Cell-Free Circulating Mitochondrial DNA: A Potential Blood-Based Marker for Atrial Fibrillation

 ,
, 

Abstract
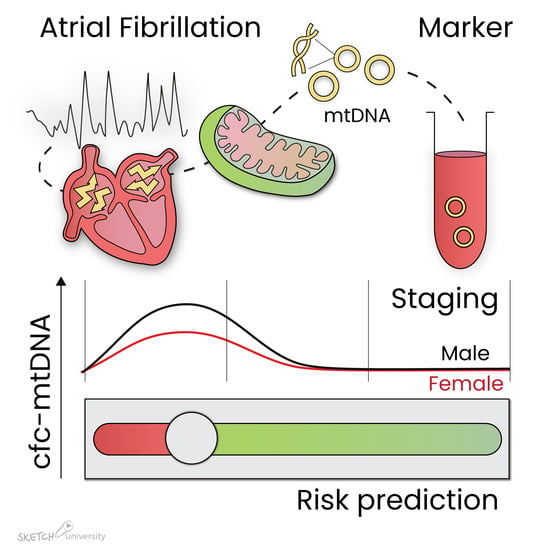
1. Introduction
2. Materials and Methods
2.1. Patient Material
2.2. HL-1 Cardiomyocyte Culture and Tachypacing
2.3. DNA Isolation and Quantitative PCR
2.4. HSP60 ELISA
2.5. Mitochondrial Dysfunction Measurements in HL-1 Atrial Cardiomyocytes
2.5.1. ATP Measurements
2.5.2. Mitochondrial Membrane Potential Analysis
2.5.3. Mitochondrial Morphology Analysis
2.5.4. Mitochondrial Calcium Transient Measurements
2.5.5. Mitochondrial Stress Analysis
2.6. Statistical Analysis
3. Results
3.1. Characteristics of the Study Population
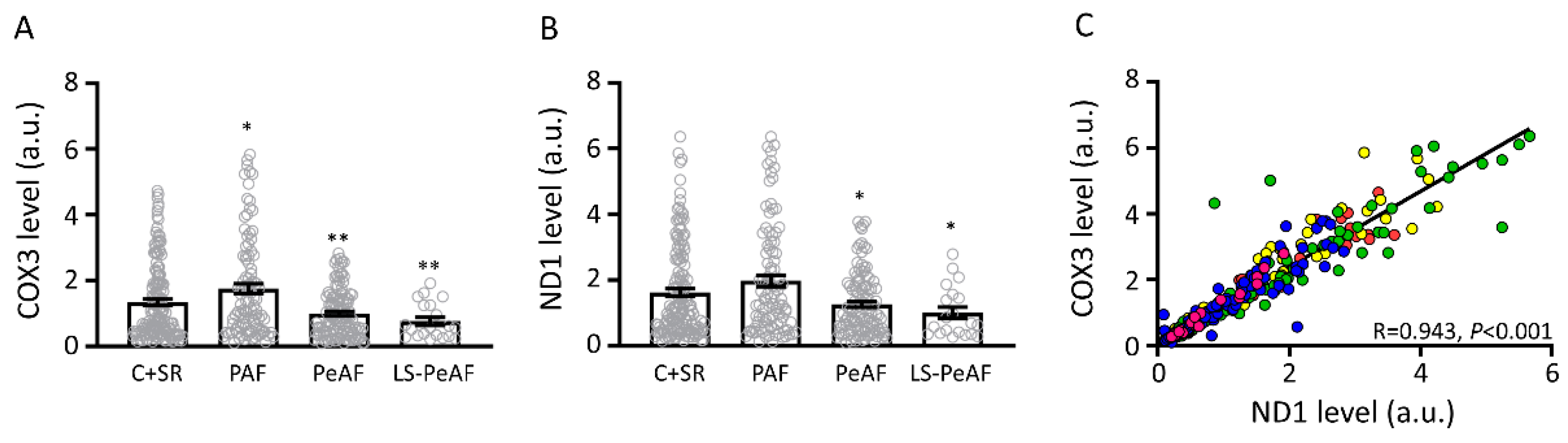
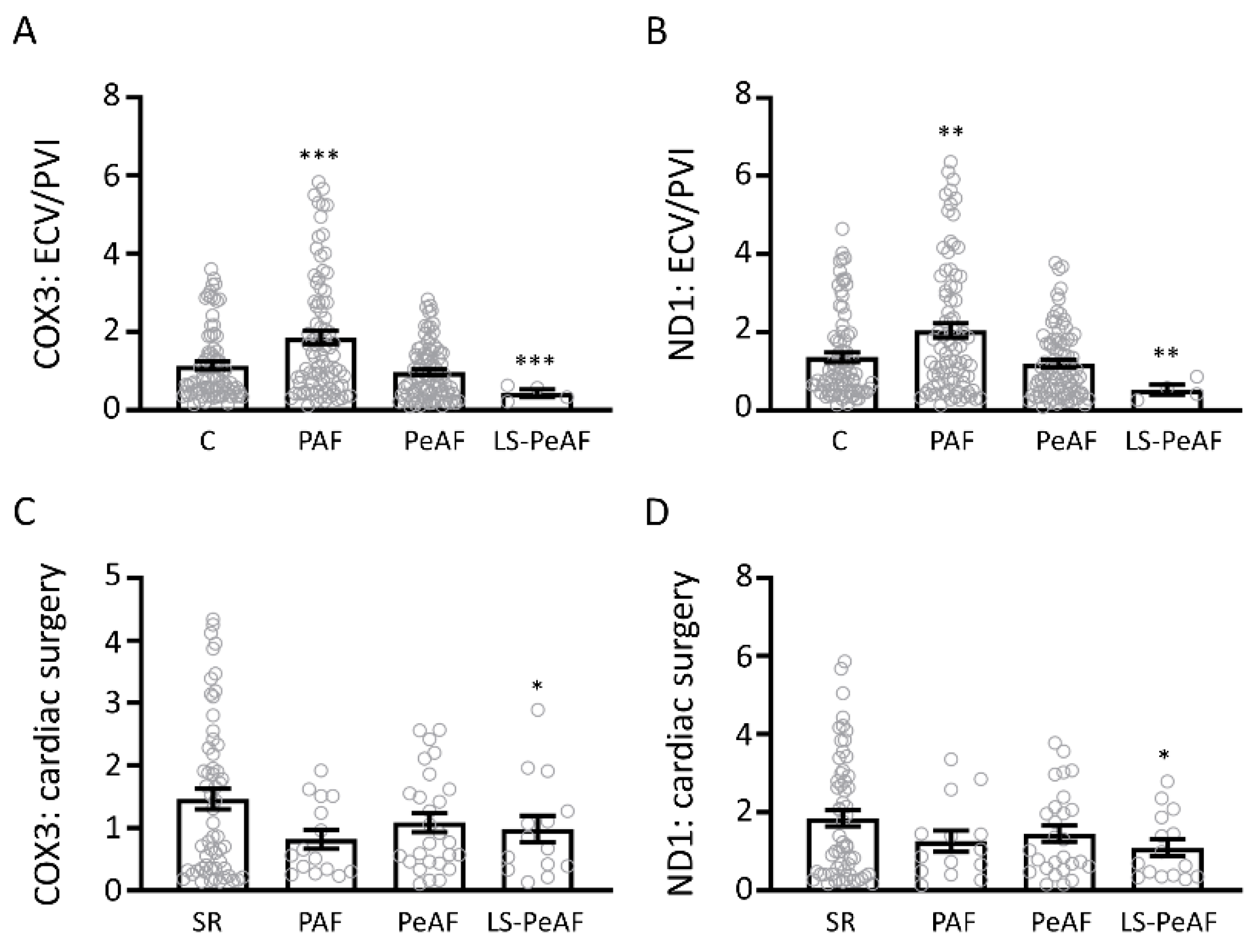
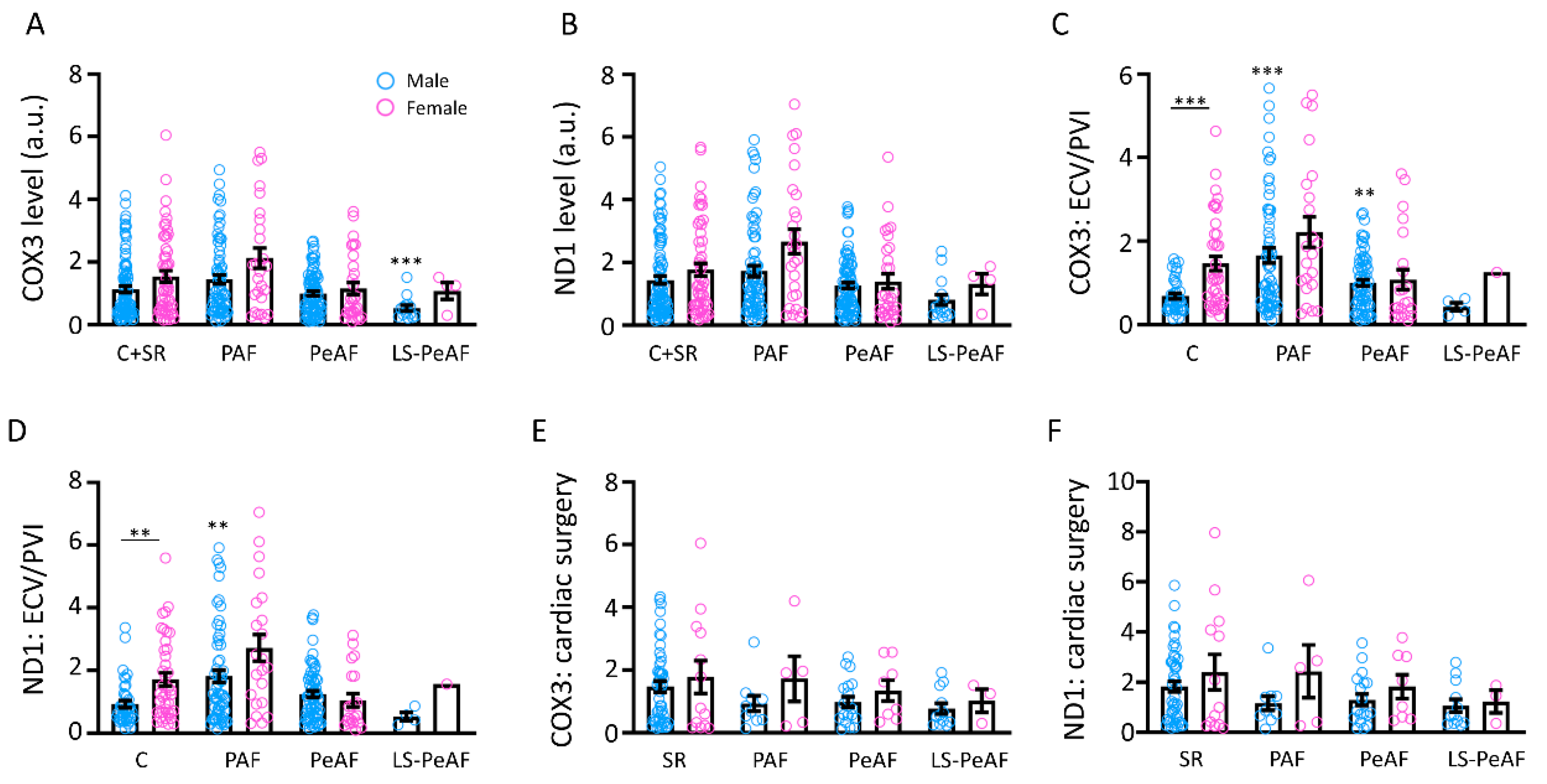
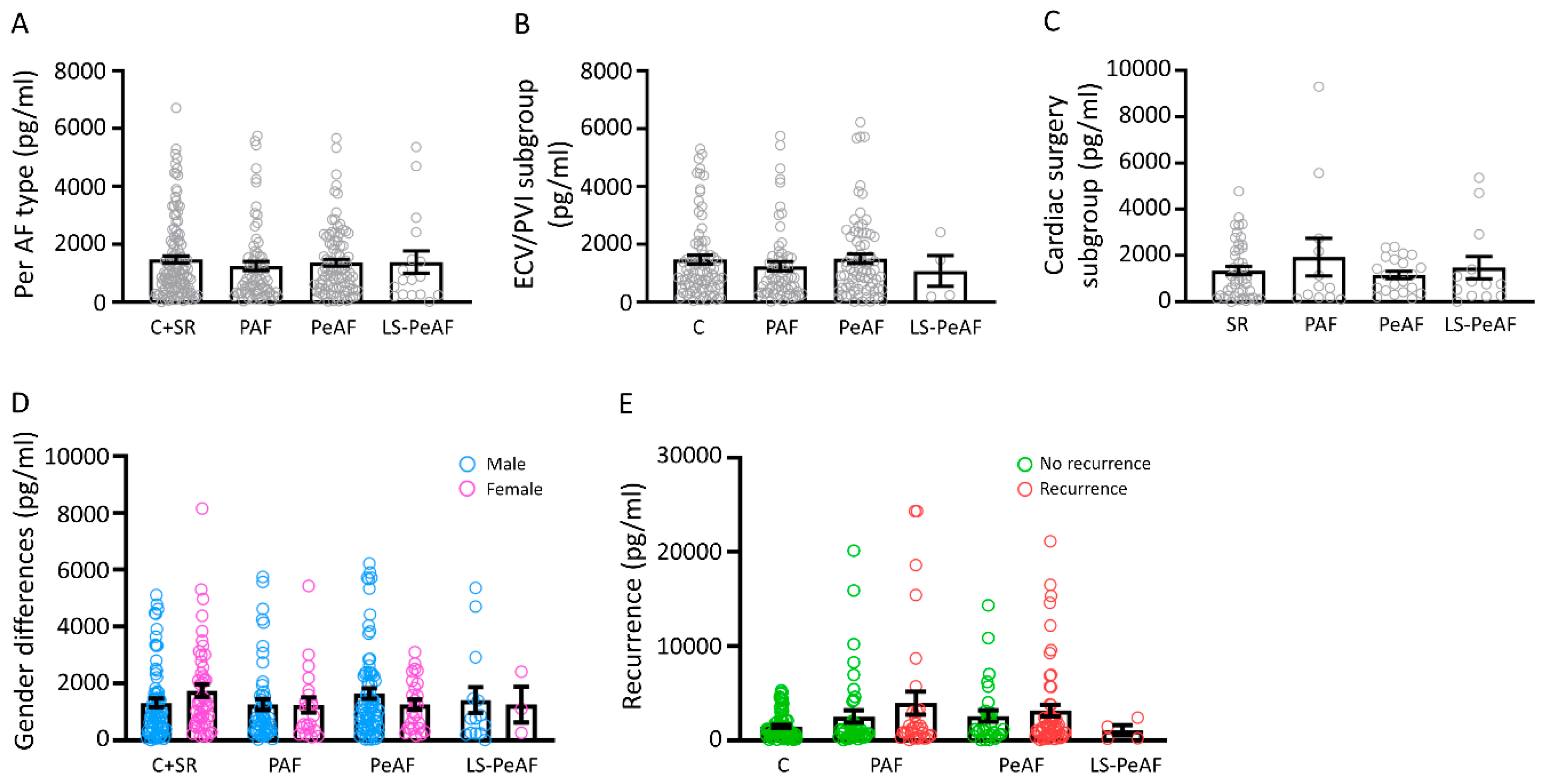
3.2. Association Between Cell-Free Circulating mtDNA and AF Stage
3.3. Cfc-mtDNA Levels in Patients Undergoing AF Treatment or Cardiac Surgery
3.4. Sex Differences in Relation to cfc-mtDNA
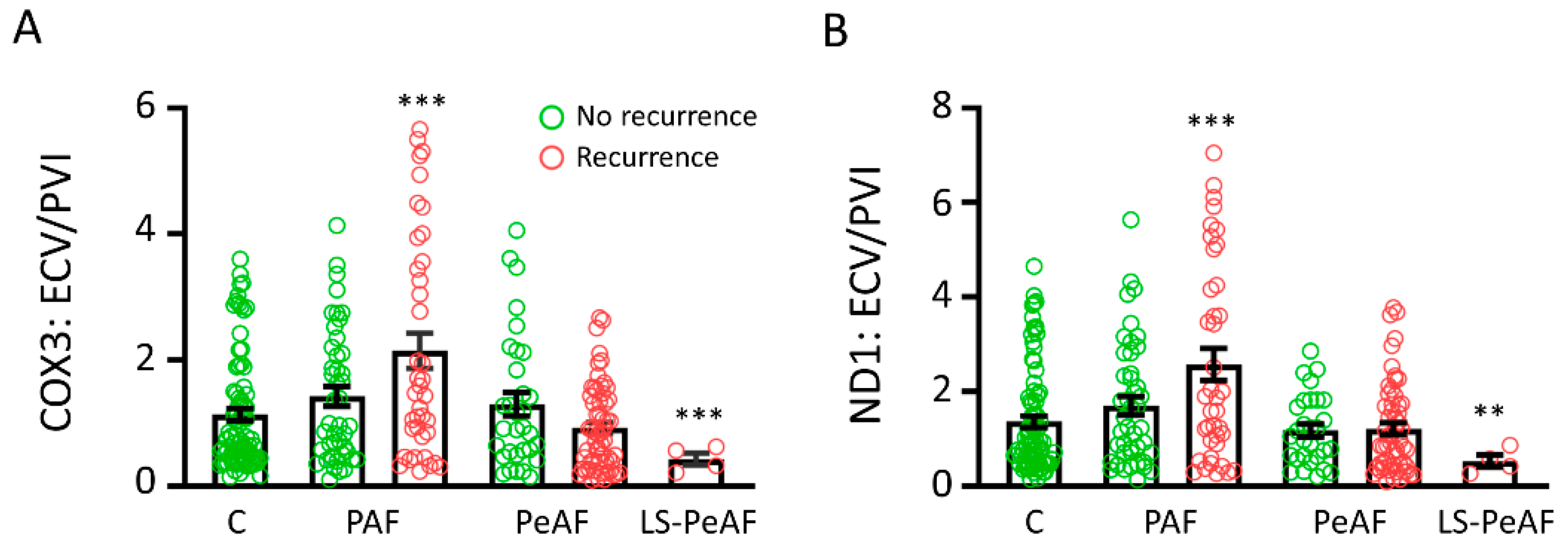
3.5. Cfc-mtDNA Levels May Indicate Recurrence after AF Treatment
3.6. HSP60 Is Not a Mitochondrial Biomarker for AF
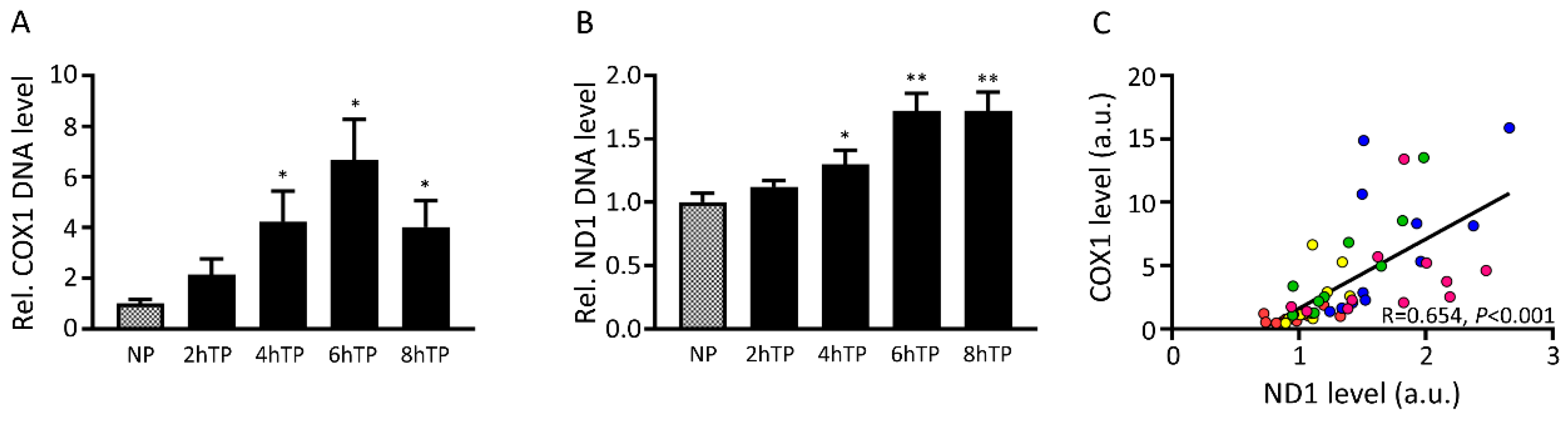
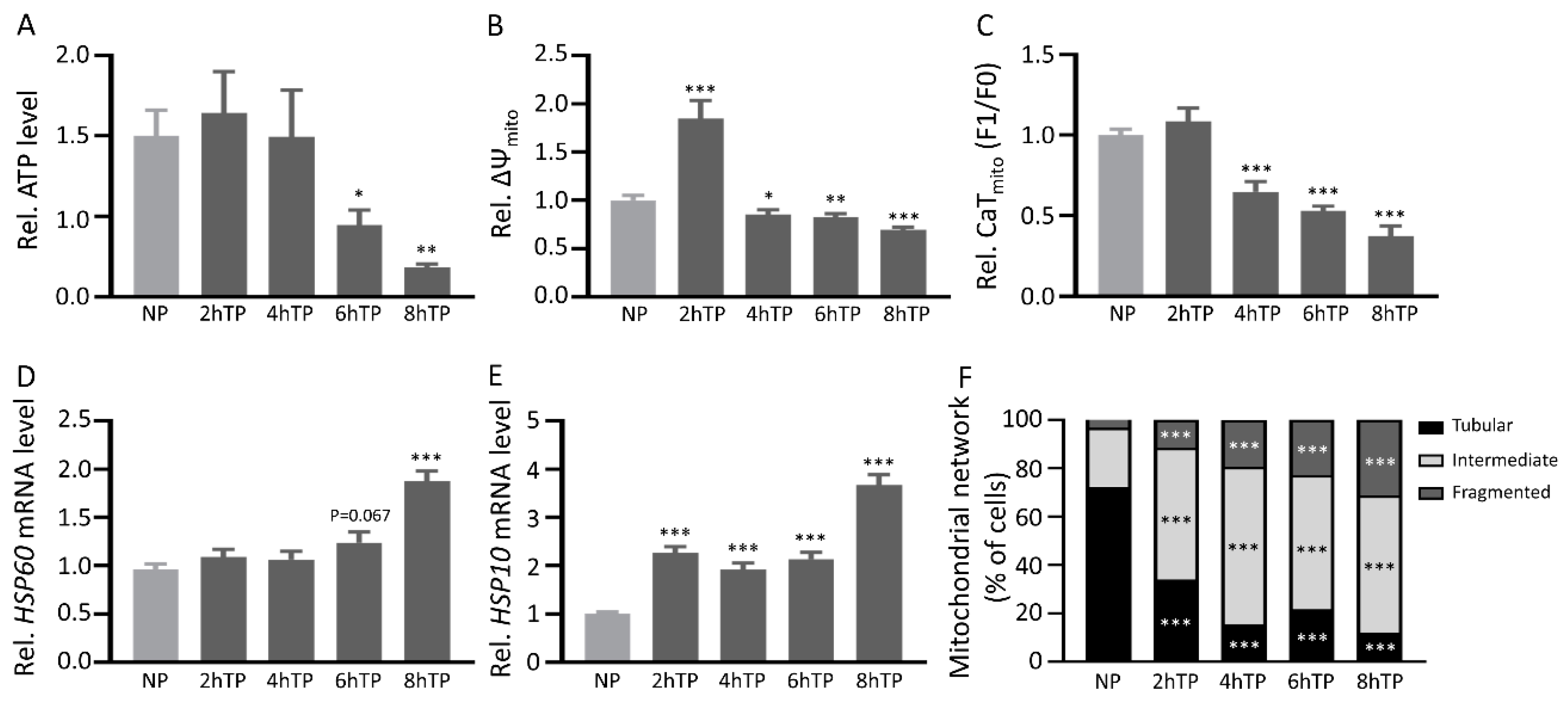
3.7. Mitochondrial DNA Is Released from HL-1 Atrial Cardiomyocytes upon Tachypacing and Is Associated with Mitochondrial Damage
4. Discussion
4.1. Current Biomarkers in AF
4.2. Clinical Implications of cfc-mtDNA in AF
4.3. The Origin of cfc-mtDNA in Serum
4.4. Cfc-mtDNA and Remodeling in AF
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heijman, J.; Guichard, J.B.; Dobrev, D.; Nattel, S. Translational Challenges in Atrial Fibrillation. Circ. Res. 2018, 122, 752–773. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Europace 2016, 18, 1609–1678. [Google Scholar] [CrossRef] [PubMed]
