Alpha Radiation as a Way to Target Heterochromatic and Gamma Radiation-Exposed Breast Cancer Cells


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blot
2.3. Irradiation
2.4. Clonogenic Survival Assay
2.5. γH2AX Immunofluorescence and Scoring
2.6. Real-Time PCR
2.7. Micronucleus Assay
2.8. Statistics and Curve Fits
3. Results
3.1. Effects of Trichostatin A (TSA) on Chromatin Structure and Clonogenic Survival after Exposure to Gamma vs. Alpha Radiation
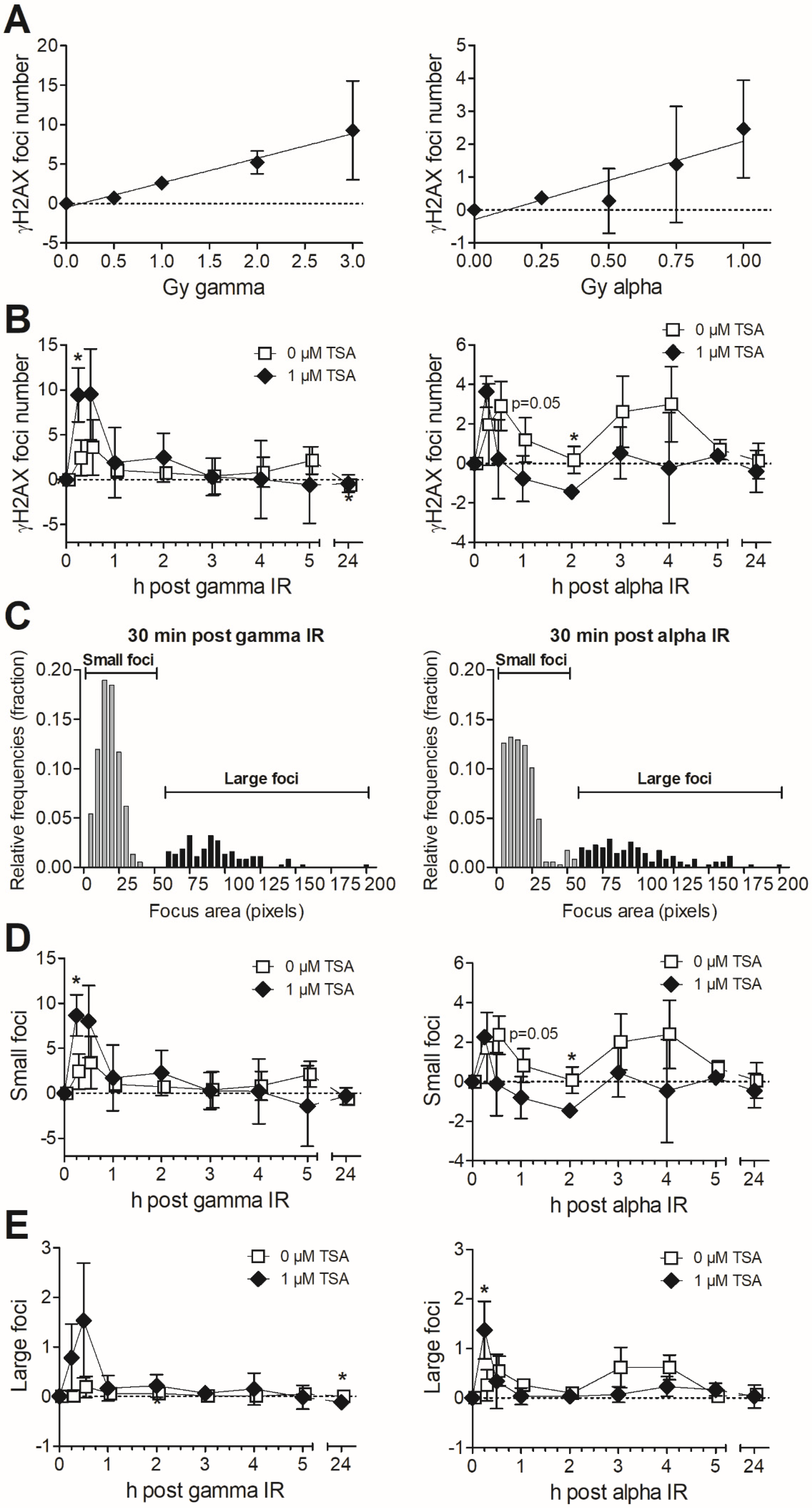
3.2. Formation and Removal of γH2AX Foci in TSA-Pretreated MDA-MB-231 Exposed to Gamma and Alpha Radiation
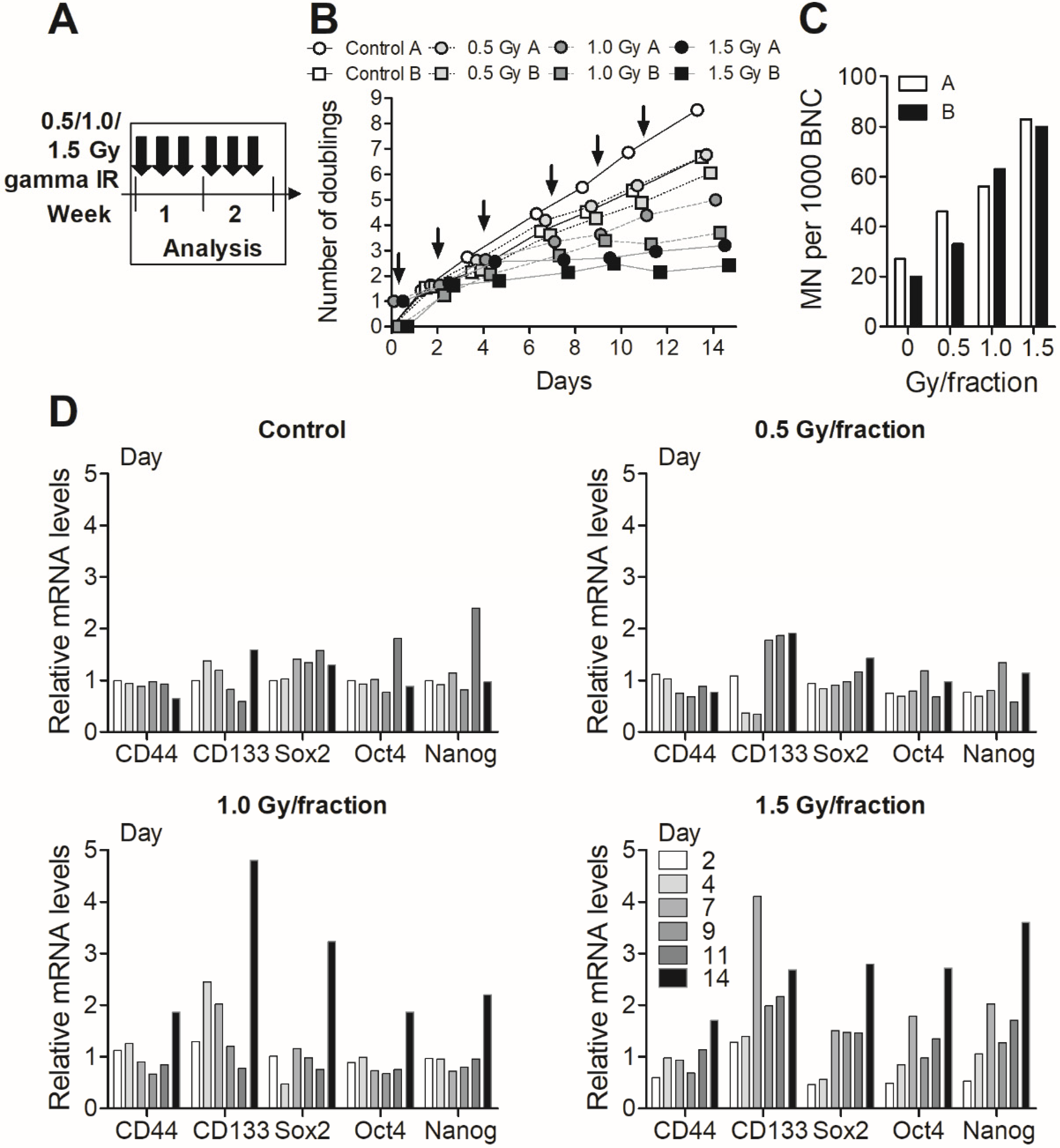
3.3. Fractionated Gamma Radiation Exposure in MDA-MB-231 Cells
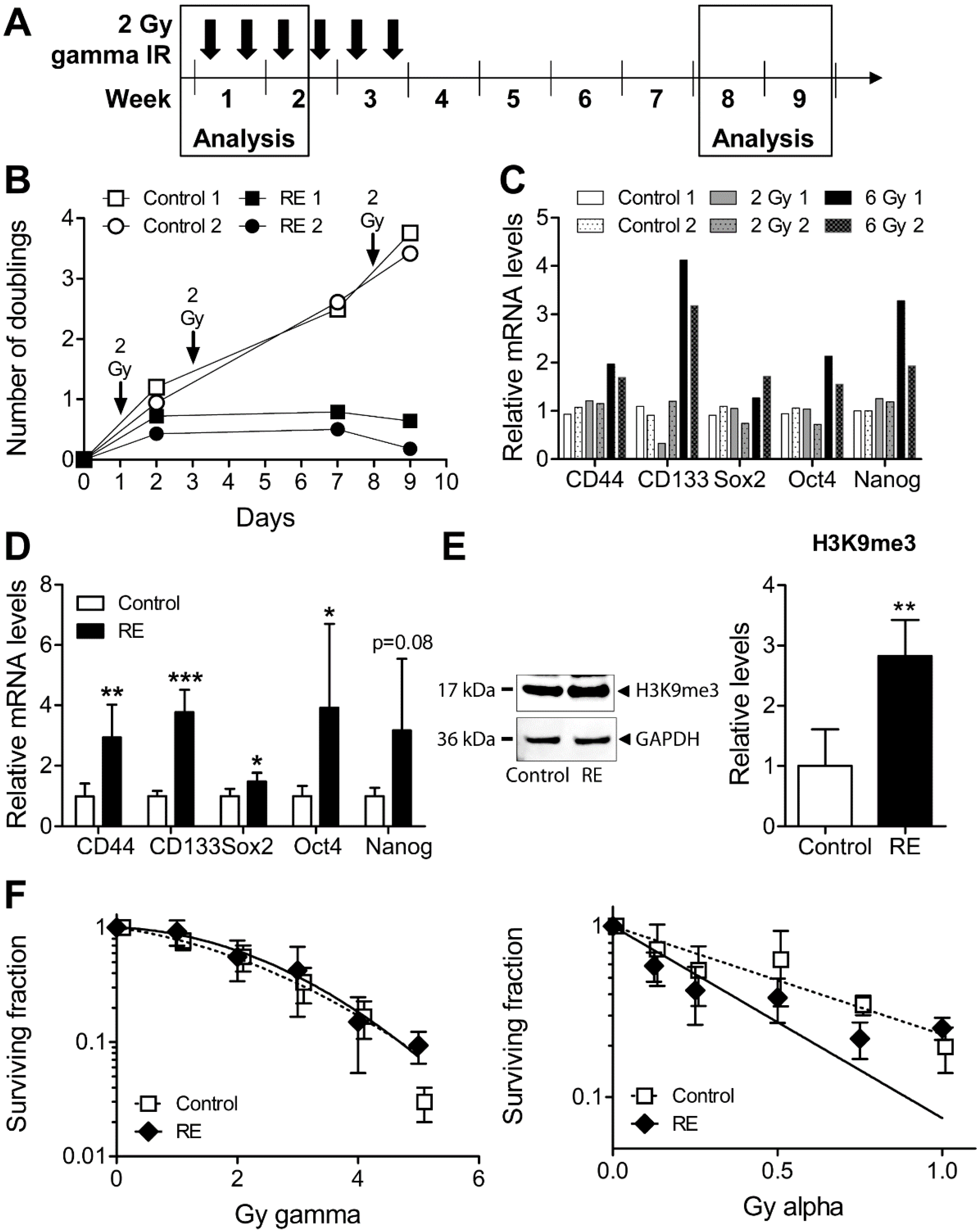
3.4. Cellular Properties and Response to Alpha Radiation in Gamma Radiation-Exposed MDA-MB-231
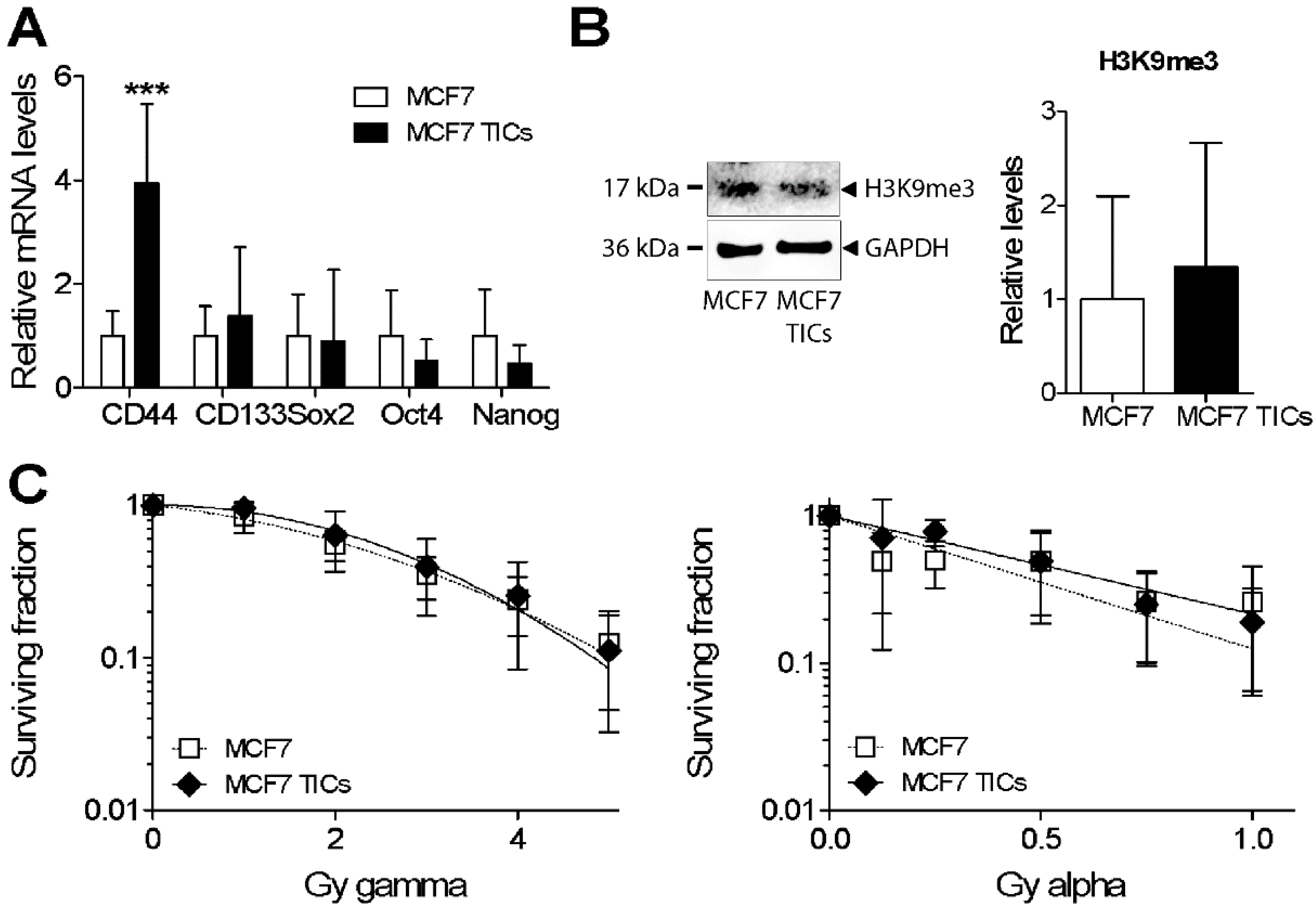
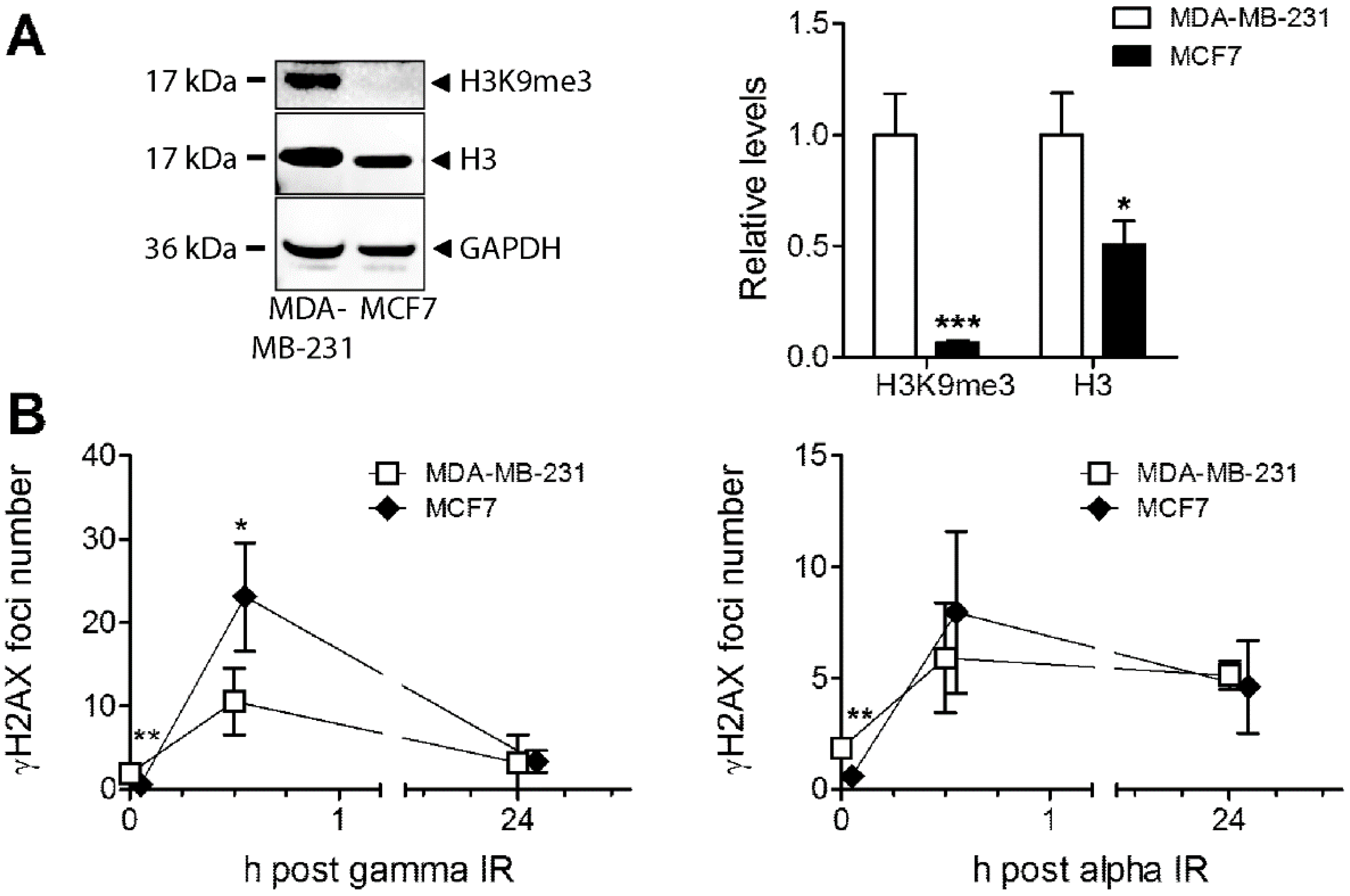
3.5. Analysis of Heterochromatin Structure and Radiation Response in Luminal Type MCF7 Compared to Triple Negative MDA-MB-231 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Langlands, F.E.; Horgan, K.; Dodwell, D.D.; Smith, L. Breast cancer subtypes: Response to radiotherapy and potential radiosensitisation. Br. J. Radiol. 2013, 86, 20120601. [Google Scholar] [CrossRef]
- Szumiel, I. Intrinsic radiation sensitivity: Cellular signaling is the key. Radiat. Res. 2008, 169, 249–258. [Google Scholar] [CrossRef]
- Gray, M.; Turnbull, A.K.; Ward, C.; Meehan, J.; Martinez-Perez, C.; Bonello, M.; Pang, L.Y.; Langdon, S.P.; Kunkler, I.H.; Murray, A.; et al. Development and characterisation of acquired radioresistant breast cancer cell lines. Radiat. Oncol. 2019, 14, 64. [Google Scholar] [CrossRef]
