Yeast-Based Genetic Interaction Analysis of Human Kinome



Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Strains, Media, Plasmids and Virus
2.2. Yeast Transformation and Spotting Assays
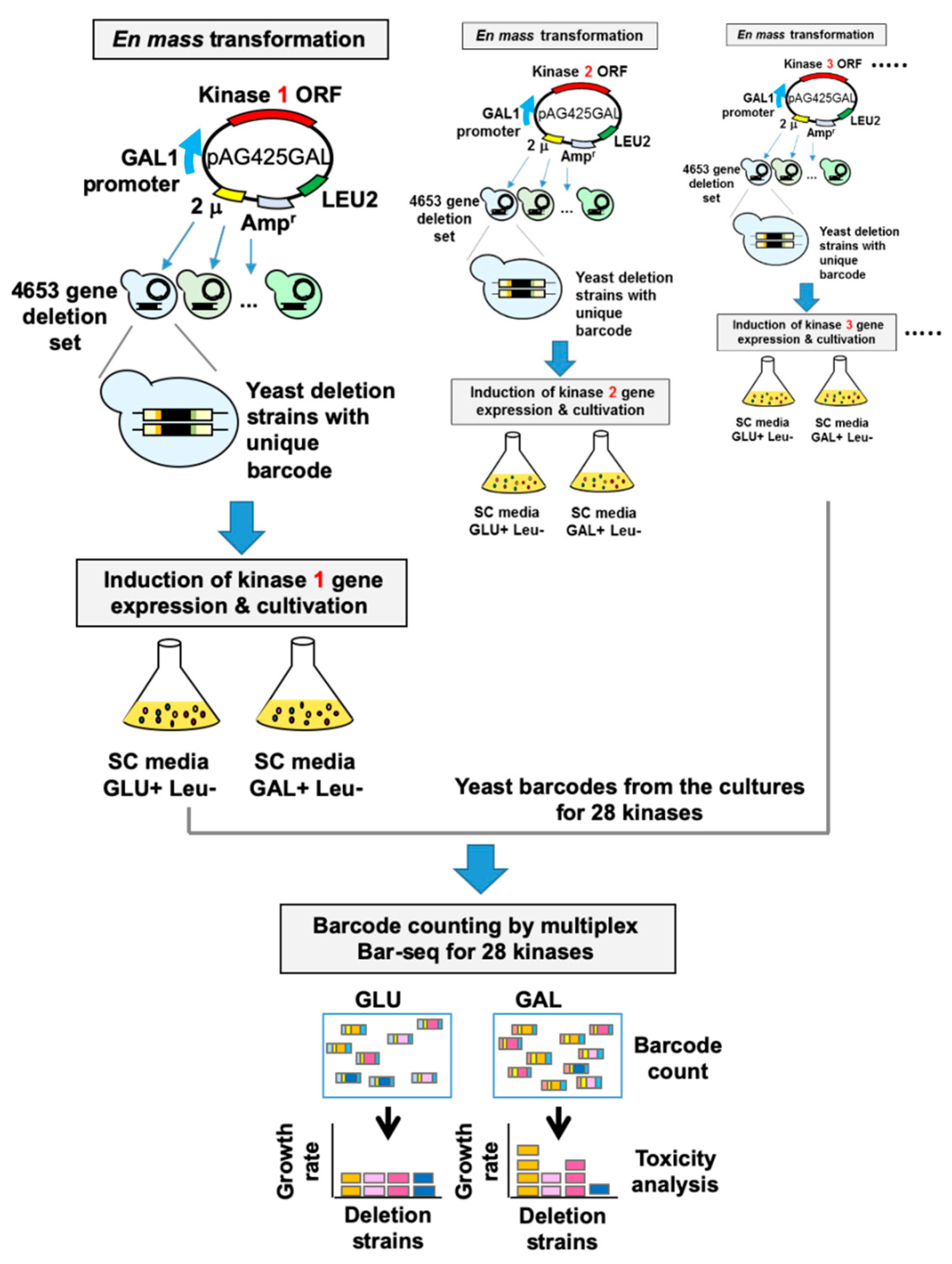
2.3. Human–Yeast Genetic Interaction Screen
2.4. Analysis of Illumina Sequencing
2.5. Bioinformatics Analysis and Network Construction
2.6. Availability of Data and Materials
3. Results
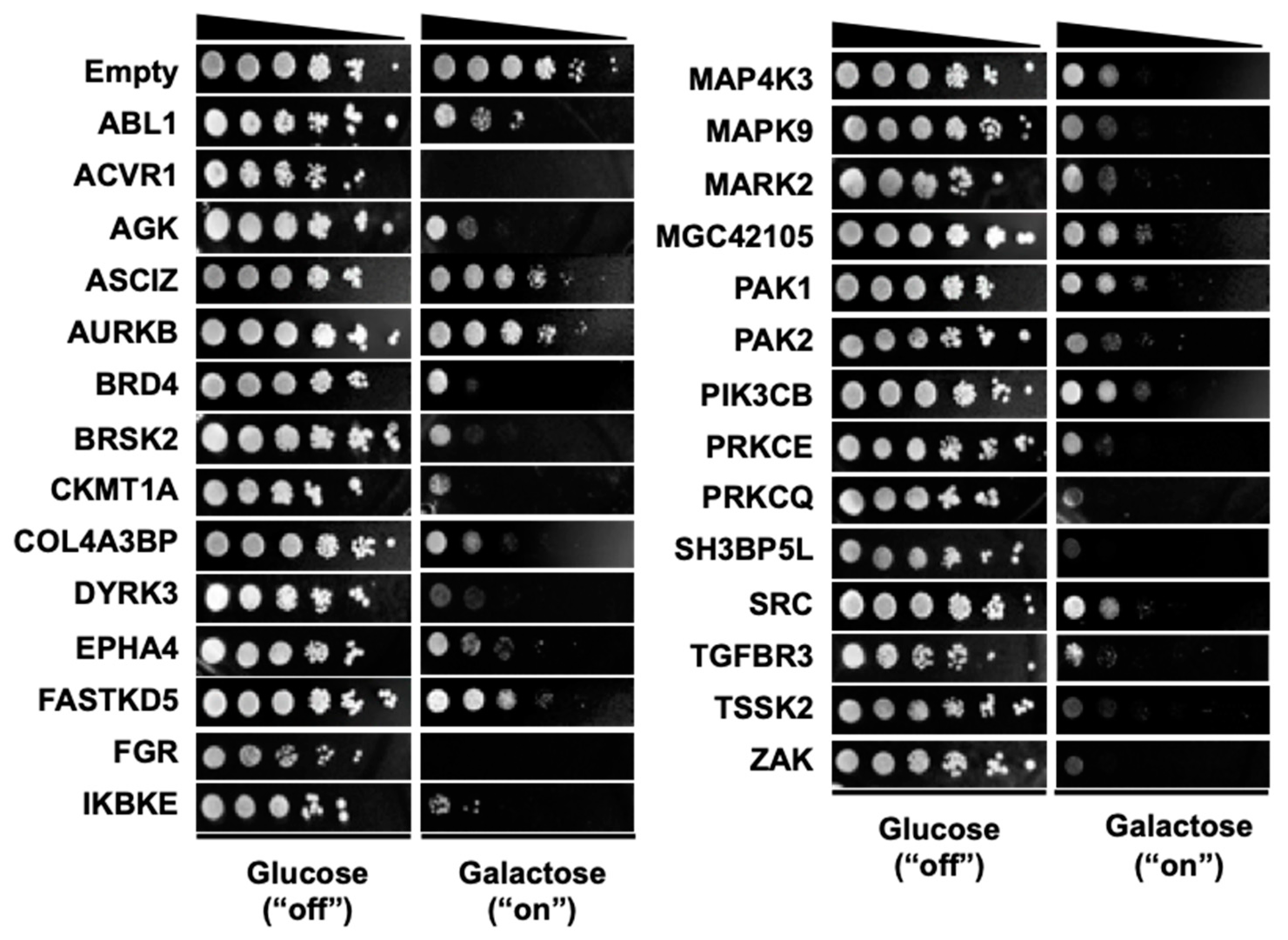
3.1. Selection of 28 Kinase Genes that are Toxic when Overexpressed in Yeast
3.2. High-Throughput Identification of Human Kinase–Yeast Genetic Interactions
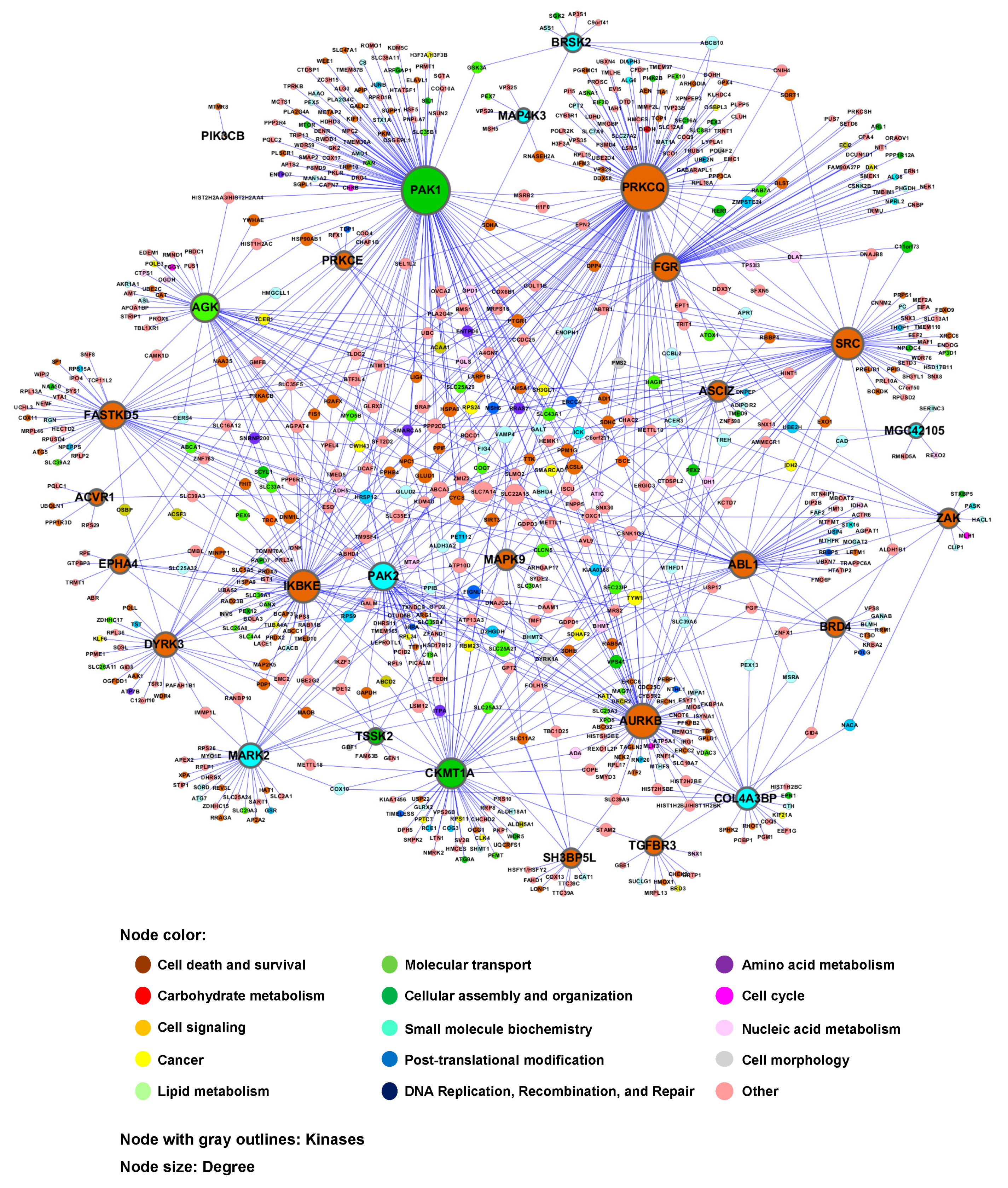
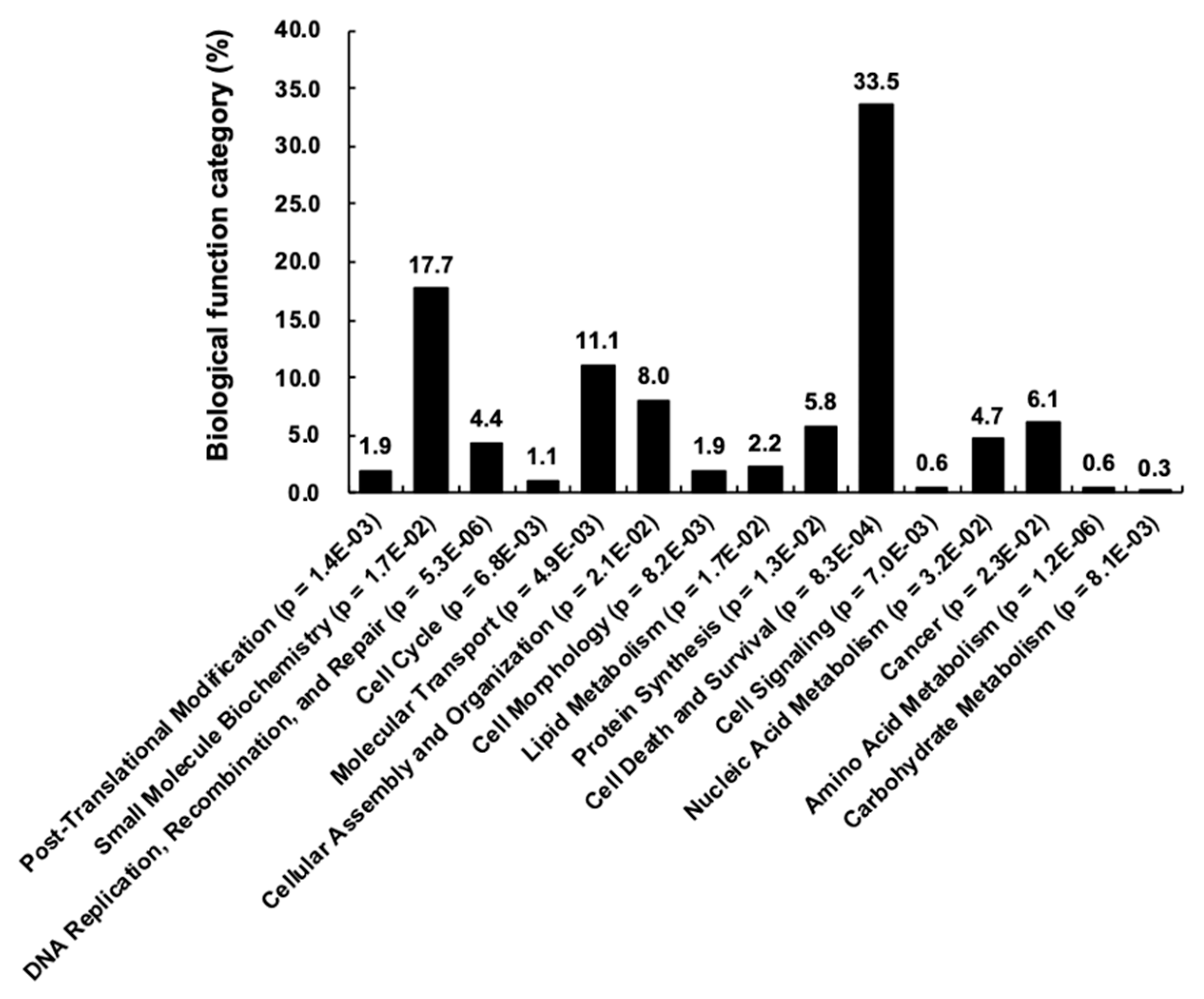
3.3. Construction of Human Kinase Genetic Interaction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gunosewoyo, H.; Yu, L.; Munoz, L.; Kassiou, M. Kinase targets in CNS drug discovery. Future Med. Chem. 2017, 9, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Zhang, J.; Fabbro, D.; Schioth, H.B. Advances in kinase targeting: Current clinical use and clinical trials. Trends Pharmacol. Sci. 2014, 35, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Colinge, J.; Cesar-Razquin, A.; Huber, K.; Breitwieser, F.P.; Majek, P.; Superti-Furga, G. Building and exploring an integrated human kinase network: Global organization and medical entry points. J. Proteomics 2014, 107, 113–127. [Google Scholar] [CrossRef]
- Sugiyama, N.; Imamura, H.; Ishihama, Y. Large-scale Discovery of Substrates of the Human Kinome. Sci. Rep. 2019, 9, 10503. [Google Scholar] [CrossRef] [PubMed]