- Zoni-Berisso, M.; Lercari, F.; Carazza, T.; Domenicucci, S. Epidemiology of atrial fibrillation: European perspective. Clin. Epidemiol. 2014, 6, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Tadros, R.; Ton, A.T.; Fiset, C.; Nattel, S. Sex differences in cardiac electrophysiology and clinical arrhythmias: Epidemiology, therapeutics, and mechanisms. Can. J. Cardiol. 2014, 30, 783–792. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef]
- O’Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E., Jr.; Chung, M.K.; de Lemos, J.A.; Ettinger, S.M.; Fang, J.C.; Fesmire, F.M.; Franklin, B.A.; et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 127, e362–e425. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kim, G.; Tak, W.Y.; Jang, S.Y.; Kweon, Y.O.; Park, J.G.; Lee, H.W.; Han, Y.S.; Chun, J.M.; Park, S.Y.; et al. Circulating exosomal non-coding RNAs as prognostic biomarkers in human hepatocellular carcinoma. Int. J. Cancer 2018. [Google Scholar] [CrossRef]
- Kamei, N.; Yamashita, M.; Nishizaki, Y.; Yanagisawa, N.; Nojiri, S.; Tanaka, K.; Yamashita, Y.; Shibata, T.; Murakoshi, M.; Suzuki, Y.; et al. Association between circulating tumor necrosis factor-related biomarkers and estimated glomerular filtration rate in type 2 diabetes. Sci. Rep. 2018, 8, 15302. [Google Scholar] [CrossRef]
- Chua, W.; Purmah, Y.; Cardoso, V.R.; Gkoutos, G.V.; Tull, S.P.; Neculau, G.; Thomas, M.R.; Kotecha, D.; Lip, G.Y.H.; Kirchhof, P.; et al. Data-driven discovery and validation of circulating blood-based biomarkers associated with prevalent atrial fibrillation. Eur. Heart J. 2019, 40, 1268–1276. [Google Scholar] [CrossRef]
- Go, A.S.; Fang, M.C.; Udaltsova, N.; Chang, Y.; Pomernacki, N.K.; Borowsky, L.; Singer, D.E.; Investigators, A.S. Impact of proteinuria and glomerular filtration rate on risk of thromboembolism in atrial fibrillation: The anticoagulation and risk factors in atrial fibrillation (ATRIA) study. Circulation 2009, 119, 1363–1369. [Google Scholar] [CrossRef]