- McDermott, N.; Meunier, A.; Mooney, B.; Nortey, G.; Hernandez, C.; Hurley, S.; Lynam-Lennon, N.; Barsoom, S.H.; Bowman, K.J.; Marples, B.; et al. Fractionated radiation exposure amplifies the radioresistant nature of prostate cancer cells. Sci. Rep. 2016, 6, 34796. [Google Scholar] [CrossRef]
- Subik, K.; Lee, J.F.; Baxter, L.; Strzepek, T.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.C.; Bonfiglio, T.; Hicks, D.G.; et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer Basic Clin. Res. 2010, 4, 35–41. [Google Scholar] [CrossRef]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef]
- Cann, K.L.; Dellaire, G. Heterochromatin and the DNA damage response: The need to relax. Biochem. Cell Biol. 2011, 89, 45–60. [Google Scholar] [CrossRef]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef]
- Leatherbarrow, E.L.; Harper, J.V.; Cucinotta, F.A.; O’Neill, P. Induction and quantification of gamma-H2AX foci following low and high LET-irradiation. Int. J. Radiat. Biol. 2006, 82, 111–118. [Google Scholar] [CrossRef]
- Schmid, T.E.; Dollinger, G.; Beisker, W.; Hable, V.; Greubel, C.; Auer, S.; Mittag, A.; Tarnok, A.; Friedl, A.A.; Molls, M.; et al. Differences in the kinetics of gamma-H2AX fluorescence decay after exposure to low and high LET radiation. Int. J. Radiat. Biol. 2010, 86, 682–691. [Google Scholar] [CrossRef]
- Sollazzo, A.; Brzozowska, B.; Cheng, L.; Lundholm, L.; Haghdoost, S.; Scherthan, H.; Wojcik, A. Alpha Particles and X Rays Interact in Inducing DNA Damage in U2OS Cells. Radiat. Res. 2017, 188, 400–411. [Google Scholar] [CrossRef]
- Goodhead, D.T. Mechanisms for the biological effectiveness of high-LET radiations. J. Radiat. Res. 1999, 40, S1–S13. [Google Scholar] [CrossRef]