- Jenardhanan, P.; Panneerselvam, M.; Mathur, P.P. Targeting Kinase Interaction Networks: A New Paradigm in PPI Based Design of Kinase Inhibitors. Curr. Top. Med. Chem. 2019, 19, 467–485. [Google Scholar] [CrossRef]
- Lun, X.K.; Szklarczyk, D.; Gabor, A.; Dobberstein, N.; Zanotelli, V.R.T.; Saez-Rodriguez, J.; von Mering, C.; Bodenmiller, B. Analysis of the Human Kinome and Phosphatome by Mass Cytometry Reveals Overexpression-Induced Effects on Cancer-Related Signaling. Mol. Cell 2019, 74, 1086–1102.e1085. [Google Scholar] [CrossRef]
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for 21st Century biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef]
- Costanzo, M.; VanderSluis, B.; Koch, E.N.; Baryshnikova, A.; Pons, C.; Tan, G.; Wang, W.; Usaj, M.; Hanchard, J.; Lee, S.D.; et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353, aaf1420. [Google Scholar] [CrossRef]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.; Toufighi, K.; Mostafavi, S.; et al. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, F.; Guidetti, P.; Nguyen, Q.; Bennett, S.C.; Muchowski, P.J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005, 37, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D. Beer and bread to brains and beyond: Can yeast cells teach us about neurodegenerative disease? Neurosignals 2008, 16, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Chung, A.Y.; Yachie, N.; Seo, M.; Jeon, H.; Nam, Y.; Seo, Y.; Kim, E.; Zhong, Q.; Vidal, M.; et al. Yeast genetic interaction screen of human genes associated with amyotrophic lateral sclerosis: Identification of MAP2K5 kinase as a potential drug target. Genome Res. 2017, 27, 1487–1500. [Google Scholar] [CrossRef]