- Hijazi, Z.; Aulin, J.; Andersson, U.; Alexander, J.H.; Gersh, B.; Granger, C.B.; Hanna, M.; Horowitz, J.; Hylek, E.M.; Lopes, R.D.; et al. Biomarkers of inflammation and risk of cardiovascular events in anticoagulated patients with atrial fibrillation. Heart 2016, 102, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Cicchillitti, L.; Corrado, G.; De Angeli, M.; Mancini, E.; Baiocco, E.; Patrizi, L.; Zampa, A.; Merola, R.; Martayan, A.; Conti, L.; et al. Circulating cell-free DNA content as blood based biomarker in endometrial cancer. Oncotarget 2017, 8, 115230–115243. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hann, H.W.; Wan, S.; Hann, R.S.; Wang, C.; Lai, Y.; Ye, X.; Evans, A.; Myers, R.E.; Ye, Z.; et al. Cell-free circulating mitochondrial DNA content and risk of hepatocellular carcinoma in patients with chronic HBV infection. Sci. Rep. 2016, 6, 23992. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.T.; Huy, N.T.; Murao, L.A.; Lan, N.T.; Thuy, T.T.; Tuan, H.M.; Nga, C.T.; Tuong, V.V.; Dat, T.V.; Kikuchi, M.; et al. Elevated levels of cell-free circulating DNA in patients with acute dengue virus infection. PLoS ONE 2011, 6, e25969. [Google Scholar] [CrossRef]
- Arnalich, F.; Codoceo, R.; Lopez-Collazo, E.; Montiel, C. Circulating cell-free mitochondrial DNA: A better early prognostic marker in patients with out-of-hospital cardiac arrest. Resuscitation 2012, 83, e162–e163. [Google Scholar] [CrossRef]
- Lee, H.K.; Song, J.H.; Shin, C.S.; Park, D.J.; Park, K.S.; Lee, K.U.; Koh, C.S. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res. Clin. Pract. 1998, 42, 161–167. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef]
- Sandler, N.; Kaczmarek, E.; Itagaki, K.; Zheng, Y.; Otterbein, L.; Khabbaz, K.; Liu, D.; Senthilnathan, V.; Gruen, R.L.; Hauser, C.J. Mitochondrial DAMPs Are Released During Cardiopulmonary Bypass Surgery and Are Associated With Postoperative Atrial Fibrillation. Heart Lung Circ. 2018, 27, 122–129. [Google Scholar] [CrossRef]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.D.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef]