- Franken, N.A.; Hovingh, S.; Ten Cate, R.; Krawczyk, P.; Stap, J.; Hoebe, R.; Aten, J.; Barendsen, G.W. Relative biological effectiveness of high linear energy transfer alpha-particles for the induction of DNA-double-strand breaks, chromosome aberrations and reproductive cell death in SW-1573 lung tumour cells. Oncol. Rep. 2012, 27, 769–774. [Google Scholar] [CrossRef]
- Falk, M.; Lukasova, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin compaction protects genomic DNA from radiation damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.A. The heterochromatic barrier to DNA double strand break repair: How to get the entry visa. Int. J. Mol. Sci. 2012, 13, 11844–11860. [Google Scholar] [CrossRef]
- Barendsen, G.W.; Koot, C.J.; Van Kersen, G.R.; Bewley, D.K.; Field, S.B.; Parnell, C.J. The effect of oxygen on impairment of the proliferative capacity of human cells in culture by ionizing radiations of different LET. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1966, 10, 317–327. [Google Scholar] [CrossRef]
- Barendsen, G.W. The relationships between RBE and LET for different types of lethal damage in mammalian cells: Biophysical and molecular mechanisms. Radiat. Res. 1994, 139, 257–270. [Google Scholar] [CrossRef]
- de Kruijff, R.M.; Wolterbeek, H.T.; Denkova, A.G. A Critical Review of Alpha Radionuclide Therapy-How to Deal with Recoiling Daughters? Pharmaceuticals (Basel) 2015, 8, 321–336. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- Bodet-Milin, C.; Kraeber-Bodere, F.; Eugene, T.; Guerard, F.; Gaschet, J.; Bailly, C.; Mougin, M.; Bourgeois, M.; Faivre-Chauvet, A.; Cherel, M.; et al. Radioimmunotherapy for Treatment of Acute Leukemia. Semin. Nucl. Med. 2016, 46, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Cordier, D.; Krolicki, L.; Morgenstern, A.; Merlo, A. Targeted Radiolabeled Compounds in Glioma Therapy. Semin. Nucl. Med. 2016, 46, 243–249. [Google Scholar] [CrossRef]
- Norain, A.; Dadachova, E. Targeted Radionuclide Therapy of Melanoma. Semin. Nucl. Med. 2016, 46, 250–259. [Google Scholar] [CrossRef]
- Fendler, W.P.; Cutler, C. More alpha Than beta for Prostate Cancer? J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 1709–1710. [Google Scholar] [CrossRef]
- Eriksson, M.; Haag, P.; Brzozowska, B.; Lipka, M.; Lisowska, H.; Lewensohn, R.; Wojcik, A.; Viktorsson, K.; Lundholm, L. Analysis of Chromatin Opening in Heterochromatic Non-Small Cell Lung Cancer Tumor-Initiating Cells in Relation to DNA-Damaging Antitumor Treatment. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 174–187. [Google Scholar] [CrossRef]
- Lundholm, L.; Haag, P.; Juntti, T.; Lewensohn, R.; Viktorsson, K. Phosphoprotein analysis reveals MEK inhibition as a way to target non-small cell lung cancer tumor initiating cells. Int. J. Radiat. Biol. 2014, 90, 718–726. [Google Scholar] [CrossRef]
- Lundholm, L.; Haag, P.; Zong, D.; Juntti, T.; Mork, B.; Lewensohn, R.; Viktorsson, K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013, 4, e478. [Google Scholar] [CrossRef]
- Staaf, E.; Brehwens, K.; Haghdoost, S.; Pachnerova-Brabcova, K.; Czub, J.; Braziewicz, J.; Nievaart, S.; Wojcik, A. Characterisation of a setup for mixed beam exposures of cells to 241Am alpha particles and X-rays. Radiat. Prot. Dosim. 2012, 151, 570–579. [Google Scholar] [CrossRef]