- Alberti, S.; Gitler, A.D.; Lindquist, S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 2007, 24, 913–919. [Google Scholar] [CrossRef]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shen, T.; Dong, B.; Creighton, C.J.; Meng, Y.; Zhou, W.; Shi, Q.; Zhou, H.; Zhang, Y.; Moore, D.D.; et al. MAPK4 overexpression promotes tumor progression via noncanonical activation of AKT/mTOR signaling. J. Clin. Invest. 2019, 129, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Stark, G.R. FER tyrosine kinase (FER) overexpression mediates resistance to quinacrine through EGF-dependent activation of NF-kappaB. Proc. Natl. Acad. Sci. USA 2011, 108, 7968–7973. [Google Scholar] [CrossRef] [PubMed]
- Le Scolan, E.; Pchejetski, D.; Banno, Y.; Denis, N.; Mayeux, P.; Vainchenker, W.; Levade, T.; Moreau-Gachelin, F. Overexpression of sphingosine kinase 1 is an oncogenic event in erythroleukemic progression. Blood 2005, 106, 1808–1816. [Google Scholar] [CrossRef][Green Version]
- Giaever, G.; Shoemaker, D.D.; Jones, T.W.; Liang, H.; Winzeler, E.A.; Astromoff, A.; Davis, R.W. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat. Genet. 1999, 21, 278–283. [Google Scholar] [CrossRef]
- Smith, A.M.; Durbic, T.; Kittanakom, S.; Giaever, G.; Nislow, C. Barcode sequencing for understanding drug-gene interactions. Methods Mol. Biol. 2012, 910, 55–69. [Google Scholar]
- Smith, A.M.; Heisler, L.E.; Mellor, J.; Kaper, F.; Thompson, M.J.; Chee, M.; Roth, F.P.; Giaever, G.; Nislow, C. Quantitative phenotyping via deep barcode sequencing. Genome Res. 2009, 19, 1836–1842. [Google Scholar] [CrossRef]