- Wiersma, M.; van Marion, D.M.S.; Wust, R.C.I.; Houtkooper, R.H.; Zhang, D.; Groot, N.M.S.; Henning, R.H.; Brundel, B. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef]
- Lindqvist, D.; Wolkowitz, O.M.; Picard, M.; Ohlsson, L.; Bersani, F.S.; Fernstrom, J.; Westrin, A.; Hough, C.M.; Lin, J.; Reus, V.I.; et al. Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacology 2018, 43, 1557–1564. [Google Scholar] [CrossRef]
- Lanters, E.A.; van Marion, D.M.; Kik, C.; Steen, H.; Bogers, A.J.; Allessie, M.A.; Brundel, B.J.; de Groot, N.M. HALT & REVERSE: Hsf1 activators lower cardiomyocyt damage; towards a novel approach to REVERSE atrial fibrillation. J. Transl. Med. 2015, 13, 347. [Google Scholar] [CrossRef]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Sarkar, M.; Das, S.; Bandyopadhaya, A.; Ray, K.; Chaudhuri, K. Upregulation of human mitochondrial NADH dehydrogenase subunit 5 in intestinal epithelial cells is modulated by Vibrio cholerae pathogenesis. FEBS Lett. 2005, 579, 3449–3460. [Google Scholar] [CrossRef]
- Armstrong, L.; Tilgner, K.; Saretzki, G.; Atkinson, S.P.; Stojkovic, M.; Moreno, R.; Przyborski, S.; Lako, M. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 2010, 28, 661–673. [Google Scholar] [CrossRef]
- Salehi, M.H.; Kamalidehghan, B.; Houshmand, M.; Yong Meng, G.; Sadeghizadeh, M.; Aryani, O.; Nafissi, S. Gene expression profiling of mitochondrial oxidative phosphorylation (OXPHOS) complex I in Friedreich ataxia (FRDA) patients. PLoS ONE 2014, 9, e94069. [Google Scholar] [CrossRef]
- Diez-Juan, A.; Rubio, C.; Marin, C.; Martinez, S.; Al-Asmar, N.; Riboldi, M.; Diaz-Gimeno, P.; Valbuena, D.; Simon, C. Mitochondrial DNA content as a viability score in human euploid embryos: Less is better. Fertil. Steril. 2015, 104, 534–541.e1. [Google Scholar] [CrossRef]
- Lumley, T.; Diehr, P.; Emerson, S.; Chen, L. The importance of the normality assumption in large public health data sets. Annu. Rev. Public Health 2002, 23, 151–169. [Google Scholar] [CrossRef]