- Brzozowska, B.; Galecki, M.; Tartas, A.; Ginter, J.; Kazmierczak, U.; Lundholm, L. Freeware tool for analysing numbers and sizes of cell colonies. Radiat. Environ. Biophys. 2019, 58, 109–117. [Google Scholar] [CrossRef]
- Staaf, E.; Brehwens, K.; Haghdoost, S.; Czub, J.; Wojcik, A. Gamma-H2AX foci in cells exposed to a mixed beam of X-rays and alpha particles. Genome Integr. 2012, 3, 8. [Google Scholar] [CrossRef]
- Chadwick, K.H.; Leenhouts, H.P. A molecular theory of cell survival. Phys. Med. Biol. 1973, 18, 78–87. [Google Scholar] [PubMed]
- Sollazzo, A.; Shakeri-Manesh, S.; Fotouhi, A.; Czub, J.; Haghdoost, S.; Wojcik, A. Interaction of low and high LET radiation in TK6 cells-mechanistic aspects and significance for radiation protection. J. Radiol. Prot. Off. J. Soc. Radiol. Prot. 2016, 36, 721–735. [Google Scholar] [CrossRef]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhasz, S.; Kunzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Lobrich, M. DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. The complexity of DNA damage: Relevance to biological consequences. Int. J. Radiat. Biol. 1994, 66, 427–432. [Google Scholar] [PubMed]
- Costes, S.V.; Boissiere, A.; Ravani, S.; Romano, R.; Parvin, B.; Barcellos-Hoff, M.H. Imaging features that discriminate between foci induced by high- and low-LET radiation in human fibroblasts. Radiat. Res. 2006, 165, 505–515. [Google Scholar] [CrossRef]
- Fenech, M.; Denham, J.; Francis, W.; Morley, A. Micronuclei in cytokinesis-blocked lymphocytes of cancer patients following fractionated partial-body radiotherapy. Int. J. Radiat. Biol. 1990, 57, 373–383. [Google Scholar]