- Smith, A.M.; Heisler, L.E.; St Onge, R.P.; Farias-Hesson, E.; Wallace, I.M.; Bodeau, J.; Harris, A.N.; Perry, K.M.; Giaever, G.; Pourmand, N.; et al. Highly-multiplexed barcode sequencing: An efficient method for parallel analysis of pooled samples. Nucleic Acids Res. 2010, 38, e142. [Google Scholar] [CrossRef]
- O’Brien, K.P.; Remm, M.; Sonnhammer, E.L. Inparanoid: A comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005, 33, D476–D480. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Timson, D.J. Galactose metabolism in Saccharomyces cerevisiae. Dyn. Biochem. Process. Biotechnol. Mol. Biol. 2007, 11, 63–73. [Google Scholar]
- Cruz, D.F.; Farinha, C.M.; Swiatecka-Urban, A. Unraveling the Function of Lemur Tyrosine Kinase 2 Network. Front. Pharmacol. 2019, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.X.; An, S.; Yang, Y.; Liu, Y.; Hao, Q.; Tang, T.; Xu, T.R. Emerging role of the Jun N-terminal kinase interactome in human health. Cell Biol. Int. 2018, 42, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Eagleson, K.L.; Xie, Z.; Levitt, P. The Pleiotropic MET Receptor Network: Circuit Development and the Neural-Medical Interface of Autism. Biol. Psychiatry 2017, 81, 424–433. [Google Scholar] [CrossRef]
- Porras, P.; Duesbury, M.; Fabregat, A.; Ueffing, M.; Orchard, S.; Gloeckner, C.J.; Hermjakob, H. A visual review of the interactome of LRRK2: Using deep-curated molecular interaction data to represent biology. Proteomics 2015, 15, 1390–1404. [Google Scholar] [CrossRef][Green Version]