- Kerr, C.R.; Humphries, K. Gender-related differences in atrial fibrillation. J. Am. Coll. Cardiol. 2005, 46, 1307–1308. [Google Scholar] [CrossRef]
- Hamelin, C.; Cornut, E.; Poirier, F.; Pons, S.; Beaulieu, C.; Charrier, J.P.; Haidous, H.; Cotte, E.; Lambert, C.; Piard, F.; et al. Identification and verification of heat shock protein 60 as a potential serum marker for colorectal cancer. FEBS J. 2011, 278, 4845–4859. [Google Scholar] [CrossRef]
- Bonanad, C.; Nunez, J.; Sanchis, J.; Bodi, V.; Chaustre, F.; Chillet, M.; Minana, G.; Forteza, M.J.; Palau, P.; Nunez, E.; et al. Serum heat shock protein 60 in acute heart failure: A new biomarker? Congest. Heart Fail. 2013, 19, 6–10. [Google Scholar] [CrossRef]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.Y.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B. Endoplasmic Reticulum Stress Is Associated With Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Li, J.; Liu, J.; Baks-Te Bulte, L.; Wiersma, M.; Malik, N.U.; van Marion, D.M.S.; Tolouee, M.; Hoogstra-Berends, F.; et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD(+) depletion in experimental atrial fibrillation. Nat. Commun. 2019, 10, 1307. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, C.T.; Qi, X.; Meijering, R.A.; Hoogstra-Berends, F.; Tadevosyan, A.; Cubukcuoglu Deniz, G.; Durdu, S.; Akar, A.R.; Sibon, O.C.; et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef]
- Michelena, H.I.; Powell, B.D.; Brady, P.A.; Friedman, P.A.; Ezekowitz, M.D. Gender in atrial fibrillation: Ten years later. Gend. Med. 2010, 7, 206–217. [Google Scholar] [CrossRef]
- Volgman, A.S.; Manankil, M.F.; Mookherjee, D.; Trohman, R.G. Women with atrial fibrillation: Greater risk, less attention. Gend. Med. 2009, 6, 419–432. [Google Scholar] [CrossRef]