- Lee, Y.H.; Liu, X.; Qiu, F.; O’Connor, T.R.; Yen, Y.; Ann, D.K. HP1beta is a biomarker for breast cancer prognosis and PARP inhibitor therapy. PLoS ONE 2015, 10, e0121207. [Google Scholar] [CrossRef]
- Saini, V.; Hose, C.D.; Monks, A.; Nagashima, K.; Han, B.; Newton, D.L.; Millione, A.; Shah, J.; Hollingshead, M.G.; Hite, K.M.; et al. Identification of CBX3 and ABCA5 as putative biomarkers for tumor stem cells in osteosarcoma. PLoS ONE 2012, 7, e41401. [Google Scholar] [CrossRef]
- Slezak, J.; Truong, M.; Huang, W.; Jarrard, D. HP1gamma expression is elevated in prostate cancer and is superior to Gleason score as a predictor of biochemical recurrence after radical prostatectomy. BMC Cancer 2013, 13, 148. [Google Scholar] [CrossRef]
- Yu, Y.H.; Chiou, G.Y.; Huang, P.I.; Lo, W.L.; Wang, C.Y.; Lu, K.H.; Yu, C.C.; Alterovitz, G.; Huang, W.C.; Lo, J.F.; et al. Network biology of tumor stem-like cells identified a regulatory role of CBX5 in lung cancer. Sci. Rep. 2012, 2, 584. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Oertel, S.; Thiemann, M.; Richter, K.; Weber, K.J.; Huber, P.E.; Perez, R.L.; Brons, S.; Bischof, M.; Kulozik, A.E.; Ehemann, V.; et al. Combination of suberoylanilide hydroxamic acid with heavy ion therapy shows promising effects in infantile sarcoma cell lines. Radiat. Oncol. 2011, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Jeynes, J.C.; Merchant, M.J.; Wera, A.C.; Barry, M.A.; Kirkby, K.J.; Suzuki, M. Radiosensitization of glioblastoma cells using a histone deacetylase inhibitor (SAHA) comparing carbon ions with X-rays. Int. J. Radiat. Biol. 2015, 91, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Yamada, S.; Hoshino, I.; Murakami, K.; Akutsu, Y.; Sakata, H.; Nishimori, T.; Usui, A.; Miyazawa, Y.; Kamada, T.; et al. Effects of carbon-ion radiotherapy combined with a novel histone deacetylase inhibitor, cyclic hydroxamic-acid-containing peptide 31 in human esophageal squamous cell carcinoma. Anticancer Res. 2009, 29, 4433–4438. [Google Scholar] [PubMed]