- Bialik, S.; Kimchi, A. The DAP-kinase interactome. Apoptosis 2014, 19, 316–328. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Gene Symbol | UniProt ID | Common Name | Note a |
|---|---|---|---|---|
| 1 | ABL1 | P00519 | c-Abl oncogene 1, non-receptor tyrosine kinase | Tyr |
| 2 | ACVR1 | Q04771 | Activin receptor type-1 | Ser/Thr |
| 3 | AGK | Q53H12 | Acylglycerol kinase, mitochondrial | Lipid |
| 4 | ASCIZ | D3DUL0 | ATM/ATR-Substrate Chk2-Interacting Zn2+-finger protein, isoform CRA_b | Putative kinase |
| 5 | AURKB | Q96GD4 | Aurora kinase B | Ser/Thr |
| 6 | BRD4 | O60885 | Bromodomain-containing protein 4 | Putative kinase |
| 7 | BRSK2 | Q8IWQ3 | BR serine/threonine kinase 2 | Ser/Thr |
| 8 | CKMT1A | P12532 | Creatine kinase U-type, mitochondrial | Creatine |
| 9 | COL4A3BP | Q9Y5P4 | Collagen type IV alpha-3-binding protein | Putative kinase |
| 10 | DYRK3 | O43781 | Dual specificity tyrosine-phosphorylation-regulated kinase 3 | Ser/Thr, Tyr |
| 11 | EPHA4 | P54764 | Ephrin type-A receptor 4 | Tyr |
| 12 | FASTKD5 | Q7L8L6 | FAST kinase domain-containing protein 5 | Putative kinase |
| 13 | FGR | P09769 | Feline Gardner-Rasheed sarcoma viral oncogene homolog | Tyr |
| 14 | IKBKE | Q14164 | Inhibitor of nuclear factor kappa-B kinase subunit epsilon | Ser/Thr |
| 15 | MAP4K3 | Q8IVH8 | Mitogen-activated protein kinase kinase kinase kinase 3 | Ser/Thr |
| 16 | MAPK9 | P45984 | Mitogen-activated protein kinase 9 | Ser/Thr |
| 17 | MARK2 | Q7KZI7 | MAP/microtubule affinity-regulating kinase 2 | Ser/Thr |
| 18 | MGC42105 | A0A024R049 | Uncharacterized protein | Putative kinase |