- Rodriguez, I.; Kilborn, M.J.; Liu, X.K.; Pezzullo, J.C.; Woosley, R.L. Drug-induced QT prolongation in women during the menstrual cycle. JAMA 2001, 285, 1322–1326. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- Baysa, A.; Fedorov, A.; Kondratov, K.; Ruusalepp, A.; Minasian, S.; Galagudza, M.; Popov, M.; Kurapeev, D.; Yakovlev, A.; Valen, G.; et al. Release of Mitochondrial and Nuclear DNA During On-Pump Heart Surgery: Kinetics and Relation to Extracellular Vesicles. J. Cardiovasc. Transl. Res. 2019, 12, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Ausma, J.; Litjens, N.; Lenders, M.H.; Duimel, H.; Mast, F.; Wouters, L.; Ramaekers, F.; Allessie, M.; Borgers, M. Time course of atrial fibrillation-induced cellular structural remodeling in atria of the goat. J. Mol. Cell. Cardiol. 2001, 33, 2083–2094. [Google Scholar] [CrossRef]
- Brundel, B.J.; Henning, R.H.; Ke, L.; van Gelder, I.C.; Crijns, H.J.; Kampinga, H.H. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell. Cardiol. 2006, 41, 555–562. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Mouse COX1 | GCCCCAGATATAGCATTCCC | GTTCATCCTGTTCCTGCTCC |
| Mouse ND1 | AAACTATGTTCTCCGCCCCAA | TGGAGTCAGTGCATTTTGGC |
| Mouse 18S rRNA | TAGAGGGACAAGTGGCGTTC | CGCTGAGCCAGTCAGTGT |
| Human COX3 | ATGACCCACCAATCACATGC | ATCACATGGCTAGGCCGGAG |
| Human ND1 | ATACCCATGGCCAACCTCCT | GGGCCTTTGCGTAGTTGTAT |
| Human ND2 [26] | TAAAACTAGGAATAGCCCCC | TTGAGTAGTAGGAATGCGGT |
| Human ND3 [26] | CACAACTCAACGGCTACATA | TTGTAGTCACTCATAGGCCA |
| Human ND4L [26] | AGCATTTACCATCTCACTTCT | GCATTGGAGTAGGTTTAGGTT |
| Human ND4 [26] | TCTTCTTCGAAACCACACTT | AAGTACTATTGACCCAGCGA |
| Human ND5 [27] | ACATCTGTACCCACGCCTTC | CAGGGAGGTAGCGATCAGAG |
| Human ND6 [28] | GTAGGATTGGTGCTGTGG | GGATCCTCCCGAATCAAC |
| Human ATP8 [29] | CTAAAAATATTAAACACAAACTACCACCTACCTC | GTTCATTTTGGTTCTCAGGGTTTGTTATAA |
| Human CYB [27] | ACATCGGCATTATCCTCCTG | GTGTGAGGGTGGGACTGTCT |
| Human 18S rRNA | AGAAACGGCTACCACATCCA | CCCTCCAATGGATCCTCGTT |
| C | SR | PAF | PeAF | LS-PeAF | |
|---|---|---|---|---|---|
| N | 84 | 59 | 100 | 116 | 20 |
| Sex | |||||