- Fortuny, A.; Polo, S.E. The response to DNA damage in heterochromatin domains. Chromosoma 2018, 127, 291–300. [Google Scholar] [CrossRef]
- Tang, N.; Bueno, M.; Meylan, S.; Incerti, S.; Tran, H.N.; Vaurijoux, A.; Gruel, G.; Villagrasa, C. Influence of chromatin compaction on simulated early radiation-induced DNA damage using Geant4-DNA. Med. Phys. 2019, 46, 1501–1511. [Google Scholar] [CrossRef]
- Lorat, Y.; Timm, S.; Jakob, B.; Taucher-Scholz, G.; Rube, C.E. Clustered double-strand breaks in heterochromatin perturb DNA repair after high linear energy transfer irradiation. Radiother. Oncol. 2016, 121, 154–161. [Google Scholar] [CrossRef]
- Kallimasioti-Pazi, E.M.; Thelakkad Chathoth, K.; Taylor, G.C.; Meynert, A.; Ballinger, T.; Kelder, M.J.E.; Lalevee, S.; Sanli, I.; Feil, R.; Wood, A.J. Heterochromatin delays CRISPR-Cas9 mutagenesis but does not influence the outcome of mutagenic DNA repair. PLoS Biol. 2018, 16, e2005595. [Google Scholar] [CrossRef]
- Qin, J.; Li, S.; Zhang, C.; Gao, D.W.; Li, Q.; Zhang, H.; Jin, X.D.; Liu, Y. Apoptosis and injuries of heavy ion beam and x-ray radiation on malignant melanoma cell. Exp. Biol. Med. (Maywood) 2017, 242, 953–960. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, D.; Guo, F.; Chen, X.; Zhu, L.; Zhang, H.; Wang, C.; Shao, C. Chromatin remodeling modulates radiosensitivity of the daughter cells derived from cell population exposed to low- and high-LET irradiation. Oncotarget 2017, 8, 52823–52836. [Google Scholar] [CrossRef]