| 19 | PAK1 | Q13153 | p21 protein (Cdc42/Rac)-activated kinase 1 | Ser/Thr |
| 20 | PAK2 | Q13177 | p21 protein (Cdc42/Rac)-activated kinase 2 | Ser/Thr |
| 21 | PIK3CB | P42338 | Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit beta isoform | Lipid |
| 22 | PRKCE | Q02156 | Protein kinase C epsilon type | Ser/Thr |
| 23 | PRKCQ | Q04759 | Protein kinase C theta | Ser/Thr |
| 24 | SH3BP5L | Q7L8J4 | SH3 domain-binding protein 5-like | Putative kinase |
| 25 | SRC | P12931 | Proto-oncogene tyrosine-protein kinase | Tyr |
| 26 | TGFBR3 | Q03167 | Transforming growth factor beta receptor type 3 | Ser/Thr |
| 27 | TSSK2 | Q96PF2 | Testis-specific serine/threonine-protein kinase 2 | Ser/Thr |
| 28 | ZAK | Q9NYL2 | Mitogen-activated protein kinase kinase kinase 20 | Ser/Thr |
| Kinase | Toxicity Suppressors | ||
| Suppressor/tested | Consistency | Average | |
| IKBKE | 10/10 | 100% | 82.9% |
| MAPK9 | 6/10 | 60% | |
| PAK1 | 32/41 | 78% | |
| PAK2 | 13/17 | 76.5% | |
| PRKCQ | 10/10 | 100% | |
| Kinase | Non-Modifiers | ||
| Non-modifier/tested | Consistency | Average | |
| IKBKE | 0/5 | 0% | 33.3% |
| MAPK9 | 2/5 | 40% | |
| PAK1 | NT a | NT | |
| PAK2 | NT | NT | |
| PRKCQ | 3/5 | 60% | |
| Kinase | Toxicity Enhancers | ||
| Enhancer/tested | Consistency | Average | |
| IKBKE | 0/3 | 0% | 43.3% |
| MAPK9 | - b | - | |
| PAK1 | - | - | |
| PAK2 | 1/1 | 100% | |
| PRKCQ | 3/10 | 30% | |
| No. | Kinase | Z-Score (>1.96) a | Presence of Human Ortholog b |
|---|---|---|---|
| 1 | ABL1 | 124 | 41 |
| 2 | ACVR1 | 23 | 6 |
| 3 | AGK | 138 | 49 |
| 4 | ASCIZ | 60 | 18 |