| Male (N, %) | 44 (52.4) | 47 (79.7) ** | 73 (73.0) * | 87 (75.0) ** | 16 (80.0) * |
| Female (N, %) | 40 (47.6) | 12 (20.3) ** | 27 (27.0) * | 29 (25.0) ** | 4 (20.0) * |
| Age (mean ± SD) | 50 ± 16 | 69 ± 11 *** | 65 ± 11 *** | 65 ± 11 *** | 71 ± 9 *** |
| Underlying heart disease, test (N, %) | |||||
| WPW | 9 (10.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| PVC | 32 (38.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Ajmaline | 43 (51.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| CAD | 0 (0.0) | 50 (84.7) | 7 (7.0) | 7 (6.0) | 5 (25.0) |
| AVD | 0 (0.0) | 14 (23.7) | 7 (7.0) | 8 (6.9) | 6 (30.0) |
| MVD | 0 (0.0) | 8 (13.6) | 4 (4.0) | 12 (10.3) | 4 (20.0) |
| Type procedure | |||||
| Cardiac surgery | 0 (0.0) | 59 (100.0) | 15 (15.0) | 27 (23.3) | 15 (75.0) |
| PVI | 0 (0.0) | 0 (0.0) | 73 (73.0) | 21 (18.1) | 2 (10.0) |
| ECV | 0 (0.0) | 0 (0.0) | 12 (12.0) | 68 (58.6) | 3 (15.0) |
| Duration of AF | - | - | 97 ± 67 | 86 ± 62 | 159 ± 96 |
| (mean±SD (months)) | |||||
| LA dilatation (> 45 mm, %) | 2 (2.4) | 13 (22.0) *** | 32 (32.0) *** | 52 (44.8) *** | 13 (65.0) *** |
| LVF (N, %) | |||||
| Normal | 68 (81.0) | 44 (74.6) | 66 (66.0) | 70 (60.3) ** | 12 (60.0) |
| Mild impairment | 9 (10.7) | 14 (23.7) | 11 (11.0) | 29 (25.0) * | 7 (35.0) * |
| Moderate impairment | 2 (2.4) | 1 (1.7) | 4 (4.0) | 14 (12.1) | 1 (5.0) |
| Severe impairment | 3 (3.6) | 0 (0.0) | 1 (1.0) | 3 (2.6) | 0 (0.0) |
| Medication (N, %) | |||||
| ACE inhibitor | 23 (27.4) | 40 (67.8) | 47 (47.0) | 57 (49.1) | 16 (80.0) |
| Statin | 14 (16.7) | 44 (74.6) | 40 (40.0) | 40 (34.5) | 15 (75.0) |
| Type I AAD | 6 (7.1) | 1 (1.7) | 33 (33.0) | 13 (11.2) | 1 (5.0) |
| Type II AAD | 26 (31.0) | 40 (67.8) | 45 (45.0) | 63 (54.3) | 14 (70.0) |
| Type III AAD | 6 (7.1) | 0 (0.0) | 45 (45.0) | 49 (42.2) | 3 (15.0) |
| Type IV AAD | 4 (4.8) | 3 (5.1) | 4 (4.0) | 8 (6.9) | 1 (5.0) |
| Digoxin | 1 (1.2) | 0 (0.0) | 7 (7.0) | 21 (18.1) | 5 (25.0) |
