genetic variation in CRISPR-based therapeutic genome editing. Nat. Med. 2017, 23, 1095–1101. [Google Scholar] [CrossRef]
- Chen, C.-L.; Rodiger, J.; Chung, V.; Viswanatha, R.; Mohr, S.E.; Hu, Y.; Perrimon, N. SNP-CRISPR: A Web Tool for SNP-Specific Genome Editing. G3: Genes Genomes Genet. 2020, 10, 489–494. [Google Scholar] [CrossRef]
- Keough, K.C.; Lyalina, S.; Olvera, M.P.; Whalen, S.; Conklin, B.R.; Pollard, K.S. AlleleAnalyzer: A tool for personalized and allele-specific sgRNA design. Genome Biol. 2019, 20, 167. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Compared Feature | FORECasT | SPROUT | inDelphi |
|---|---|---|---|
| Indel frequency | YES | YES | YES |
| Average indel length | NO | YES | NO |
| Sequences of predictions | YES | NO | YES |
| Alignment of predicted sequences | YES | NO | YES |
| Frequency count for every predicted sequence | YES | NO | YES |
| Frameshift/ in frame frequency | YES | NO | YES |
| MH strength of target site | NO | NO | YES |
| Level of homogeneity of predictions | NO | NO | YES |
| Distinction between MH and MH-less deletions | NO | NO | YES |
| Cell type enable to choose | NO | NO | YES (HEK293, HCT116, K562, mESC, U2OS) |
| Batch mode | Available in command line tool | NO | YES |
| Gene mode | NO | NO | YES (for human and mouse) |
| Shareable link to results | NO | NO | YES |
| Summary statistics for download | YES | NO | YES |
| References | [3] | [82] | [7] |
| Compared Feature | CRISPResso2 | Cas-Analyzer | CRISPR-GA | TIDE/TIDER |
|---|---|---|---|---|
| Type of analysis | NGS | NGS | NGS | Sanger sequencing |
| File type | FASTQ | FASTQ | FASTQ | ABI |
| Output | -indel sizes and positions -HDR/NHEJ frequency -sequence alignment with reference -allele specific quantification | -indel sizes and positions -HDR/NHEJ frequency -sequence alignment with reference | -indel sizes and positions -HDR/NHEJ frequency | -indel sizes and positions -HDR/NHEJ frequency |
| Batch functionality | YES | NO | NO | NO |
| Base editing experiments | YES | NO | NO | NO |
| Template-mediated editing | YES | YES | YES | YES (TIDER) |
| Supported nucleases | Cas9, Cpf1 | SpCas9, StCas9, NmCas9, SaCas9, CjCas9, AsCpf1/LbCpf1, paired nucleases: ZFNs, TALENs, Cas9 nickases, dCas9-FokI | Cas9 | SpCas9, SaCas9, St1Cas9, NmCas9, AsCpf1, FnCpf1, LbCpf1 |
| Need to upload dataset to a server | YES (up to 100Mb) | NO | YES | YES |
| Command line interface tool available | YES | NO | NO | NO |
| References | [90] | [88] | [89] | [49,92] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sledzinski, P.; Nowaczyk, M.; Olejniczak, M. Computational Tools and Resources Supporting CRISPR-Cas Experiments. Cells 2020, 9, 1288. https://doi.org/10.3390/cells9051288
Sledzinski P, Nowaczyk M, Olejniczak M. Computational Tools and Resources Supporting CRISPR-Cas Experiments. Cells. 2020; 9(5):1288. https://doi.org/10.3390/cells9051288
Chicago/Turabian StyleSledzinski, Pawel, Mateusz Nowaczyk, and Marta Olejniczak. 2020. "Computational Tools and Resources Supporting CRISPR-Cas Experiments" Cells 9, no. 5: 1288. https://doi.org/10.3390/cells9051288
APA StyleSledzinski, P., Nowaczyk, M., & Olejniczak, M. (2020). Computational Tools and Resources Supporting CRISPR-Cas Experiments. Cells, 9(5), 1288. https://doi.org/10.3390/cells9051288