| 5 | AURKB | 203 | 75 |
| 6 | BRD4 | 40 | 14 |
| 7 | BRSK2 | 27 | 9 |
| 8 | CKMT1A | 164 | 56 |
| 9 | COL4A3BP | 54 | 24 |
| 10 | DYRK3 | 120 | 33 |
| 11 | EPHA4 | 35 | 11 |
| 12 | FASTKD5 | 138 | 45 |
| 13 | FGR | 134 | 46 |
| 14 | IKBKE | 189 | 66 |
| 15 | MAP4K3 | 22 | 5 |
| 16 | MAPK9 | 45 | 17 |
| 17 | MARK2 | 103 | 32 |
| 18 | MGC42105 | 10 | 5 |
| 19 | PAK1 | 402 | 131 |
| 20 | PAK2 | 135 | 45 |
| 21 | PIK3CB | 14 | 1 |
| 22 | PRKCE | 31 | 9 |
| 23 | PRKCQ | 322 | 126 |
| 24 | SH3BP5L | 25 | 12 |
| 25 | SRC | 211 | 63 |
| 26 | TGFBR3 | 33 | 11 |
| 27 | TSSK2 | 23 | 6 |
| 28 | ZAK | 53 | 13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Seo, Y.; Jo, M.; Jeon, H.; Lee, W.-H.; Yachie, N.; Zhong, Q.; Vidal, M.; Roth, F.P.; Suk, K. Yeast-Based Genetic Interaction Analysis of Human Kinome. Cells 2020, 9, 1156. https://doi.org/10.3390/cells9051156
Kim J-H, Seo Y, Jo M, Jeon H, Lee W-H, Yachie N, Zhong Q, Vidal M, Roth FP, Suk K. Yeast-Based Genetic Interaction Analysis of Human Kinome. Cells. 2020; 9(5):1156. https://doi.org/10.3390/cells9051156
Chicago/Turabian StyleKim, Jae-Hong, Yeojin Seo, Myungjin Jo, Hyejin Jeon, Won-Ha Lee, Nozomu Yachie, Quan Zhong, Marc Vidal, Frederick P. Roth, and Kyoungho Suk. 2020. "Yeast-Based Genetic Interaction Analysis of Human Kinome" Cells 9, no. 5: 1156. https://doi.org/10.3390/cells9051156
APA StyleKim, J.-H., Seo, Y., Jo, M., Jeon, H., Lee, W.-H., Yachie, N., Zhong, Q., Vidal, M., Roth, F. P., & Suk, K. (2020). Yeast-Based Genetic Interaction Analysis of Human Kinome. Cells, 9(5), 1156. https://doi.org/10.3390/cells9051156