| Hypertension (N, %) | 21 (25.0) | 38 (64.4) *** | 52 (52.0) *** | 59 (50.9) *** | 11 (55.0) * |
| Diabetes Mellitus (N, %) | 5 (6.0) | 17 (28.8) *** | 11 (11.0) | 15 (12.9) | 6 (30.0) ** |
| BMI (N, %) | |||||
| Underweight (< 18.50) | 1 (1.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Normal (18.5–25) | 45 (53.6) | 13 (22.0) *** | 34 (34.0) * | 30 (25.9) *** | 3 (15.0) ** |
| Overweight (25–30) | 29 (34.5) | 26 (44.1) | 46 (46.0) | 52 (44.8) | 10 (50.0) |
| Obese class I (30–35) | 7 (8.3) | 17 (28.8) ** | 17 (17.0) | 23 (19.8) | 6 (30.0) * |
| Obese class II (35–40) | 2 (2.4) | 3 (5.1) | 3 (3.0) | 10 (8.6) | 0 (0.0) |
| Obese class III (> 40) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.9) | 1 (5.0) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiersma, M.; van Marion, D.M.S.; Bouman, E.J.; Li, J.; Zhang, D.; Ramos, K.S.; Lanters, E.A.H.; de Groot, N.M.S.; Brundel, B.J.J.M. Cell-Free Circulating Mitochondrial DNA: A Potential Blood-Based Marker for Atrial Fibrillation. Cells 2020, 9, 1159. https://doi.org/10.3390/cells9051159
Wiersma M, van Marion DMS, Bouman EJ, Li J, Zhang D, Ramos KS, Lanters EAH, de Groot NMS, Brundel BJJM. Cell-Free Circulating Mitochondrial DNA: A Potential Blood-Based Marker for Atrial Fibrillation. Cells. 2020; 9(5):1159. https://doi.org/10.3390/cells9051159
Chicago/Turabian StyleWiersma, Marit, Denise M.S. van Marion, Emma J. Bouman, Jin Li, Deli Zhang, Kennedy S. Ramos, Eva A.H. Lanters, Natasja M.S. de Groot, and Bianca J.J.M. Brundel. 2020. "Cell-Free Circulating Mitochondrial DNA: A Potential Blood-Based Marker for Atrial Fibrillation" Cells 9, no. 5: 1159. https://doi.org/10.3390/cells9051159
APA StyleWiersma, M., van Marion, D. M. S., Bouman, E. J., Li, J., Zhang, D., Ramos, K. S., Lanters, E. A. H., de Groot, N. M. S., & Brundel, B. J. J. M. (2020). Cell-Free Circulating Mitochondrial DNA: A Potential Blood-Based Marker for Atrial Fibrillation. Cells, 9(5), 1159. https://doi.org/10.3390/cells9051159