- Marques, I.A.; Neves, A.R.; Abrantes, A.M.; Pires, A.S.; Tavares-Da-Silva, E.; Figueiredo, A.; Botelho, M.F. Targeted alpha therapy using Radium-223: From physics to biological effects. Cancer Treat. Rev. 2018, 68, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Ho, S.Y.; Lin, P.; Wang, Y.J. Combination of the novel histone deacetylase inhibitor YCW1 and radiation induces autophagic cell death through the downregulation of BNIP3 in triple-negative breast cancer cells in vitro and in an orthotopic mouse model. Mol. Cancer 2016, 15, 46. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svetličič, M.; Bomhard, A.; Sterr, C.; Brückner, F.; Płódowska, M.; Lisowska, H.; Lundholm, L. Alpha Radiation as a Way to Target Heterochromatic and Gamma Radiation-Exposed Breast Cancer Cells. Cells 2020, 9, 1165. https://doi.org/10.3390/cells9051165
Svetličič M, Bomhard A, Sterr C, Brückner F, Płódowska M, Lisowska H, Lundholm L. Alpha Radiation as a Way to Target Heterochromatic and Gamma Radiation-Exposed Breast Cancer Cells. Cells. 2020; 9(5):1165. https://doi.org/10.3390/cells9051165
Chicago/Turabian StyleSvetličič, Maja, Anton Bomhard, Christoph Sterr, Fabian Brückner, Magdalena Płódowska, Halina Lisowska, and Lovisa Lundholm. 2020. "Alpha Radiation as a Way to Target Heterochromatic and Gamma Radiation-Exposed Breast Cancer Cells" Cells 9, no. 5: 1165. https://doi.org/10.3390/cells9051165
APA StyleSvetličič, M., Bomhard, A., Sterr, C., Brückner, F., Płódowska, M., Lisowska, H., & Lundholm, L. (2020). Alpha Radiation as a Way to Target Heterochromatic and Gamma Radiation-Exposed Breast Cancer Cells. Cells, 9(5), 1165. https://doi.org/10.3390/cells9051165